
Method Article
Uma plataforma de Metil-Seq de Rato para identificar alterações epigenéticas associadas à exposição ao estresse
Neste Artigo
Resumo
Aqui, descrevemos o protocolo e a implementação de metil-Seq, uma plataforma de epigenomic, usando um modelo do rato para identificar alterações epigenéticas associadas com exposição de estresse crônico. Os resultados demonstram que a plataforma de metil-Seq rato é capaz de detectar diferenças de metilação que surgem a partir da exposição de estresse em ratos.
Resumo
Como genomas de uma ampla variedade de animais se tornam disponíveis, há uma crescente necessidade de ferramentas que pode capturar alterações epigenéticas dinâmicas nestes modelos animais. O rato é um animal de modelo específico, onde uma ferramenta epigenética pode complementar muitos estudos farmacológicos e comportamentais para fornecer informação esclarecedora mecanicista. Para este fim, adaptamos o SureSelect capturar sistema alvo (referido como metil-Seq) para o rato, que pode avaliar os níveis de metilação do DNA através do genoma do rato. O design do rato alvo promotores, consoles de CpG, margens da ilha e regiões de GC-ricos de todos os genes RefSeq.
Para implementar a plataforma em um experimento de rato, ratos Sprague Dawley masculinos foram expostos ao estresse crônico de variável por 3 semanas, após o qual foram colhidas amostras de sangue para extração de DNA genômica. Metil-Seq bibliotecas foram construídas a partir das amostras de DNA de rato por cisalhamento, ligadura de adaptador, enriquecimento de alvo, conversão de bissulfito e multiplexação. As bibliotecas foram sequenciadas em uma plataforma de sequenciamento de última geração e as leituras sequenciais foram analisadas para identificar DMRs entre DNA de ratos estressados e átonos. DMRs candidato independente foram validadas por bissulfito pyrosequencing confirmar a robustez da plataforma.
Os resultados demonstram que a plataforma de metil-Seq de rato é uma útil ferramenta epigenética que pode capturar as alterações de metilação induzidas pela exposição ao estresse.
Introdução
Avanços no sequenciamento do elevado-throughput conduziram a uma riqueza de sequências genomic para modelo e organismos não-modelo. A disponibilidade de tais sequências facilitou a investigação em genética e genómica comparativa transcriptomics. Por exemplo, sequências genômicas disponíveis são altamente úteis para o alinhamento de dados de sequenciamento de ChIP-Seq experimentos que enriquecem o DNA com base em sua associação com modificações de histona1ou sequenciamento de bissulfito, que mede a metilação do DNA por detecção de uracil formado a partir de conversão de bissulfito de citosinas unmethylated2. No entanto, tem havido atrasos na implementação de plataformas de epigenomic que incorporam dados de sequenciamento genômico disponíveis na sua concepção, devido à falta de dados anotados de sequências reguladoras específicas que podem influenciar a função dos genes.
Em particular, metilação do DNA é uma das mais amplamente estudadas modificações epigenéticas no DNA que pode alavancar dados genômicos disponíveis para a construção de uma plataforma de methylomic. Um exemplo é uma plataforma baseada em array para o methylome humano3, que tem sido amplamente utilizado em diversas disciplinas de Oncologia a psiquiatria4,5. Infelizmente, plataformas semelhantes para modelos animais não-humanos são escassas, como não há praticamente nenhuma plataforma amplamente utilizado que aproveitou a sequência genômica em seu projeto inicial.
Um método comum para apreciar a paisagem de methylomic de modelos de animais não-humanos é reduzida representação bissulfito sequenciamento (RRBS)6. Esta abordagem supera o custo do sequenciamento do genoma inteiro bissulfito que, proporcionando uma paisagem de methylomic abrangente, fornece cobertura de leitura-profundidade inferior devido ao custo e informação funcional limitada em grandes áreas pobres em genes do genoma2 . RRBS envolve restrição digest e seleção de tamanho de DNA genômico de enriquecer para altamente rico em GC sequências como consoles de CpG que são comumente encontrados perto de promotores de genes e pensados para jogar um papel no gene Regulamento7. Enquanto o método RRBS tem sido usado em um número de importantes estudos, sua dependência de enzimas de restrição não é sem limitações e desafios notáveis. Por exemplo, o enriquecimento de sequências de GC-ricos em RRBS é totalmente dependente da presença de sequências específicas, reconhecida pela enzima de restrição e seleção de tamanho subsequente por eletroforese. Isto significa que quaisquer áreas genômicas que não contêm esses sites de restrição são excluídas durante a seleção do tamanho. Além disso, comparações entre espécies estão desafiando a menos que nos mesmos sítios de restrição estão presentes no mesmos Locus entre as diferentes espécies.
Uma abordagem para superar as limitações do RRBS é usar um método de enriquecimento que tira proveito da sequência genômica publicado no design da plataforma. A plataforma humana baseada em matriz usa sondas cartilha projetadas contra CpGs específico para alelo-específico (CG vs TG após a conversão de bissulfito) alvo recozimento e primer a extensão. Seu design reflete não só a sequência genômica humana disponível, mas regiões reguladoras verificados experimentalmente adquiridas de várias linhas de investigação, tais como ENCODE e ENSEMBL8. Apesar de sua ampla utilização em investigações de methylomic humana, uma plataforma semelhante não existe para os animais de modelo. Além disso, o formato baseado em matriz coloca restrição significativa na área da superfície disponível para a colocação da sonda. Nos últimos anos, têm-se esforços para combinar a alvo-especificidade conferida pela captura sonda design e o recurso de alta produtividade da próxima geração de sequenciamento. Tal um esforço resultou no sistema alvo baseada no sequenciamento de enriquecimento para o genoma do rato (mouse de metil-Seq), que foi usado para identificar diferenças específicas do cérebro ou induzida por glicocorticoide na metilação9,10. Plataformas semelhantes para outros modelo e não do modelo animais são necessários para facilitar a pesquisa de epigenomic nesses animais.
Aqui, demonstramos a implementação desta novela plataforma para realizar análise de methylomic no rato. O rato tem servido como um importante modelo animal em farmacologia, metabolismo, Neuroendocrinologia e comportamento. Por exemplo, há uma crescente necessidade de compreender os mecanismos subjacentes que dão origem a toxicidade de drogas, obesidade, estresse ou toxicodependência. Uma plataforma de alta produtividade capaz de captar mudanças methylomic associadas a essas condições iria aumentar a nossa compreensão dos mecanismos. Uma vez que o genoma do rato ainda carece de anotação para regiões reguladoras, nós incorporado promotores não-redundante, consoles de CpG, ilha margens11e identificado previamente sequências GC-rico no rato metil-Seq plataforma12.
Para avaliar o projeto bem sucedido e implementação da plataforma de SureSelect alvo enriquecimento (genericamente referido como metil-Seq) para o genoma do rato, utilizamos um modelo do rato de estresse crônico variável (CVS)13 para identificar diferencialmente metilados regiões entre animais átonas e tônicas. Nosso projeto da plataforma, protocolo e implementação podem ser útil para os investigadores que queiram conduzir uma investigação epigenética abrangente e imparcial sobre um organismo cuja sequência genômica já está disponível, mas continua a ser mal anotado.
Protocolo
Todos os experimentos foram concluídos em conformidade e em conformidade com todas as orientações regulamentares e institucionais relevantes, incluindo o Comité de uso no Johns Hopkins School of Medicine e institucional Cuidado Animal.
1. os animais
- Obter ratos Sprague-Dawley adolescentes masculinos em 4 semanas de idade. Os animais em gaiolas de rato de policarbonato em uma temperatura de casa- e a sala de controle de umidade em um 12h, 12h escuro ciclo de luz com luz início às 0600 h. fornecer os animais com ad libitum acesso à água.
- Permitem que ratos se aclimate por 1 semana reduzir o stress associado com transporte. Par-casa dos animais (N = 16), impede o estresse de isolamento, e em 5 semanas de idade, começar a variável crônica estresse regime (CVS) por 3 semanas.
2. crônico estresse variável
- Administre o regime CVS uma vez de manhã (9-11:00) e uma vez na parte da tarde (1 – 15:00) em horários irregulares para manter a rotina imprevisível. Incorpore a estressores suaves durante a noite. O CVS esquema inclui: 1) 3 h em um cilindro de contenção; 2) 10 min de natação; gaiola 3) 3 h de inclinação 4) 1h lento tremendo plataforma; e 5) 1 h na sala de frio de 4 ° C.
Nota: Overnight estressores incluem a exclusão social (5 por gaiola), isolamento social, roupa de cama molhada, restrição alimentar e luzes acesas. Uma agenda semanal típica do regime de estresse é fornecida na tabela 1.
3. endócrinos ensaios
-
Determinar os níveis de corticosterona (CORT) usando amostras de sangue (~ 50 mL) de cauda coletados ao mesmo tempo (09:00) duas vezes por semana durante todo o experimento, antes do regime de CVS para estabelecer os níveis de hormônio de linha de base (dia 0), uma vez durante o meio do CVS do semanal (dias 4,11 e 18), depois de CVS a cada 7 dias (dias 7 e 14) e na conclusão do CVS (dia 21). Colete amostras de sangue antes do regime de estresse diário.
- Recolha uma amostra de sangue do tronco final durante a eutanásia (dia 25) para extração de DNA genômica e RIA.
- Todas as amostras de sangue (600 x g, 4 ° C, 10 min.) para separar as células do sangue do plasma do centrifugador. Pipetar para fora do plasma (sobrenadante) e armazenar as amostras a-80 ° C.
- Descongelar e usar o plasma para determinar níveis CORT por radioimunoensaio (RIA). Certifique-se de que os níveis CORT 3 semana plasma são elevados nos animais forçados para verificar a robustez do regime de estresse.
4. comportamento
- Após o regime CVS (dias 23 – 24), avaliar cada animal para o comportamento de ansiedade sobre a elevada mais labirinto (EPM)14.
- Usando uma câmera de vídeo, registro dos animais sobre o aparelho EPM para 300 s e pontuação, o tempo gasto no centro, fechados braços e braços abertos.
5. projeto do rato metil-Seq
- Usando o UCSC Genome Browser, obter coordenadas genômicas não-redundante (rato Nov de 2004 rn4 montagem) para consoles de CpG e margens da ilha (± 1 kb flanqueando consoles de CpG), promotores (± 1 kb de cada TSS) de cada gene RefSeq e outras sequências que podem estar disponíveis em Literatura relevante.
Nota: O rato metil-Seq, adicionais sequências de GC-ricos de uma plataforma baseada em array metilação anterior foi adicionado12. Para regiões superiores a 5 kbps, regiões alternadas de 500 bps foram amostradas seguido de 1 kbps que foram ignorados. O projeto de metil-Seq rato final consiste em 111 Mbps, CpGs 2,3 milhões; e um tamanho médio da região de 594 bps. Alveja 228.800 únicos Locus. - Entra uma lista compilada de genômicas coordenadas em um software de desenho de captura alvo comercialmente disponíveis para o projeto sonda apropriado.
6. construção da biblioteca de metil-Seq rato de DNA genômico
Nota: Para eliminar os efeitos do lote, processar amostras múltiplas ao mesmo tempo e intensificar as mixagens mestre nesse sentido. Extrair DNA usando um kit de extração de DNA disponível comercialmente. Coluna ou precipitação-com base em ambos os métodos produzem DNA genômico de alta qualidade (relação de 260/280 ~ 1.8). Utilização de métodos baseados em fenol não são recomendados. Eluir ou resuspenda o DNA no buffer de baixo TE (10mm TE, 0,1 mM EDTA, pH 8.0).
- Preparação da amostra
Nota: Para cada passo usando DNA-ligando grânulos magnéticos, certifique-se os grânulos estão acostumados à temperatura ambiente pelo menos 30 min e bem misturados antes do uso.- DNA de cisalhamento
- Use um dados para determinar a concentração de DNA dupla-hélice inicial de cada amostra. Diluir > 1 µ g de gDNA de 50 µ l, com buffer de baixo TE (TE, 0,1 mM EDTA, pH 8.0 de 10 mM) em baixos tubos microcentrifuga de DNA-ligando.
- Shear amostras usando um sonicador isotérmico (10% ciclo de dever, intensidade 5, 200 ciclos por explosão, 6 ciclos de 60 s, Varrição de frequência, 4 ° C).
- Avalie a qualidade do DNA usando um sistema baseado em eletroforese que mede a quantidade e tamanho de DNA.
Nota: A quantidade de DNA recomendada é de 1 µ g ou 3 µ g. Se há limitado material começar, deve ser a menor quantidade de entrada > 500 ng, como montantes inferiores afectará negativamente a quantidade e a qualidade das bibliotecas geradas.
- Reparação DNA termina.
- Use o rato kit de metil-Seq para preparar a mistura final-reparo mestre no gelo. Adicionar 52 µ l de mistura para cada amostra e incubá-los em um termociclador sem tampa aquecida (20 ° C por 30 min, espera de 4 ° C).
Final-reparação Master Mix (por exemplo):
35,2 µ l de água
10 µ l de tampão de reparação final (10x)
1,6 µ l de dNTP Mix
1 μL de T4 DNA Polymerase
2 µ l de Klenow polimerase de DNA
2.2 µ l da quinase de polinucleotido T4 - Purifica amostras usando 180 µ l de grânulos magnéticos de DNA-ligando e 400 µ l de etanol a 70% recentemente preparada por amostra. Adicionar 180 µ l de grânulos para cada amostra e incubar durante 5 min à temperatura ambiente. Grânulos de Pelotas, remover o sobrenadante e ressuspender em 200 µ l de etanol 70%. Remover o etanol e repetir a lavagem uma vez.
- Use uma placa magnética para grânulos de pelotas e remover tanto etanol quanto possível. Seco em um 37 ° C heatblock para 3-5 min até que o sedimento do grânulo é completamente seca. Resuspenda em 44 µ l de água livre de nuclease e coletar aproximadamente 42 µ l do sobrenadante.
Ponto de parada: Após reparação DNA termina, amostras podem ser seladas e armazenadas a-20 ° C.
- Use o rato kit de metil-Seq para preparar a mistura final-reparo mestre no gelo. Adicionar 52 µ l de mistura para cada amostra e incubá-los em um termociclador sem tampa aquecida (20 ° C por 30 min, espera de 4 ° C).
- Adenilato a 3' termina.
- Prepare Adenylation Master Mix no gelo. Adicionar 9 mistura µ l de cada amostra e incubá-los em um termociclador sem tampa aquecida (37 ° C por 30 min, espera de 4 ° C).
Adenylation Master Mix (por exemplo):
5 µ l de tampão de Klenow
1 µ l de dATP
3 µ l de Klenow polimerase de DNA - Purifica amostras usando 90 µ l de grânulos magnéticos de DNA-ligando e 400 µ l de etanol a 70% recentemente preparada por amostra. Adicione 90 µ l dos grânulos de cada amostra e incubar durante 5 min à temperatura ambiente. Grânulos de Pelotas, remover o sobrenadante e ressuspender em 200 µ l de etanol 70%. Remover o etanol e repetir a lavagem uma vez.
- Use uma placa magnética para grânulos de pelotas e remover tanto etanol quanto possível. Seco em um 37 ° C heatblock para 3-5 min até que o sedimento do grânulo é completamente seca. Resuspenda em 35 µ l de água livre de nuclease e coletar aproximadamente 33,5 µ l do sobrenadante.
- Prepare Adenylation Master Mix no gelo. Adicionar 9 mistura µ l de cada amostra e incubá-los em um termociclador sem tampa aquecida (37 ° C por 30 min, espera de 4 ° C).
- Ligar o adaptador metilado.
- Prepare a ligadura Master Mix no gelo e adicionar 16,5 µ l de mistura para cada amostra. Incube em um termociclador sem tampa aquecida (20 ° C por 15 min, espera de 4 ° C).
Ligadura da Master Mix (por exemplo):
2,5 µ l de água
2,5 µ l de metil-Seq metilado adaptador
10 μL de T4 DNA Ligase Buffer (5x)
1,5 μL de T4 DNA Ligase - Purifica amostras usando 90 µ l de grânulos magnéticos de DNA-ligando e 400 µ l de etanol a 70% recentemente preparada por amostra. Adicione 90 µ l dos grânulos de cada amostra e incubar durante 5 min à temperatura ambiente. Grânulos de Pelotas, remover o sobrenadante e ressuspender em 200 µ l de etanol 70%. Remover o etanol e repetir a lavagem uma vez.
- Use uma placa magnética para grânulos de pelotas e remover tanto etanol quanto possível. Seco em um 37 ° C heatblock para 3-5 min até que o sedimento do grânulo é completamente seca. Resuspenda em 22 µ l de água livre de nuclease e coletar aproximadamente 22 µ l do sobrenadante. Avalie a qualidade usando um bioanalyzer.
Nota: Se a quantidade total de DNA é inferior a 500 ng, cisalhamento e processo de DNA adicionais antes de prosseguir com as etapas subsequentes. Se o tamanho médio de DNA não aumenta por mais de 30 bps, verifique se que os reagentes são novos, como T4 DNA polimerase, Klenow e/ou T4 ligase pode ser velho.
Ponto de parada: Depois de ligar o adaptador metilado, amostras podem ser seladas e armazenadas a-20 ° C.
- Prepare a ligadura Master Mix no gelo e adicionar 16,5 µ l de mistura para cada amostra. Incube em um termociclador sem tampa aquecida (20 ° C por 15 min, espera de 4 ° C).
- DNA de cisalhamento
- Hibridização
- Transferir as amostras para baixos tubos de ligação a DNA microcentrifuga e usar um concentrador de vácuo aquecido para reduzir o volume de amostra para menos de 3,4 µ l. reconstituir as amostras a 3,4 µ l.
Nota: Concentre-se as amostras a cerca de 3 ~ µ l para assegurar que as amostras são removidas do concentrador de vácuo antes de todo o líquido evapora. - Prepare o tampão de hibridização em temperatura ambiente e misture de bloco metil-Seq no gelo. Adicionar 5,6 µ l de mistura de metil-Seq bloco para cada amostra e incubar em termociclador (95 ° C por 5 min, 65 ° C por 2 min, preensão de 65 ° C).
Tampão de hibridização (por exemplo):
6,63 µ l de metil-Seq Hyb 1
0,27 µ l de metil-Seq Hyb 2
2.65 µ l de metil-Seq Hyb 3
3.45 µ l de metil-Seq Hyb 4
Metil-Seq bloco Mix (por exemplo):
2,5 µ l de metil-Seq indexação bloco 1
2,5 µ l de metil-Seq bloco 2
0,6 µ l de metil-Seq bloco 3 - Prepare o RNase bloco Mix e o mix de hibridação de capturar biblioteca. Adicionar 20 µ l de capturar Mix de hibridação de biblioteca para cada amostra e incubar a 65 ° C, durante pelo menos 16 h.
RNase bloco Mix (por exemplo):
0,5 µ l de RNase bloco
1,5 µ l de água
Capture o Mix de hibridação de biblioteca (por exemplo):
13 µ l de tampão de hibridização
2 µ l de RNase bloco Mix
Biblioteca de captura de 5 µ l de rato metil-Seq
Nota: manter reações a 65 ° C ao adicionar a mistura de hibridização para evitar inespecificas. - Alíquotas de 50 µ l de estreptavidina grânulos magnético por amostra em um tubo de tiras de 8 poços novos. Lave os grânulos com 200 µ l de tampão de ligação metil-Seq. Use a placa magnética para grânulos de pelotas e remover o sobrenadante entre cada lavagem para um total de 3 lavagens. Após a lavagem final, resuspenda streptavidin grânulos em 200 µ l de tampão de ligação metil-Seq.
- Adicionar as amostras a 200 µ l de grânulos magnético streptavidin lavados e incubar a temperatura ambiente por 30 min, utilizando um misturador rotativo. Enquanto mistura, alíquota 200 µ l de metil-Seq Wash Buffer 2 em triplicado poços de uma placa de 96 poços, por exemplo e o lugar em um termociclador para pré-aquecer a 65 ° C.
- Após a incubação, pelota streptavidin grânulos magnético usando placa magnética e Resuspenda os grânulos em 200 µ l metil-Seq Wash Buffer 1. Incube durante 15 minutos à temperatura ambiente. Use uma placa magnética para pelotas e descartar o sobrenadante.
- Lave os grânulos 3 vezes com metil-Seq lavar Buffer 2: grânulo Ressuspender em 200 µ l de lavar Buffer 2 (pré aquecido na etapa 6.2.5.), incubar grânulos no termociclador (65 ° C, 10 min) e grânulos de Pelotas. Desprezar o sobrenadante após cada lavagem, usando uma placa magnética.
Nota: Manter as reacções de hibridização a 65 ° C ao adicionar Wash Buffer 2 para evitar a ligação não-específica. - Adicionar 20 µ l de tampão de eluição de metil-Seq aos talões lavados e incubar a temperatura ambiente por 20 min. Use uma placa magnética para grânulos de pelota e transferência sobrenadante para um novo tubo de strip-tease. Descarte os grânulos.
Nota: Durante a incubação, prepare o reagente de conversão do bissulfito.
- Transferir as amostras para baixos tubos de ligação a DNA microcentrifuga e usar um concentrador de vácuo aquecido para reduzir o volume de amostra para menos de 3,4 µ l. reconstituir as amostras a 3,4 µ l.
- Conversão de bissulfito
Nota: Execute a conversão de bissulfito do ssDNA eluted usando reagentes apropriados e instruções de um kit de conversão de bissulfito comercialmente disponíveis.- Adicione o reagente de conversão 130 µ l bissulfito preparado ao sobrenadante da etapa anterior. Divida cada uma das 150 reações µ l igualmente em dois poços. Incube em um termociclador (64 ° C por 2,5 h, espera de 4 ° C).
Nota: A reação de 150 µ l é dividida igualmente em dois poços separados para assegurar a temperatura homogénea. Após incubação por 2,5 h, proceda imediatamente para a próxima etapa. - Vincule as amostras girar colunas adicionando 600 µ l de tampão de ligação e lave uma vez com 100 µ l de tampão de lavagem. Centrifugar colunas (15.000 x g, 1 min) entre todas as etapas de conversão de bissulfito e descartar fluir.
- Desulphonate amostras adicionando-se 200 µ l de tampão de Desulphonation de colunas. Incubar a temperatura ambiente por 15-20 min. repetir centrifugação e descartar fluir.
- Lave as colunas duas vezes com 200 µ l de tampão de lavagem. Eluir cada amostra adicionando 10 µ l de tampão de eluição para a coluna, a incubação por 3 min à temperatura ambiente e centrifugação (15.000 x g, 1 min). Repita a etapa de eluição para um total de 20 µ l.
- Prepare a reação de PCR Master Mix 1 no gelo. Adicione 82 µ l de mistura para cada amostra. Incube em um termociclador com o programa a seguir.
Reação PCR Master Mix 1 (por exemplo):
30 µ l de água
50 µ l de metil-Seq PCR Master Mix
1 µ l de metil-Seq PCR1 Primer F
1 µ l de metil-Seq PCR1 Primer R
Termociclador programa:
Fase 1, 1 ciclo: 95 ° C 2 min
Fase 2, 8 ciclos: 95 ° C 30 s, 60 ° C 30 s, 72 ° C 30 s
Fase 3, 1 ciclo: 72 ° C 7 min
Fase 4, 1 ciclo: 4 ° C Hold - Purifica amostras usando 180 µ l de grânulos magnéticos de DNA-ligando e 400 µ l de etanol a 70% recentemente preparada por amostra. Adicionar 180 µ l de grânulos para cada amostra e incubar durante 5 min à temperatura ambiente. Grânulos de Pelotas, remover o sobrenadante e ressuspender em 200 µ l de etanol 70%. Remover o etanol e repetir a lavagem uma vez.
- Use uma placa magnética para grânulos de pelotas e remover tanto etanol quanto possível. Seco em um 37 ° C heatblock para 3-5 min até que o sedimento do grânulo é completamente seca. Resuspenda em 21 µ l de água livre de nuclease e coletar aproximadamente 19,5 µ l do sobrenadante.
- Adicione o reagente de conversão 130 µ l bissulfito preparado ao sobrenadante da etapa anterior. Divida cada uma das 150 reações µ l igualmente em dois poços. Incube em um termociclador (64 ° C por 2,5 h, espera de 4 ° C).
- Indexação
- Prepare a reação de PCR Master Mix 2 no gelo. Adicionar µ l 25.5 Master Mix 2 para cada amostra. Adicionar 5 µ l comercial indexação primers para amostras individuais e incubá-los em um termociclador.
PCR reação Master Mix 2 (por exemplo):
25 µ l de metil-Seq PCR Master Mix
0,5 µ l Primer indexação comum de metil-Seq
Termociclador programa:
Fase 1, 1 ciclo: 95 ° C 2 min
Fase 2, 6 ciclos: 95 ° C 30 s, 60 ° C 30 s, 72 ° C 30 s
Fase 3, 1 ciclo: 72 ° C 7 min
Fase 4, 1 ciclo: 4 ° C Hold
Nota: Ciclos adicionais (2-3) podem ser necessários se a concentração inicial de DNA está abaixo dos valores recomendados. - Purifica amostras usando 90 µ l de grânulos magnéticos de DNA-ligando e 400 µ l de etanol a 70% recentemente preparada por amostra. Adicione 90 µ l dos grânulos de cada amostra e incubar durante 5 min à temperatura ambiente. Grânulos de Pelotas, remover o sobrenadante e ressuspender em 200 µ l de etanol 70%. Remover o etanol e repetir a lavagem uma vez.
- Use uma placa magnética para grânulos de pelotas e remover tanto etanol quanto possível. Seco em um 37 ° C heatblock para 3-5 min até que o sedimento do grânulo é completamente seca. Resuspenda em 24 µ l de água livre de nuclease e coletar aproximadamente 24 µ l do sobrenadante.
- Avalie a concentração e tamanho de bp usando os reagentes de detecção de DNA de alta sensibilidade em um bioanalyzer.
Nota: Se o bioanalyzer falhar detectar a presença da biblioteca de DNA, repita as etapas de preparação com DNA adicional.
Parando de ponto: após a purificação, amostras indexadas podem ser seladas e armazenadas a-20 ° C. - Pool de amostras para a plataforma apropriada da próxima geração de sequenciamento usada.
- Usando os dados de concentração do bioanalyzer, que determina a molaridade do DNA baseada no tamanho da biblioteca e a quantidade em um determinado volume, diluir com tampão de baixa TE (6.1.1.1) e combinar todas as amostras para uma concentração final de 15 pM.
Nota: Um método mais sensível de quantificar a biblioteca é por PCR quantitativo em tempo real, usando as primeiras demão que destino os adaptadores ligados. - Execute em pool de amostras sobre o número de pistas suficientes para 4 amostras por raia em um sequenciador de última geração.
Nota: por exemplo, se 16 amostras de biblioteca excepcionalmente foram indexadas e combinadas, execute as bibliotecas mais 4 pistas, equivalentes a 4 amostras por raia.
- Usando os dados de concentração do bioanalyzer, que determina a molaridade do DNA baseada no tamanho da biblioteca e a quantidade em um determinado volume, diluir com tampão de baixa TE (6.1.1.1) e combinar todas as amostras para uma concentração final de 15 pM.
- Prepare a reação de PCR Master Mix 2 no gelo. Adicionar µ l 25.5 Master Mix 2 para cada amostra. Adicionar 5 µ l comercial indexação primers para amostras individuais e incubá-los em um termociclador.
7. sequenciamento de um sequenciador de última geração
- Envie as amostras para o núcleo de sequenciamento institucional para clustering da biblioteca de metil-Seq, seguida por sequenciamento em uma máquina de sequenciamento de nova geração.
8. análise para identificar DMRs
- Implemente Bismark15, que invoca Bowtie 2.0 como uma sequência interna alinhador16,17, para alinhar crus entradas leituras de bissulfito-convertido, plus-vertente do genoma. Após o alinhamento, use o Bismark_methylation_extractor para executar o controle de qualidade e atribuir um valor estimado de metilação para cada CpG.
- Gera uma lista de DMRs com o BS-Seq pacote18 em Bioconductor. Filtro do DMRs baseia tendo maior que 3 CpGs consecutivos e P-valor < 0,05.
Nota: Para gerar uma lista DMR que inclui coordenadas genômicas, distância para o gene RefSeq mais próxima, número de CpGs dentro cada DMR, média % valor de metilação CpG em toda a DMR para os grupos de dois comparação (por exemplo, salientado vs átonas), o P-valor, e o valor FDR (taxa falsa da descoberta). Use a lista DMR, i. e., coordenadas genômicas, para projetar primers de pyrosequencing para validação.
9. validação por bissulfito Pyrosequencing
-
Projeto da primeira demão
- Desenho de primers para PCR de bissulfito e pyrosequencing. Desenha dois conjuntos de iniciadores de PCR (exteriores e aninhados) para que o PCR aninhado amplificará 150 – 400 bps de um DMR.
Nota: em geral, primers projetados são pelo menos 24 bases de comprimento com pelo menos 4 – 5 não-consecutivos do G (o C para o primer reverso) conta para reduziu a temperatura da perda de complexidade da sequência do recozimento. Um dos primers aninhados será rotulado de biotina e HPLC-purificado. No entanto, as primeiras demão padrão devem ser ordenadas primeiro a otimizar a etapa PCR resolvendo as reações em um gel de agarose.- Projeto do pyrosequencing do ensaio da primeira demão para que alveja o biotinilado complementar vertente apenas 1-2 bases montante das CpGs para ser analisada. Design múltiplo pyrosequencing cartilhas, se necessário, como cada cartilha pyrosequencing pode confiantemente do ensaio 30 bps a jusante.
- Para o Rt1-m4, use o seguinte:
rRT1M4 fora – F TGTAYGATTTTGGTTATYGTAAAT
rRT1M4 fora – R AACTTACAAATTTCACCAACTCA
rRT1M4 Nested – F GTGGGTTAYGTGGATAATATATAG
rRT1M4 Nested-R AATCACTTACCATTCTCTCTCTAACTA
rRT1M4 Pyro1 TAYGTGGATAATATATAGAT
rRT1M4 Pyro2 GATAGTTATTTGGYGAGTTAG
rRT1M4 Pyro3 GAGTATTTGGAGGAGTTGAT
rRT1M4 Pyro4 GGATTTTAATATTTGGT
- Desenho de primers para PCR de bissulfito e pyrosequencing. Desenha dois conjuntos de iniciadores de PCR (exteriores e aninhados) para que o PCR aninhado amplificará 150 – 400 bps de um DMR.
-
Use um kit comercialmente disponíveis para a conversão de bissulfito de gDNA de sangue de rato.
Nota: As etapas de conversão de bissulfito foram adaptadas do kit comercialmente disponíveis com as seguintes modificações: na etapa 1, adicione 50 – 100 ng de gDNA de sangue e diluir com água até 20 µ l. Na etapa 9, eluir 20 µ l por amostra.- Preparar o reagente de conversão bissulfito de acordo com o protocolo do fabricante e combinar com gDNA diluído. Incube em termociclador (64 ° C por 2,5 h, espera de 4 ° C).
- Adicione ligação Buffer gDNA convertido em colunas de rotação e centrífuga (15.000 x g, 1 min). Colunas de lavagem uma vez então adicionem Desulphonation Buffer para as colunas e incubam por 15 min à temperatura ambiente. Centrífuga (15.000 x g, 1 min).
- Lave a coluna com amortecedor da lavagem e a centrifugação (15.000 x g, 1 min). Repita a etapa de lavagem com centrifugação (15.000 x g, 2 min). Adicionar 20 µ l tampão de eluição e centrífuga (15.000 x g, 1 min) para eluir.
-
Amplificação por PCR
- Prepare-se fora do PCR Master Mix. Adicionar 21,5 µ l do Master Mix para 3,5 µ l bissulfito-convertido gDNA e executar programa de termociclador.
Exterior do PCR Master Mix:
16.25 µ l de água
2,5 µ l de tampão de Polymerase [10x]
0,5 µ l de dNTP [10 mM]
1 µ l de Primer para a frente [0.1 µM]
1 µ l de Primer reverso [0.1 µM]
0,25 µ l de Taq DNA polimerase [5000 U/mL].
Programa cycler térmico:
Fase 1, 1 ciclo: 94 ° C 4 min
Fase 2, 47 ciclos: 94 ° C min 1, 53 ° C 30 s, 72 ° C 1 min
Fase 3, 1 ciclo: 72 ° C 8 min, 4 ° C Hold - Prepare a mistura de mestre do PCR aninhado. Adicionar 23 µ l do Master Mix a 2 µ l da amostra de PCR fora e repetir o programa de termociclador PCR fora. Avalie a qualidade do produto do PCR através de eletroforese em gel (1 x TAE buffer, gel de agarose 1%).
PCR aninhado Master Mix:
17,75 µ l de água
2,5 µ l de tampão de Polymerase [10x]
0,5 µ l de dNTP [10 mM]
1 µ l de Primer para a frente [0.1 µM]
1 µ l de Primer reverso [0.1 µM]
0,25 µ l de Taq DNA polimerase [5000 U/mL]
Nota: Para PCR aninhado, o avanço ou o reverso primer deve ser biotinilado.
- Prepare-se fora do PCR Master Mix. Adicionar 21,5 µ l do Master Mix para 3,5 µ l bissulfito-convertido gDNA e executar programa de termociclador.
-
Pyrosequencing
- Faça uma mistura de mestre contendo 38 µ l de tampão de ligação, 35 µ l de água e 2 µ l de grânulos de sepharose streptavidin-revestido por amostra. Em uma placa de 96 poços, adicione 75 µ l do mix mestre e 5 µ l de produto PCR aninhado. Agitar um agitador de placa por 15-60 min.
- Agitando, adicione 12 µ l de primer (0,5 µM, diluído em tampão de recozimento) em poços de uma placa de ensaio pyrosequencing.
- Após agitação, execute as etapas de lavagem usando buffers de lavagem de reação de ligação. Coloque a ferramenta vácuo na gamela cheia de água, em seguida, recolher amostras de placa. Submergi o vácuo ferramenta em cochos meio-cheio, contendo tampão de acetato de Tris (10 mM, pH 7,4), NaOH (0,2 M) e 70% de etanol. Desconecte-se da ferramenta de vácuo vácuo e lugar na placa de ensaio HS para transferir contas.
- Colocar a placa no bloco de calor e incubar a 80 ° C por 2 min. Allow placa esfriar por 5 min, em seguida, iniciar o programa de pyro.
Resultados
Uma implementação bem sucedida da plataforma de metil-Seq rato varia de acordo com vários critérios. A Figura 1 mostra o fluxo de trabalho geral do estudo e destaca as etapas de controle de qualidade específico (QC) que são necessários antes de avançar. Um dos primeiros fatores a considerar é a robustez do modelo animal e o regime de estresse, que determinam a magnitude de mudanças epigenéticas que ocorrem em todo o methylome. Desde que nosso trabalho animal baseia-se na nossa observação anterior de que a exposição de corticosterona (CORT) pode levar a mudanças na metilação de DNA19,20, nosso regime de estresse crônico variável (CVS) precisava ser de rigor suficiente para produzir salientou ratos com níveis de elevados de plasma CORT. Um típico regime semanal de CVS é mostrado na tabela 1 e consistia de estressores diários pela manhã, à tarde, e durante a noite que são alteradas constantemente para evitar habituação e diminuída resposta ao estresse. Durante todo o regime de 3 semanas, os animais forçados exibiram níveis significativamente elevados de plasma média CORT [dias 4-21, controlam: 32,7 3.7 ng/mL, Stress: 103,0 11,9 ng/mL (SEM média), P = 2,2 x 10-4, Figura 2A] sobre aquelas de átonas, controle de animais. Consistentemente, estes animais também mostraram maior comportamento de ansiedade sobre a elevada além de labirinto (EPM), conforme indicado pelo significativamente mais tempo gasto nos braços fechados da EPM e menos tempo nos braços abertos (Figura 2B). Estes resultados demonstram que a exposição CVS levou a significativa endócrinas e alterações comportamentais, levando-na investigar se estas mudanças foram associadas com assinaturas de metilação de DNA específicas.
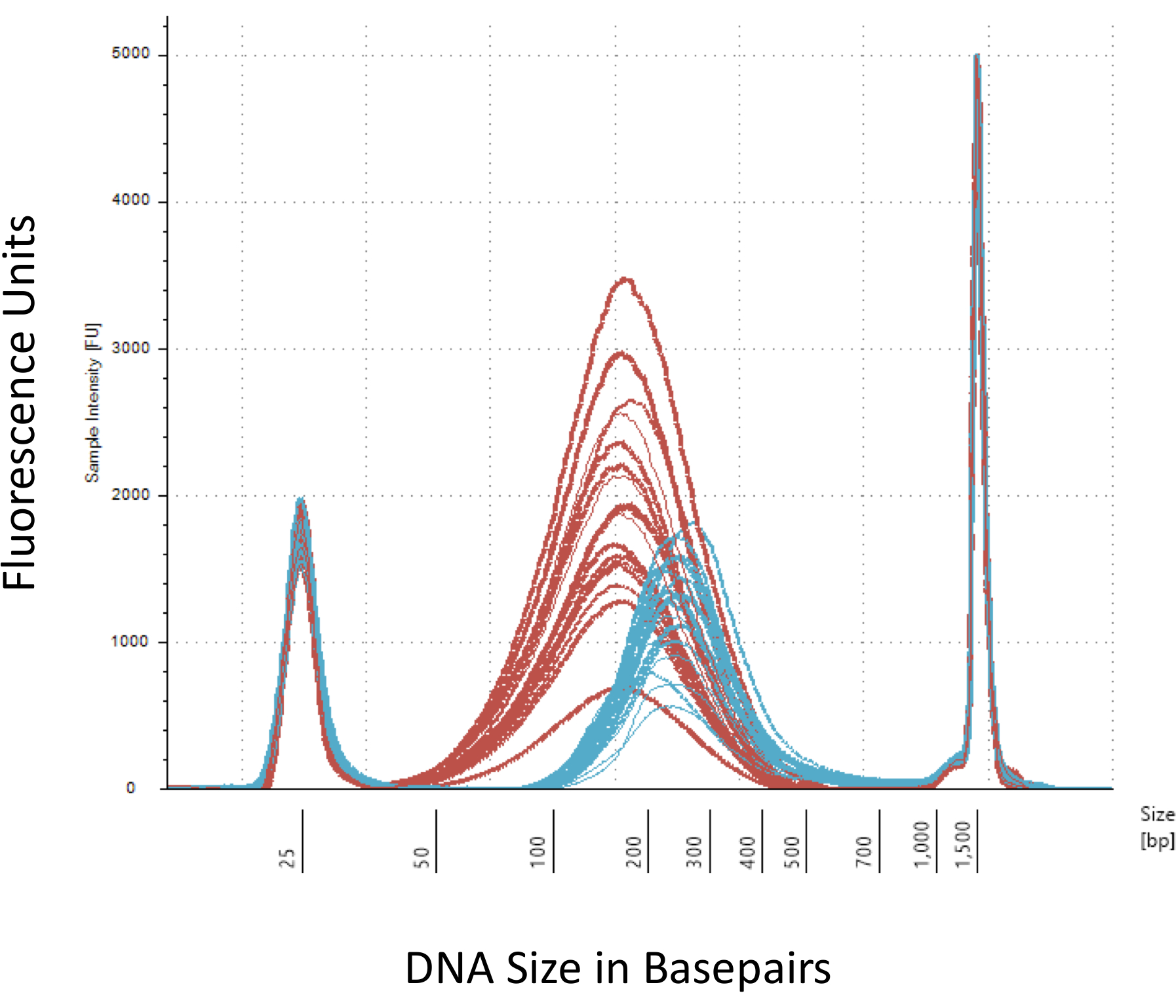
Ressaltamos que vários pontos de verificação que são cruciais para a construção bem-sucedida da biblioteca de metil-Seq. Começando com uma quantidade suficiente de DNA é necessário, como sonication, várias lavagem/purificação, enriquecimento do alvo, e as etapas de conversão de bissulfito sucessivamente reduzem a quantidade de DNA na biblioteca acabada. Apesar de várias etapas de amplificação da PCR aliviar a perda do molde do ADN, número excessivo de ciclo PCR pode introduzir maiores leituras duplicadas. Para o atual estudo em ratos metil-Seq, utilizou-se 2 g de sangue gDNA por rato. Notamos que bibliotecas de metil-Seq podem ser feitas com quantidade de DNA de partida tão baixa quanto 500 ng. Menores, a partir do material permite aos usuários gerar bibliotecas de DNA isolado por FACS (classificação de fluorescência-ativado da pilha) ou agulha socos, apesar de haver aumento do risco de produzir uma quantidade insuficiente de bibliotecas para sequenciamento subsequente. QC é executado pela electroforese de 1 L da amostra em um bioanalyzer, que fornece a molaridade, quantidade e peso molecular DNA. Três passos críticos que exigem o uso de bioanalyzer são: 1) seguindo passo sonication para garantir suficiente cisalhamento do DNA (~ 170 bp, vermelho, Figura 3); 2) após a etapa de ligadura adaptador indicaram por uma mudança no tamanho médio do DNA cortado (~ 200 bp, azul, Figura 3) para garantir a sua subsequente amplificação por PCR; e 3) após a etapa de purificação biblioteca final para garantir a quantidade e o tamanho da biblioteca para o sequenciamento.
Os R-pacotes BSSeq e BSmooth em Bioconductor foram usados para analisar o bissulfito de sequenciamento de dados18. Eles incluem ferramentas e métodos para alinhar as leituras de sequência, realizando o controle de qualidade, e identificando diferencialmente metilado regiões (DMRs). BSmooth software invoca Bowtie 2.016,17 como um alinhador de sequência interno para obter resumos de medição de nível CpG, pelo alinhamento de leituras entradas brutos de sequências genomic bissulfito-convertido. O lê alinhado então é filtrado através de procedimentos de controle de qualidade rigoroso que procuram identificar sequenciação sistemática e base-chamada erros que podem distorcer as análises a jusante. Uma série de parcelas são gerados para visualmente auxiliar neste processo de filtragem. Métricas de sequenciamento também são geradas para documento informações relevantes como o número de leituras alinhadas, % alvo e por cobertura de CpG, entre outros (tabela 2). Uma vez que os dados são filtrados, um algoritmo de suavização/normalização é realizado, onde cada CpG é atribuído um valor de metilação estimado com base em QC todos lê de cada amostra e estimativas das vizinhas CpGs para garantir mais preciso chamando de metilação estatuto, mesmo nos casos onde a cobertura de sequência é baixa. Esse valor fornece uma estimativa suavizada da probabilidade de metilação em cada local de CpG. Comparando-se a média das estimativas de cada amostra entre os dois grupos de tratamento e ranking de regiões genômicas do mais significativamente diferentes para menos metilação suavizados, é gerada uma lista de DMRs (tabela 3).
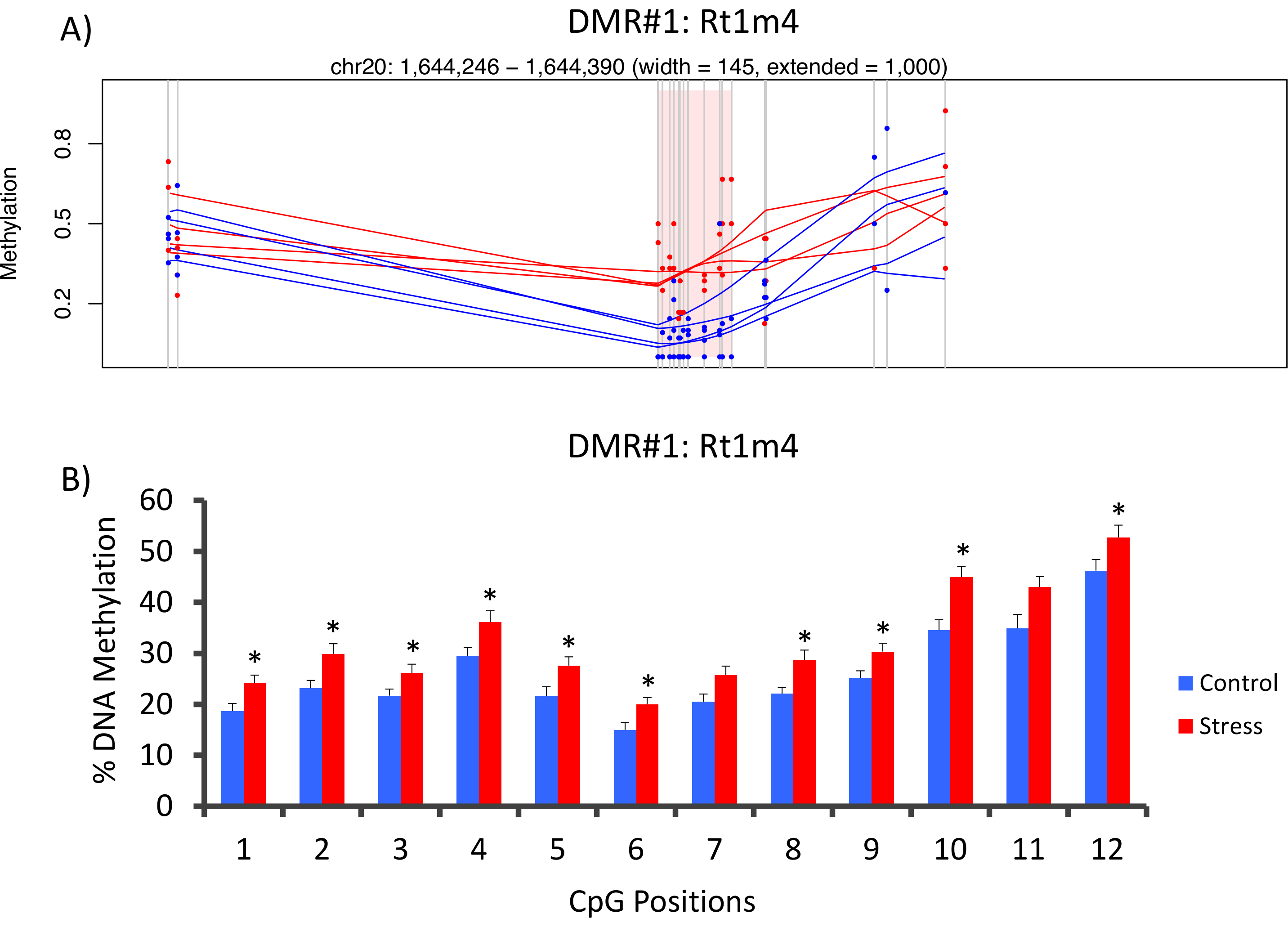
O top com que DMR entre grupos tônicas e átonos localizava-se no promotor do gene de histocompatibilidade do rato Rt1-m4, estressada animais exibem níveis mais elevados de metilação através de todas as CpGs que animais átonos (Figura 4A). Para confirmar o sucesso da implementação da plataforma metil-Seq e análise dos dados, primers foram desenhados contra o DMR e os níveis de metilação do DNA de sangue a coorte de inteira de animais estressados e átonos (8 sequenciado como metil-Seq e 8 não sequenciado) foram avaliados pelo bissulfito pyrosequencing. Resultados demonstraram aumento significativo na metilação do DNA em 10 fora as 12 CpGs analisadas (alteração 5.1 – 10,4% metilação, P < 0.037, Figura 4B). KEGG pathway análise foi realizada em todas as DMRs nominalmente significativas para identificar caminhos associados com o stress. Consistentemente, vias associado DMR implicado doenças associadas com a exposição de estresse crônico, tais como diabetes, doenças cardiovasculares e câncer (tabela 4). 21 , 22 , 23 para demonstrar uma associação entre os dados epigenéticos e o grau de exposição ao estresse, níveis de metilação no CpG-10 foram comparados com os níveis CORT 3-semana médios para cada animal. Os resultados mostraram uma modesta correlação entre os dados do sistema endócrino e metilação (R2= 0,54, P = 0,001, Figura 5).

Figura 1: fluxo de trabalho geral esquemático para a plataforma de rato metil-Seq. G um do DNA genômico extraído do sangue do estressado e ratos controle é processado primeiro para a construção das bibliotecas de metil-Seq para sequenciamento, análise e identificação do alvo. Outros 100 ng de DNA é usado para validação independente dos alvos epigenéticas identificados pelo bissulfito pyrosequencing. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: a exposição ao estresse crônico variável (CVS) leva a alterações endócrinas e comportamentais em ratos. (A) amostras múltiplas de corticosterona (CORT) demonstram a robustez da semana 3 regime de CVS. Foram colhidas amostras de sangue de manhã antes do regime de estresse diário. (B) animais estressados passaram mais tempo nos braços fechados e menos tempo nos braços abertos da elevada além de labirinto (EPM). Boxplots com ponto de dados para cada animal são mostrados. Realizou-se o teste T de Student para a significância estatística. * P < 0.05, * * P < 0,01, e * * * P < 0,001. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: quantificação de rato cortado e adaptador-ligado DNA em um bioanalyzer. As curvas vermelhas e azuis mostram a quantidade e o tamanho do DNA genômico (vermelho), após o corte em um sonicador isotérmico e ligadura do adaptador, respectivamente. Cada linha representa uma amostra e o vermelho e azuis curvas refletem tanto a perda de DNA durante as diversas etapas (final-reparação, 3'-adenylation e limpeza de amostra) e aumentam de tamanho bp devido a ligadura dos adaptadores. Afiados picos a 25 bp e 1500 bp são marcadores padrão que foram adicionados para o buffer de carregamento. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: alterações epigenéticas induzida pelo CVS são detectadas pelo rato metil-Seq. (A) análise do rato metil-Seq dados implicado o promotor do gene da Rt1m4 como uma região diferencialmente metilada (DMR) entre estressado (vermelho) e ratos controle (azul). A saída gráfica para o Rt1m4 DMR (região sombreada rosa) exibe cada CpG (linha vertical cinza), quatro amostras de cada grupo (as linhas vermelha ou azuis) e os níveis de metilação % para cada animal (ponto vermelho ou azul). (B) doze CpGs dentro o DMR foram validadas pela pyrosequencing bissulfito. A barra de gráficos são representadas como dizer SEM, e um de T-Student foi realizada para a significância estatística. * P < 0,05. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: análise de Regressão Linear mostrou uma modesta correlação entre % DNA metilação no CpG-10 de Rt1m4 e a semana 3 significa plasma níveis CORT de ambos estressado e controlam os animais (N = 16). Dados de animais forçados são representados por círculos vermelhos. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Semana | Dia 1 | Dia 2 | Dia 3 | Dia 4 | Dia 5 | Dia 6 | Dia 7 |
| AM | Retenção | Nadar | Quarto frio | Nadar | Retenção | Abanador | Nadar |
| PM | Abanador | Inclinação de gaiola | Retenção | Abanador | Quarto frio | Retenção | Quarto frio |
| Durante a noite | Restringir a comida | Molhar a roupa de cama | Isolamento | Luz acesa | Apinhamento | Luz acesa | Molhar a roupa de cama |
Tabela 1: Uma agenda semanal típica do regime variável de estresse crônico (CVS).
| Métricas de sequenciamento | Stress1 | Controle1 |
| (n = 4) | (n = 4) | |
| Emparelhado final leituras (por) | 89,290,397 | 80,165,674 |
| Final emparelhado com exclusividade mapeada lê (UMPER) | 39,200,255 | 35,013,406 |
| Eficiência de taxa/mapeamento de alinhamento (UMPER / PER) | 44% | 44% |
| Lê de duplicata (% de UMPER) | 73% | 65% |
| Do UMPER | 10,481,031 | 12,306,018 |
| Média de cobertura de profundidade de leitura (x) (ARDC) | 6 x | 6 x |
| CpGs (N) | 12,056,878 | 12,056,878 |
| ARDC (x) de CpGs | 2 x | 2 x |
| CpGs pelo menos 10 leituras (N) | 481.383 | 595.850 |
| ARDC (X) de CpGs pelo menos 10 leituras | 19 | 19 |
| No alvo CpGs (sobreposição completa com sonda regiões-alvo) | 1.923.872 | 2.007.638 |
| No alvo ARDC (x) de CpGs | 7 x | 8 x |
| No alvo CpGs pelo menos 10 leituras (N) | 428.249 | 531.419 |
| Na ARDC alvo (x) de CpGs pelo menos 10 lê | 18 x | 18 x |
| No alvo (PER com sobreposição de 1 ou mais pares de Base com sonda regiões-alvo) (UMPER) | 8.277.715 | 9.369.523 |
| % No alvo (do UMPER) | 78% | 77% |
| No alvo (Bases Total mapeadas) Mb | 125 mb | 128 mb |
| Cobertura de profundidade no alvo leitura média (x) (ARDC) | 9 x | 10 x |
| 1 Sequenciamento de métricas baseadas em médias de sujeitos em cada grupo |
Tabela 2: Sequenciamento métricas obtidas a partir da plataforma de metil-Seq de rato.
| Chr | início | final | Gene | distância | areaStat | meanDiff | stress | controle | direção |
| chr20 | 1.644.246 | 1.644.390 | RT1-M4 | in_gene | 93.03 | 0.22 | 0,33 | 0.11 | ganho de |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | -0.19 | 0.72 | 0.91 | perda de |
| chr3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0.21 | 0.94 | 0.72 | ganho de |
| chr2 | 143,064,811 | 143,065,010 | Ufm1 | 8569 | -59.48 | -0.11 | 0.13 | 0,24 | perda de |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57,04 | 0.21 | 0.94 | 0,73 | ganho de |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0.13 | 0,74 | 0,88 | perda de |
| chr7 | 47,101,725 | 47,101,930 | PAWR | in_gene | -50.54 | -0.12 | 0.64 | 0.76 | perda de |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0.11 | 0.85 | 0,96 | perda de |
| chr11 | 80,640,132 | 80,640,356 | DGKG | in_gene | -50.07 | -0.16 | 0,73 | 0,89 | perda de |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0.17 | 0,58 | 0.75 | perda de |
Tabela 3: Top 10 diferencialmente metilado regiões. Para cada DMR, a tabela de saída mostra da esquerda para a coluna da direita: localização cromossômica (chr), coordena (início/término), nome do gene, distância da transcrição iniciar site, estatísticas de área diferencial entre estressados e controlam grupos (areaStat), quer dizer metilação diferencial (meanDiff), os níveis de metilação média em cada DMR para estressados e grupos de controle (stress/controle) e direção de metilação mudam de controles.
| KEGG Pathway termos | Contagem de gene | % | P-valor | Benjamini |
| Diabetes | ||||
| Diabetes mellitus tipo II | 12 | 0.1 | 3,6 x 10-4 | 9,8 x 10-3 |
| Doença cardiovascular | ||||
| Contração do músculo liso vascular | 18 | 0.1 | 1.6 x 10-3 | 3,6 x 10-2 |
| Arritmogênica do direito cardiomiopatia ventricular (ARVC) | 13 | 0.1 | 4.0 x 10-3 | 7,1 x 10-2 |
| Cardiomiopatia dilatada | 14 | 0.1 | 7,6 x 10-3 | 1.2 x 10-1 |
| Função do neurônio | ||||
| Potenciação de longa duração | 11 | 0.1 | 1.5 x 10-2 | 1.4 x 10-1 |
| Sinalização | ||||
| Via de sinalização MAPK | 35 | 0.2 | 2,4 x 10-4 | 9,9 x 10-3 |
| Via de sinalização de cálcio | 22 | 0.1 | 1.2 x 10-2 | 1.4 x 10-1 |
| Via de sinalização do chemokine | 21 | 0.1 | 1.2 x 10-2 | 1,3 x 10-1 |
| Câncer | ||||
| Percursos em câncer | 42 | 0.3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Glioma | 15 | 0.1 | 4.4 x 10-5 | 2,4 x 10-3 |
| Câncer de pulmão de células não pequenas | 10 | 0.1 | 7,9 x 10-3 | 1.1 x 10-1 |
| Câncer colorretal | 13 | 0.1 | 8,4 x 10-3 | 1.1 x 10-1 |
| Leucemia mieloide crônica | 12 | 0.1 | 1.2 x 10-2 | 1,3 x 10-1 |
Tabela 4: Análise KEGG Pathway do DMRs identificadas a partir do rato metil-Seq
Discussão
Neste estudo, nós concebido e implementado a plataforma de metil-Seq para o genoma do rato. Demonstrando sua utilidade com um modelo do rato de stress, demonstrámos que o pipeline experimental e analítico pode fornecer diferencialmente metiladas regiões entre dois grupos de comparação.
Para garantir uma implementação bem-sucedida da plataforma, várias etapas críticas precisam ser observados. Primeiro, inicial, quantidade e qualidade do DNA tem um impacto significativo sobre a qualidade e quantidade da biblioteca de metil-Seq final. Usamos um dados, ao invés de um espectrofotômetro, para garantir que nossa medida de DNA reflete a quantidade de DNA de cadeia dupla presente. O bioanalyzer foi usado para medir o tamanho molecular e a quantidade de DNA após o corte e após ligadura do adaptador. Verificar o tamanho molecular "mudança" entre estas etapas é crucial para confirmar a presença de adaptadores nas extremidades de cada fragmento de DNA que passará por PCR mediada por adaptador nas etapas subsequentes. A quantidade de DNA restante no final da etapa de ligadura adaptador também é importante, desde pelo menos 100 ng do produto biblioteca é necessário nesta etapa para assegurar quantidade suficiente disponível após as etapas de conversão de enriquecimento e bissulfito de alvo. Uma medição de alta sensibilidade a final foi realizada na biblioteca de metil-Seq construída para que a biblioteca pode ser devidamente diluída para clustering subsequentes sobre o sequenciador de última geração. Finalmente, bissulfito pyrosequencing foi empregado como um método altamente quantitativa, independente para avaliar a precisão do pipeline analítica. A validação final usando as amostras originais e replicação usando animais adicionais são etapas cruciais para garantir que a experiência pode detectar mudanças biologicamente significativas na metilação do DNA.
Nós também incluímos várias orientações no caso de desvio de protocolo ou se problemas são encontrados. Primeiro, é possível perder muito DNA durante o fim-reparação, ligadura de adaptador ou etapas de purificação magnética do grânulo. Alternativamente, a partir de quantidades de DNA pode ser pequena (< 200 ng) devido à disponibilidade limitada de tecido/DNA ou a aplicação de diferentes métodos de enriquecimento como fluorescência ativada celular classificação. Aumentando o número de ciclo durante as etapas de amplificação de dois biblioteca pode ser capaz de compensar a perda excessiva de DNA ou baixa quantidade de DNA em todo o protocolo de construção de biblioteca a começar. No entanto, não mais do que um adicional 2 – 3 ciclos são recomendados, como amplificação excessiva modelo é susceptível de conduzir a um aumento no número de leituras duplicados sendo sequenciado. Estas duplicatas são excluídas durante a etapa de alinhamento para evitar viés nos cálculos da porcentagem de metilação. Em segundo lugar, se o tamanho médio de DNA não aumenta por mais de 30 bps, verifique se que os reagentes são novos, como T4 DNA polimerase, Klenow e/ou T4 ligase pode ser velho. Reagentes de substituição disponível comercialmente podem ser usados.
Além disso, é possível que a predita DMRs não podem validar por pyrosequencing, onde as diferenças de metilação de DNA não existem ou são significativamente menos do que aqueles preditos pela análise. Validação de pobre das regiões do candidato é um problema muito comum para muitas análises de todo o genoma, tais como quando pyrosequencing resultados não confirmam a metilação diferencial ou o tamanho do efeito é muito menor do que o previsto pela análise. BSmooth é um pacote analítico que os níveis de metilação "suaviza" através de uma janela de múltiplos CpGs. Para a experiência atual, BSmooth implicado um DMR cujos níveis de metilação foram validados pela pyrosequencing bissulfito. No entanto, provavelmente haverá discrepâncias entre os níveis de metilação preditos por BSmooth e aqueles verificados por pyrosequencing. As discrepâncias surgem a partir da função de suavização que estima os valores médios de metilação através de todas as CpGs dentro um DMR, incluindo consecutivos CpGs que podem diferir na metilação do DNA por mais de 50% ou CpGs cujos valores de metilação foram excluídos devido a limite de sub leitura de profundidade. R-pacotes como MethylKit24 podem ser usados para identificar janelas menores de CpGs ou mesmo única CpGs cujos níveis de metilação correlacionam fortemente com aqueles validados pelo pyrosequencing. Implementação de pacotes diferentes e testando suas regiões previstas ou CpGs de metilação diferencial por pyrosequencing garantem robustez dos dados. Alternativamente, bibliotecas de metil-Seq originais podem ser reordenadas e adicionadas aos arquivos de leitura para aumentar a profundidade de leitura. Desde que a determinação dos níveis de metilação são semi-quantitativa e ditada pelo número de leituras [(# of CpGs) / (n º de TpGs + CpGs)], aumento da profundidade de leitura para um determinado CpG aumentará a precisão do seu valor percentual de metilação. Neste estudo, consideramos apenas CpGs cujos valores de metilação foram determinados pelo menos dez leituras e alcançado uma cobertura global de leitura de 19 x para cada CpG.
A plataforma de metil-Seq de rato não é sem suas limitações. Enquanto é mais rentável do que o sequenciamento do genoma inteiro bissulfito, é consideravelmente mais caro do que outros métodos. No entanto, a maioria do custo foi para adquirir pistas sobre o sequencer e não para o sistema de captura. Dependendo da profundidade de leitura necessária, com comparações de cruz-tecido que exigem menos devido a diferenças de grande (25% a 70%)12 na metilação do DNA, o custo pode ser reduzido por multiplexação mais amostras por raia e usando uma plataforma de maior capacidade. Além disso, a preparação da amostra é mais demorada do que outros métodos. Embora semelhante a outras abordagens de pulldown que incorporam o sequenciamento de nova geração, as etapas de conversão e purificação de bissulfito adicionado adicionem à carga de trabalho. Em geral, a plataforma de metil-Seq é uma alternativa econômica para o sequenciamento do genoma inteiro e fornece resolução de pares de base em mais de 2,3 milhões de CpGs, que é consideravelmente mais do que aqueles analisada por plataformas microarray. Até à data, os humanos comercialmente disponíveis e mouse metil-Seq plataformas têm sido usadas para documentar as alterações dependentes de álcool no cérebro de macaco25,26, desenvolvimento neurológico genes no cérebro de rato9e sangue-cérebro alvos de glicocorticoides10. Além disso, a capacidade de direcionar a regiões específicas, independentemente do reconhecimento de sequência por enzimas de restrição torna uma plataforma ideal para comparações entre espécies. Para este estudo, nós projetamos a plataforma de metil-Seq para o rato, para o qual muitas experiências farmacológicas, metabólicas e comportamentais são executadas sem o benefício de uma ferramenta de methylomic de todo o genoma. Nossos dados mostram que ele pode ser usado para detectar DMRs em um modelo do rato de stress e correlacionados outros parâmetros fisiológicos, tais como níveis de plasma global CORT.
A plataforma de metil-Seq é ideal para epigenéticos experimentos em animais com genomas sequenciados que podem não ter suficiente evidência experimental documentando regiões reguladoras. Quando tais regiões são disponibilizadas, regiões adicionais podem ser personalizados e anexadas à versão atual. Além disso, a plataforma é ideal para genómica comparativa, uma vez que o enriquecimento de destino não é restringido por reconhecimento de enzima de restrição. Por exemplo, a região promotora de qualquer gene de interesse pode ser capturada independentemente se ele abriga um sítio de restrição específica. Da mesma forma, qualquer regiões reguladoras, tais como aquelas identificadas no mouse ou seres humanos, que são conservados no genoma de interesse podem ser capturados.
Divulgações
O manuscrito é parte de um prémio do concurso da Agilent Technologies.
Agradecimentos
Este estudo foi financiado pelo NIH grant MH101392 (RSL) e apoio dos seguintes prêmios e fundações: um NARSAD Young Investigator Award, Margaret Ann preço investigador Fund, fundo de estudioso de James Wah humor distúrbios através do Charles T. Bauer Foundation, Baker Fundação e a Fundação de partida do projeto (RSL).
Materiais
| Name | Company | Catalog Number | Comments |
| Radioimmuno assay (RIA) | MP Biomedicals | 7120126 | Corticosterone, 125I labeled |
| Master Pure DNA Purification Kit | Epicentre/Illumina | MC85200 | |
| Thermal-LOK 2-Position Dry Heat Bath | USA Scientific | 2510-1102 | Used with 1.5 mL tubes |
| Vortex Genie 2 | Fisher | 12-812 | Vortex Mixer |
| Ethyl alcohol, Pure | Sigma-Aldrich | E7023 | 100% Ethanol, molecular grade |
| Centrifuge 5424 R | Eppendorf | - | Must be capable of 20,000 x g |
| Qubit 2.0 | ThermoFisher Scientific | Q32866 | Fluorometer |
| Qubit dsDNA BR Assay Kit | ThermoFisher Scientific | Q32850 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Q32851 | High sensitivity DNA detection reagents |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| SureSelectXT Rat Methyl-Seq Reagent Kit | Agilent Technologies | G9651A | Reagents for preparing the Methyl-Seq library |
| SureSelect Rat Methyl-Seq Capture Library | Agilent Technologies | 931143 | RNA baits for enrichment of rat targets |
| IDTE, pH 8.0 | IDT DNA | 11-05-01-09 | 10 mM TE, 0.1 mM EDTA |
| DNA LoBind Tube 1.5 mL | Eppendorf | 22431021 | |
| Covaris E-series or S-series | Covaris | - | Isothermal sonicator |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm (25) | Covaris | 520045 | |
| Water, Ultra Pure (Molecular Biology Grade) | Quality Biological | 351-029-721 | |
| Veriti 96 Well-Thermal Cycler | Applied Biosystems | 4375786 | |
| AMPure XP Beads | Beckman Coulter | A63880 | DNA-Binding magnetic beads |
| 96S Super Magnet | ALPAQUA | A001322 | Magnetic plate for purification steps |
| 2200 TapeStation | Agilent Technologies | G2965AA | Electrophresis-based bioanalyzer |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 | |
| D1000 ScreenTape High Sensitivity | Agilent Technologies | 5067-5584 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 Reagents High Sensitivity | Agilent Technologies | 5067-5585 | |
| DNA110 SpeedVac | ThermoFisher Scientific | - | Vacuum Concentrator |
| Dynabeads MyOne Streptavidin T1 magnetic beads | Invitrogen | 65601 | Streptavidin magnetic beads |
| Labquake Tube Rotator | ThermoFisher Scientific | 415110Q | Nutator Mixer is also acceptable |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5006 | Bisulfite conversion kit. Contains Binding, Wash, Desulphonation, and Elution buffers |
| Illumina Hi-Seq 2500 | Illumina | - | Next-generation sequencing machine |
| PCR and Pyrosequencing Primers | IDT DNA | Variable | |
| Taq DNA Polymerase with ThermoPol Buffer - 2,000 units | New England BioLabs | M0267L | |
| Deoxynucleotide (dNTP) Solution Set | New England BioLabs | N0446S | |
| Pyromark MD96 | QIAGEN | - | Pyrosequencing machine |
| Ethyl Alcohol 200 Proof | Pharmco-Aaper | 111000200 | 70% Ethanol solution |
| Sodium Hydroxide Pellets | Sigma-Aldrich | 221465 | 0.2 M NaOH denature buffer solution |
| Tris (Base) from J.T. Baker | Fisher Scientific | 02-004-508 | 10 mM Tris Acetate Buffer wash buffer solution |
| PyroMark Gold Q96 Reagents (50x96) | QIAGEN | 972807 | Reagents required for pyrosequencing |
| PyroMark Annealing Buffer | QIAGEN | 979009 | |
| PyroMark Binding Buffer (200 mL) | QIAGEN | 979006 | |
| Streptavidin Sepharose High Performance Beads | GE Healthcare | 17-5113-01 | Streptavidin-coated sepharose beads |
| PyroMark Q96 HS Plate | QIAGEN | 979101 | Pyrosequencing assay plate |
| Eppendorf Thermomixer R | Fisher Scientific | 05-400-205 | Plate mixer. 96-well block sold separately (cat. No 05-400-207) |
| SureDesign Website | Agilent Technologies | - | Target capture design software (https://earray.chem.agilent.com/suredesign/) |
| UCSC Genome Browser | University of California Santa Cruz | - | rat Nov 2004 rn4 assembly |
| Agilent Methyl-Seq Protocol | Agilent Technologies | - | https://www.agilent.com/cs/library/usermanuals/public/G7530-90002.pdf |
Referências
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Meissner, A., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454 (7205), 766-770 (2008).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Naumov, V. A., et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 8 (9), 921-934 (2013).
- Wockner, L. F., et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational Psychiatry. 4, e339 (2014).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research. 33 (18), 5868-5877 (2005).
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A., Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48 (3), 226-232 (2009).
- Slieker, R. C., et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 6 (1), 26 (2013).
- Hing, B., et al. Adaptation of the targeted capture Methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 10 (7), 581-596 (2015).
- Seifuddin, F., et al. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 12 (8), 637-652 (2017).
- Irizarry, R. A., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 41 (2), 178-186 (2009).
- Lee, R. S., et al. Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics. 6 (11), 1378-1390 (2011).
- Jankord, R., et al. Stress vulnerability during adolescent development in rats. Endocrinology. 152 (2), 629-638 (2011).
- Pellow, S., Chopin, P., File, S. E., Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 14 (3), 149-167 (1985).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 10 (3), R25 (2009).
- Hansen, K. D., Langmead, B., Irizarry, R. A. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology. 13 (10), R83 (2012).
- Lee, R. S., et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 151 (9), 4332-4343 (2010).
- Lee, R. S., et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. , (2011).
- Bose, M., Olivan, B., Laferrere, B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Current Opinion in Endocrinology, Diabetes, and Obesity. 16 (5), 340-346 (2009).
- Brydon, L., Magid, K., Steptoe, A. Platelets, coronary heart disease, and stress. Brain, Behavior, and Immunity. 20 (2), 113-119 (2006).
- McKlveen, J. M., et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry. 80 (10), 754-764 (2016).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology. 13 (10), R87 (2012).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Translational Psychiatry. 7 (1), e994 (2017).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 60, 103-113 (2017).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados