
Method Article
Sequenciamento de nova geração de RNA e um fluxo de análise de bioinformática para identificar expressão de LINE-1s a nível locus-específico
Neste Artigo
Resumo
Aqui, apresentamos uma abordagem Bioinformatica e análises para identificar a expressão da linha 1 no nível específico do locus.
Resumo
Os elementos INterspersed longos-1 (LINEs/L1s) são elementos repetitivos que podem copiar e inserir aleatòria no genoma tendo por resultado a instabilidade e a mutagenese genomic. Compreender os padrões de expressão de Locos L1 no nível individual irá emprestar para a compreensão da biologia deste elemento mutagênico. Este elemento autônomo compõe uma parcela significativa do genoma humano com mais de 500.000 cópias, embora 99% são truncados e defeituosos. Entretanto, sua abundância e número dominante de cópias defeituosas fazem-lhe o desafio identificar autenticamente expressado L1s das seqüências L1-relacionadas expressadas como parte de outros genes. Também é desafiador identificar qual Locus L1 específico é expresso devido à natureza repetitiva dos elementos. Superando esses desafios, apresentamos uma abordagem Bioinformatica de RNA-Seq para identificar a expressão de L1 no nível específico do locus. Em resumo, nós coletamos o RNA cytoplasmic, selecionamos para transcritos poliadenilado, e utilizamos análises Strand-specific do RNA-Seq para mapear excepcionalmente leituras ao loci L1 no genoma humano da referência. Nós visualmente cura cada Locus L1 com leituras excepcionalmente mapeadas para confirmar a transcrição de seu próprio promotor e ajustar as leituras de transcrição mapeadas para dar conta da mappability de cada Locus L1 individual. Esta aproximação foi aplicada a uma linha celular do tumor da próstata, DU145, para demonstrar a habilidade deste protocolo de detectar a expressão de um número pequeno dos elementos L1 full-length.
Introdução
Retrotransposons são elementos repetitivos do ADN que podem "saltar" no genoma em um mecanismo da copiar-e-pasta através dos intermediários do RNA. Um subconjunto de retrotransposons é sabido como elementos INterspersed Long-1 (LINEs/L1s) e compõe um sexto do genoma humano com sobre 500, 0000 cópias1. Apesar de sua abundância, a maioria dessas cópias são defeituosas e truncados com apenas um estimado 80-120 L1 elementos pensado para ser ativo2. Um L1 de comprimento total é de cerca de 6 KB de comprimento com 5 ' e 3 ' regiões não traduzidas, um promotor interno e promotor anti-Sense associado, dois quadros de leitura aberta não sobrepostas (ORFS), e um sinal e cauda Polya3,4,5 . Nos seres humanos, L1s são compo das subfamílias distinguidas pela idade evolutiva com as famílias mais velhas que acumularam umas mutações mais originais da seqüência sobre o tempo comparada à subfamília a mais nova, L1HS6,7. L1s são os únicos retrotransposons autônomos, humanos e seus ORFS codificam uma transcriptase reversa, um endonuclease, e um rnps com as atividades do RNA-ligação e do acompanhante exigidas retrotranspose e inserção no genoma em um processo referido como alvo-aprontado transcrição reversa8,9,10,11,12.
O retrotransposição de L1s foi relatado para causar doenças humanas do germline por uma variedade de mecanismos que incluem o mutagenesis do insertional, as eliminações do local de destino, e os rearranjos13,14,15, 16. recentemente, foi supor que L1s pode desempenhar um papel na oncogênese e/ou progressão tumoral como o aumento da expressão e eventos de inserção deste elemento mutagênico têm sido observados em uma variedade de cânceres epiteliais17,18 . Estima-se que haja uma nova inserção de L1 em cada 200 nascimentos19. Portanto, é imperativo compreender melhor a biologia do expressar ativamente L1s. A natureza repetitiva e abundância de cópias defeituosas encontradas dentro de transcrições de outros genes tornaram este nível de análise desafiador.
Felizmente, com o advento de tecnologias de sequenciamento de alta taxa de transferência, foram feitos avanços para analisar e identificar autenticamente expressando L1s no nível específico do locus. Há umas filosofias de deferimento em como identificar melhor expressado L1s usando o sequenciamento da próxima geração do RNA. Houve apenas duas abordagens razoáveis sugeridas para mapear transcrições L1 no nível Locus-specific. Um centra-se somente na transcrição potencial que lê através do sinal da poliadenilação L1 e em seqüências flanqueando20. Nossa aproximação aproveita-se das diferenças pequenas da seqüência entre elementos L1 e mapeia somente aqueles RNA-Seq lê esse mapa excepcionalmente a um locus21. Ambos os métodos têm limitações em termos de quantificação dos níveis de transcrição. A quantitation pode ser melhorada potencialmente adicionando uma correção para a "mappability original" de cada lócus L121, ou usando algoritmos mais complexos que redistribuir as leituras de vários mapeados que não puderam ser mapeadas exclusivamente para um locus específico22. Aqui, iremos detalhar de forma passo a passo a extração de RNA e o protocolo de sequenciamento e bioinformática da próxima geração para identificar elementos L1 expressos no nível locus-específico. Nossa aproximação toma a vantagem máxima de nosso conhecimento da biologia de elementos funcionais de L1. Isso inclui saber que os elementos funcionais L1 devem ser gerados a partir do promotor L1, iniciado no início do elemento L1, deve ser traduzido no citoplasma e que suas transcrições devem ser colineares com o genoma. Momentaneamente, nós coletamos o RNA fresco, cytoplasmic, selecionamos para transcritos poliadenilado, e utilizamos análises Strand-specific do RNA-Seq para mapear excepcionalmente leituras ao loci L1 no genoma humano da referência. Estas leituras alinhadas então ainda exigem o curadoria manual extensivo para determinar se as leituras do Transcript originam do promotor L1 antes de designar um locus como um L1 autenticamente expressado. Nós aplicamos esta aproximação na amostra da linha celular do tumor da próstata DU145 para demonstrar como identifica relativamente poucos membros L1 ativamente transcritos da massa de cópias inativas.
Protocolo
1. extração de RNA citoplasmático
- Obter células através dos seguintes métodos.
- Colete células ao vivo de 2,75% – 100% confluentes, frascos T-75.
- Lave o balão 2 vezes em 5 mL de PBS frio e, na última lavagem, raspe as células e transfira para um tubo cônico de 15 mL. Centrifugue durante 2 min a 1.000 x g e 4 ° c e retire e elimine cuidadosamente o sobrenadante (tabela de materiais).
- Colete células de espécimes de tecido.
- Prepare o tecido para extração de RNA citoplasmático dentro de uma hora de ser dissecado e sempre manter no gelo. Para o armazenamento a longo prazo, use soluções do inibidor do RNA para armazenar o tecido por até 72 horas após a dissecção que segue o protocolo do fabricante (tabela dos materiais).
- Dados uma amostra de 10 μm3 e homogeneizar a amostra fresca com 5 ml de PBS frio em um homogeneizador de com estéril, transferência para um tubo cônico de 15 ml, centrífuga por 2 min a 1.000 x g a 4 ° c, e remover e descartar cuidadosamente o sobrenadante (tabela de materiais < /C8 >).
- Colete células ao vivo de 2,75% – 100% confluentes, frascos T-75.
- Adicione 2 mL de tampão de Lise ao pellet-Mix celular e incubar no gelo por 5 min.
- Prepare a reserva de Lise fresca com 150 mM de NaCl, 50 mM de HEPES (pH 7,4) e 25 μg/mL de digitonina (tabela de materiais).
- Como a concentração mínima de digitonina no tampão de Lise necessária para penetrar na membrana plasmática pode variar de acordo com o tipo de célula, confirme microscopicamente que as células tratadas com tampão de Lise perdem a membrana plasmática e mantêm a membrana nuclear intacta.
- Pouco antes de usar, adicionar 1.000 U/mL inibidor de RNase (tabela de materiais).
- Centrifugue por 1 minuto em 1.000 x g e 4 ° c, e colete o sobrenadante.
- Adicionar sobrenadante a pré-refrigerados 7,5 mL de Trizol e 1,5 mL de clorofórmio. Todos os passos que necessitam de clorofórmio devem ser feitos dentro de uma capa química limpa (tabela de materiais).
- Centrifugador para 35 min em 3.220 x g e 4 ° c.
- Transfira a porção aquosa (camada superior) para um tubo de 15 mL pré-refrigerado fresco.
- Adicionar 4,5 mL de clorofórmio e Vortex.
- Centrifugador por 10 min a 3.220 x g e 4 ° c.
- Transfira a porção aquosa para o tubo pré-refrigerado fresco.
- Adicionar 4,5 mL de isopropanol, agitar bem e incubar a-80 ° c durante a noite (tabela de materiais).
- Centrifugador em 3.220 x g e 4 ° c por 45 minutos.
- Remover isopropanol, adicione 15 mL de 100% de etanol (tabela de materiais).
- Centrifugador a 3.220 x g durante 10 min.
- Remova o etanol, escorra e seque por aproximadamente 1 h.
- Use um cotonete de algodão estéril para apagar qualquer etanol restante (tabela de materiais).
- Resuspenda a amostra em 100 a 200 μL de água livre de RNase dependendo do tamanhoda pelota (tabela de materiais).
- Fractionate amostras usando a tecnologia da electroforese para determinar a qualidade e a concentração das amostras de acordo com intructions do fabricante23 (tabela dos materiais).
- As amostras se qualificam para análise de RNA-Seq se RIN > 824.
2. sequenciamento da próxima geração
- Envie amostras de RNA citoplasmáticos a serem sequenciadas usando a plataforma de sequenciamento de próxima geração destinada a gerar pelo menos 50 milhões leituras de 100 BP de extremidade emparelhada.
- Selecione para RNAs poli-adeniladas e sequenciamento específico da vertente.
3. criar anotações (opcional se um tiver uma anotação existente)
- Crie uma anotação L1 de comprimento total ou faça o download da anotação L1 de comprimento total (arquivo suplementar 1a-b).
- Transfira anotações do Masker da repetição para os elementos LINE-1 do navegador do genoma de UCSC com a ferramenta do navegador da tabela (https://genome.ucsc.edu/cgi-bin/hgTables). Especifique o clado de mamíferos, o genoma humano, o conjunto hg19 (ou hg38 para um genoma mais atualizado) e filtre para "LINE1" em nome da classe. Baixe como um arquivo. GTF e Rotule como FL-L1-BLAST. GTF.
- Execute uma pesquisa BLAST local da primeira 300 BP do elemento L1 de comprimento total L 1.3 abrangendo a região promotora no genoma humano e adicione 6.000 BP downstream para criar um fim das coordenadas L1 para o arquivo de anotação. Salve em um arquivo GTF e Rotule como FL-L1-RM. GTF.
- Intercepta a anotação RepeatMasker e a anotação L1 baseada em promotor usando bedtools e Rotule como FL-L1-BLAST_RM. txt (pacotes de software).
- Use este comando no terminal Linux: bedtools Intersect-a fl-L1-Blast. GTF-b FL-L1-RM. gtf > FL-L1-BLAST_RM. txt.
- Separe a anotação FL-L1 intersectada pela vertente superior e inferior.
- Copie sobre o FL-L1-BLAST_RM. txt em software de planilha e classificar pela "menos" e "mais" vertente e, em seguida, ordenar por localização do cromossomo.
- Crie dois novos documentos de planilha, um com as coordenadas intersectadas para o comprimento total L1s na vertente menos e uma na vertente inferior e salve como FL-L1-BLAST_RM_minus. xls e FL-L1-BLAST_RM_plus. xls.
- Guarde os dois novos documentos como ficheiros. txt.
- Use o programa mac2unix para converter os arquivos. txt para os arquivos de anotação corretos (pacotes de software).
- Use este comando no terminal: Mac2unix.sh FL-L1-BLAST_RM_minus. GFF.
- Use este comando no terminal: Mac2unix.sh FL-L1-BLAST_RM_plus. GFF.
- Salve novos arquivos com a extensão. GFF.
- Alternativamente, use AWK para filtrar linhas associadas com a vertente + e –.
- Use o seguinte comando para obter o + vertente: awk '/+/' FL-L1_BLAST_RM. gtf > FL-L1_BLAST_RM_plus. GTF.
- Use a seguinte linha de comando para obter o-Strand: awk '/-/' FL-L1_BLAST_RM. gtf > FL-L1_BLAST_RM_minus. GTF.
4. Leia o pipeline de alinhamento para identificar as L1s expressas
| Opção | Descrição |
| – p | Isso detalha o número de threads que o computador deve usar executando o alinhamento. Maior memória do computador permitirá mais threads e deve ser empiricamente d. |
| – m 1 | Isso informa ao programa para aceitar apenas leituras que têm uma correspondência no genoma que é melhor do que qualquer outra correspondência de genoma. |
| – y | Este é o interruptor de tryhard que faz a busca do mapeamento para todas as correspondências possíveis e para não permitir que feche depois que um número fixo de fósforos é alcangado. |
| – v 3 | Isso só permite que o programa utilize memória para leituras mapeadas com 3 ou menos correspondências para o genoma. |
| – X 600 | Isso permite somente leituras emparelhadas que mapeiam dentro de 600 bases umas das outras. Isto certifica-se que os pares lidos são co-lineares no genoma e seleciona de encontro a s que envolvem moléculas processadas do RNA. |
| – chunkmbs 8184 | Esse comando atribui memória extra para manipular a grande quantidade de alinhamentos possíveis para cada leitura relacionada a L1. |
Tabela 1: opções de linha de comando para o bowtie.
- Execute os arquivos fastq de sequenciamento de fim emparelhado de alinhamento com a amostra de RNA-Seq de interesse usando bowtie.
Nota: Bowtie1 deve ser usado e não Bowtie2 porque os parâmetros necessários para o alinhamento exclusivo são especificamente encontrados apenas nesta versão do bowtie (pacotes de software). Bowtie é usado sobre alinhadores Splice-Aware como STAR em ordem avaliar concordante, contíguo lê mais relevante para a biologia L1 e expressão.- Use esta linha de comando no terminal Linux: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_sample_1. FQ-2 hg_sample_2. FQ | samtools View-hbuS-| samtools Sort – hg_sample_sorted. bam. Consulte a tabela 1 para obter uma descrição das opções de linha de comando para o bowtie.
- Vertente separar o arquivo de saída Bam usando samtools (pacotes de software) e os seguintes comandos do Linux. Observe que os valores de sinalizador real podem variar se um não estiver usando protocolos de seqüenciamento de próxima geração padrão.
- Use esta linha de comando para selecionar para a vertente superior: samtools View-h hg_sample_sorted. bam | awk ' substr ($ 0, 1, 1) = = "@" | | $2 = = 83 | | $2 = = 163 {Print} ' | samtools View-bS-> hg_sample_sorted_topstrand. bam.
- Use esta linha de comando para selecionar para a vertente inferior: samtools View-h hg_sample_sorted. bam | awk ' substr ($ 0, 1, 1) = = "@" | | $2 = = 99 | | $2 = = 147 {Print} ' | samtools View-bS-> hg_sample_sorted_bottomstrand. bam.
- Gere contagens de leitura contra anotações para L1 loci usando bedtools (pacotes de software).
- Use esta linha de comando para gerar contagens de leitura para L1s na direção do sentido na vertente superior: bedtools Coverage-Abam FL-L1-BLAST_RM_plus. GFF-b hg_sample_sorted_topstrand. bam > hg_sample_sorted_bowtie_tryhard_plus_top. txt.
- Use esta linha de comando para gerar contagens de leitura para L1s na direção do sentido na vertente inferior: bedtools Coverage-Abam FL-L1-BLAST_RM_minus. GFF-b hg_sample_sorted_bottomstrand. bam > hg_sample_sorted_bowtie_tryhard_minus_bottom. txt.
- Indexar o arquivo Bam da etapa 5.1.1 para torná-lo visível no Visualizador de genômica Integrativa (IGV)25 (pacotes de software).
- Use esta linha de comando: samtools index hg_sample_sorted. bam
- Para usar um modo de lote para aumentar o número de amostras de RNA-Seq canalizada por um tempo, use um script de supercomputador para concluir a etapa 4,1 chamada human_bowtie. sh, um script para concluir as etapas 4.2-4.3 foi criado chamado human_L1_pipeline. sh e um script para concluir etapa 4,4 foi criado chamado bam_index. sh. Esses scripts podem ser encontrados no arquivo suplementar 2 com comandos supercomputador associados para executar os scripts.
5. Curação manual
- Crie uma planilha para leituras mapeadas para cada Locus L1 anotado.
- Cópia sobre hg_sample_sorted_bowtie_tryhard_minus_bottom. txt criado na etapa 4.3.2 e página de rótulo como "menos-inferior."
- Classifique todas as colunas com base no número mais alto para o menor de leituras encontradas na coluna J.
- Copie hg_sample_sorted_bowtie_tryhard_plus_top. txt criado na etapa 4.3.1 e Rotule como "Top-Plus" em outra planilha.
- Classifique todas as colunas com base no número mais alto para o menor de leituras encontradas na coluna J.
- Crie uma terceira página rotulada como "combinada" e adicione todos os loci com dez ou mais leituras das páginas "menos-inferior" e "Plus-Top".
- Classifique todas as colunas com base no número mais alto para o menor de leituras encontradas na coluna J.
- Carregue os seguintes arquivos no IGV25 (pacotes de software): 1) genoma de referência de interesse para visualizar genes anotados, 2) FL-L1-BLAST_RM. GFF para visualizar a anotação L1, 3) hg_sample_sorted. bam para visualizar transcrições mapeadas de amostra de interesse, e 4) hg_genomicDNA_sorted. bam para avaliar a mappability de regiões genomic.
- Remova a cobertura e as linhas de junção associadas a cada arquivo Bam.
- Comprimir hg_sample_sorted. Bam e hg_genomicDNA_sorted. bam para que todas as faixas IGV caber em uma tela.
- Cópia sobre hg_sample_sorted_bowtie_tryhard_minus_bottom. txt criado na etapa 4.3.2 e página de rótulo como "menos-inferior."
- Manualmente curate.
- Usando as coordenadas de loci listado na planilha "combinado" página, ver chamado loci em IGV25 (pacotes de software).
- Curate um locus a ser expressado autenticamente fora de seu próprio se não há nenhuma leitura ascendente na direção L1 até 5 KB.
- Rotule a linha verde na cor e anote porque é um L1 autenticamente expressado.
Nota: uma excepção a esta regra existe se a região a montante do L1 não é mappable. Se for esse o caso, rotule a linha de cor vermelha e observe que a expressão da região a montante do promotor L1 não pode ser avaliada e, portanto, a expressão L1's não é capaz de ser determinada com confiança.
- Rotule a linha verde na cor e anote porque é um L1 autenticamente expressado.
- Curate um locus para não ser autenticamente expressa fora de seu próprio promotor, se houver leituras upstream até 5 KB.
- Rotule a linha de cor vermelha e observe por que não é um L1 autenticamente expresso.
- Curate um locus como o falso se é expressado dentro de um intron de um gene expresso no mesmo sentido com leituras a montante do L1, se é a jusante de um gene expresso no mesmo sentido com leituras a montante do L1, ou para os testes padrões un-anotated da expressão com re anúncios a montante do L1.
Observação: uma exceção a essa regra se aplica quando há leituras mínimas diretamente sobrepondo o local de início do promotor L1, mas ligeiramente a montante do L1. Se não há nenhum outro lê a montante de um caso L1 como este, considere este L1 para ser expressado autenticamente. Rotule a cor verde da linha e observe por que ele é um L1 autenticamente expresso.
- Curate um locus L1 como provável ser falso se o teste padrão de leituras traçadas ao Locus não se correlacionar com as regiões L1's específicas do mappability.
Nota: por exemplo, se um L1 é altamente mapeável mas tem somente uma pilha acima das leituras em uma região condensada dentro do L1, é menos provável estar relacionada à expressão L1 fora de seu próprio promotor e mais provável ser das fontes un-anotated como exons ou ltrs. Em casos como este, cura o loci como a laranja e anote porque o locus é suspeito. Verifique as fontes de pilha suspeitos, verificando a localização L1 no navegador do genoma UCSC. - Curate um locus para não ser autenticamente expressado se está dentro de um ambiente genomic de regiões un-anotated esporadicamente expressas
Nota: por exemplo, leituras podem ser expressas 10 KB upstream do L1, mas cada 10 KB ou assim há leituras mapeadas e algumas dessas leituras alinhar com o L1. Estes L1s são menos propensos a ser expressa fora de seu próprio promotor, e mais propensos a ter mapeado leituras devido a un-anotado padrões de expressão genômica. Em casos como este, cura o loci como a laranja e anote porque o locus é suspeito.
6. Leia a estratégia do alinhamento para avaliar o mappability no genoma da referência (opcional se um tem um conjunto de dados genomic alinhado existente do ADN)
- Transfira arquivos inteiros da seqüência do ADN do genoma e converta-os aos arquivos. FQ
- Vá para o site NCBI encontrado aqui: https://www.ncbi.nlm.nih.gov/sra
- Digite WGS HeLa final emparelhado.
- Selecione para Homo sapiens resultados por táxon.
- Selecione um exemplo que está emparelhado final e tem leituras com 100 ou mais BP como o exemplo a seguir: https://www.ncbi.nlm.nih.gov/sra/ERX457838 [ACCN]
- Confirme o comprimento de leitura selecionando executar e, em seguida, metadados como mostrado aqui: https://Trace.NCBI.nlm.nih.gov/TRACES/Sra/?Run=ERR492384
- Para baixar todos os dados da sequência de DNA do genoma, digite este comando no terminal Linux: sratoolkit. 2.9.2-mac64/bin/prefetch-X 100g ERR492384
Nota: a função de pré-busca do Toolkit SRA transfere o número de adesão "ERR492384" encontrado no site NCBI (pacotes de software). O "100G" limita a quantidade de dados baixados para 100 gigabytes. - Digite este comando no terminal Linux: fastq-dump--Split-files ERR492384
Observação: isso divide o conjunto de dados de DNA genômica baixado em dois arquivos fastq.
- Execute o alinhamento usando bowtie.
- Use este comando no Linux para alinhamento: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_genomicDNA_1. FQ-2 hg_genomicDNA_2. FQ | samtools View-hbuS-| samtools Sort – hg_genomicDNA_sorted. bam.
- Consulte a etapa 4,1 para entender os parâmetros usados no alinhamento do bowtie (pacotes de software).
- Transfira o arquivo Bam genômico alinhado para avaliar o mappability disponível em cima do pedido do autor.
- Use este comando no Linux para alinhamento: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_genomicDNA_1. FQ-2 hg_genomicDNA_2. FQ | samtools View-hbuS-| samtools Sort – hg_genomicDNA_sorted. bam.
- Indexar o arquivo Bam da etapa 4.2.1 usando samtools para torná-lo visível no IGV25 (pacotes de software) para informar ainda mais a Curação manual.
- Use esta linha de comando no Linux: samtools index hg_genomicDNA_sorted. bam
- Avaliar a mappability de cada L1 loci
- Determine o número de leituras mapeadas exclusivamente para L1 loci usando o programa bedtools, a anotação FL-L1 e os dados de sequência genômica alinhados (pacotes de software).
- Use esta linha de comando no Linux: bedtools Coverage-Abam FL-L1-BLAST_RM. GTF – b hg_genomicDNA_sorted. bam ≫ L1_Mappability_hg_genomicDNA. txt.
- Designe um locus L1 para ter o mappability cheio da cobertura quando 400 leituras originais são alinhadas a ela.
- Determine o fator exigido para escalar acima ou para baixo ADN genomic alinhado lê a 400 para cada L1 individual.
- Para ter uma medida dimensionada de expressão de acordo com a mappability individual do locus L1, multiplique o fator determinado na etapa 6.4.3 ao número de leituras do transcrito do RNA que alinham ao L1s autenticamente expressado determinado nas seções 4 – 5.
- Determine o número de leituras mapeadas exclusivamente para L1 loci usando o programa bedtools, a anotação FL-L1 e os dados de sequência genômica alinhados (pacotes de software).
Resultados
As etapas descritas acima e descritas graficamente na Figura 1 foram aplicadas a uma linha celular de tumor prostático humano DU145. A amostra do RNA era citomicamente preparado e era Next-Gen seqüenciada em um poli-a selecionou, vertente-específico, protocolo do par-fim. Usando o bowtie, os arquivos de sequenciamento de fim pareado foram alinhados, permitindo apenas correspondências exclusivas nas quais a leitura de extremidade emparelhada correspondeu melhor a uma localização genômica em comparação a qualquer outro local genômico. Os arquivos de seqüência DU145 foram alinhados ao genoma de referência humana criando um arquivo Bam, que está disponível mediante solicitação do autor. Usando bedtools, os dados foram extraídos dos arquivos Bam separados por vertente DU145 no número de leituras que mapeadas para o comprimento total L1s. Essas leituras foram classificadas em uma planilha de maior a menor e manualmente curadoria examinando o ambiente genómico em torno de cada lócus L1 em IGV para confirmar sua autenticidade (tabela suplementar 1). Se uma amostra foi curada para ser autenticamente expressada, era verde codificado por cores com uma explicação para sua aceitação na coluna mais à direita. Exemplos de loci L1 aceitos para serem expressos autenticamente seguindo as diretrizes descritas na seção de métodos são mostrados na Figura 2a-b. Se uma amostra foi rejeitada para ser autenticamente expressa, ele foi codificado por cores como vermelho com o motivo da rejeição na coluna mais à direita. Exemplos de loci L1 rejeitados por causa da expressão de um promotor diferente de suas próprias diretrizes descritas na seção de métodos são detalhados na Figura 2c-e.
Aqui, somente o L1s completo com uma região intacta do promotor foi estudado. Se esta distinção não é feita, uma grande fonte do ruído transcricional que origina de L1s truncado é introduzida. Exemplos de L1s truncados em DU145 são mostrados na Figura 3a-b onde foram identificados como tendo leituras de RNA-Seq exclusivamente mapeadas. Em IGV, entretanto, é aparente que aquelas transcrições não estiveram iniciadas do L1 truncado, mas da inclusão da seqüência L1 em um gene ou a jusante de um gene expressado.
Globalmente, em DU145, a percentagem de loci L1 de comprimento total e leituras que são rejeitadas como autenticamente expressas L1s após a Curação manual é de aproximadamente 50% (tabela suplementar 2) demonstrando o alto nível de leituras de transcrição mapeada L1 que seria caso contrário, ser gravado como falsos positivos sem Curação manual. Especificamente, em DU145 havia 114 total Full-Length L1 loci para ter exclusivamente mapeado leituras na direção sentido com um total de 3.152 leituras, mas havia apenas 60 loci identificado para ser expressa fora de seu próprio promotor após a Curação manual com 1.879 leituras ( Tabela suplementar 1). Este é o caso, mesmo quando as medidas foram tomadas para reduzir a expressão irrelevante para a biologia L1, selecionando para mRNA citoplasmática. Note-se que o locus com o mais alto nível de transcrições mapeadas em DU145 foi rejeitado por não ser um L1 autenticamente expresso (Figura 4). Em geral, o número de transcrições mapeadas para intervalos específicos de L1 localizam similarmente entre os Locos L1 aceitos e rejeitados como autenticamente expressos após a Curação manual (Figura 4).
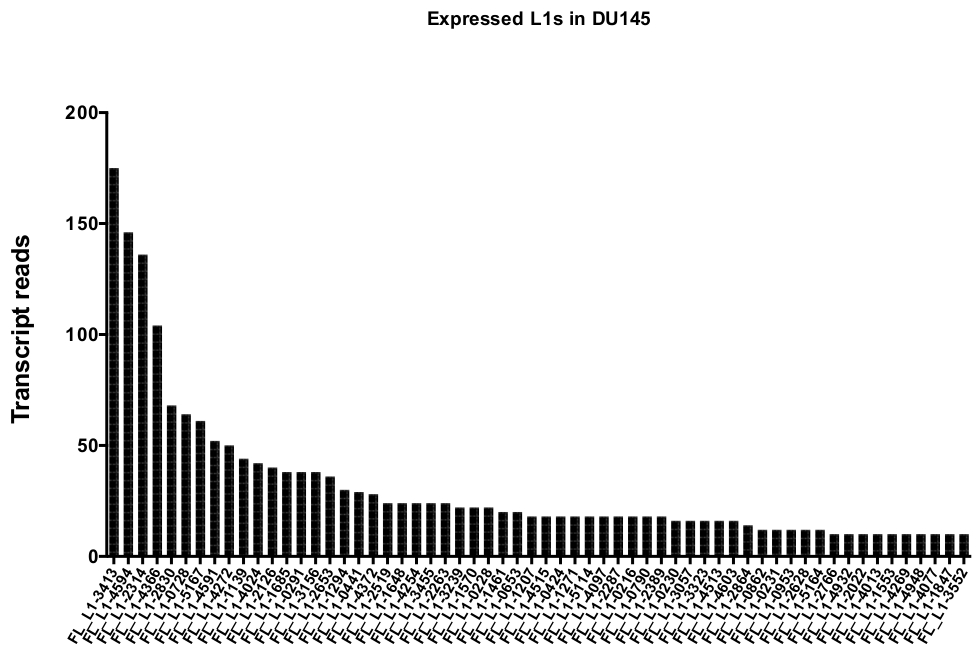
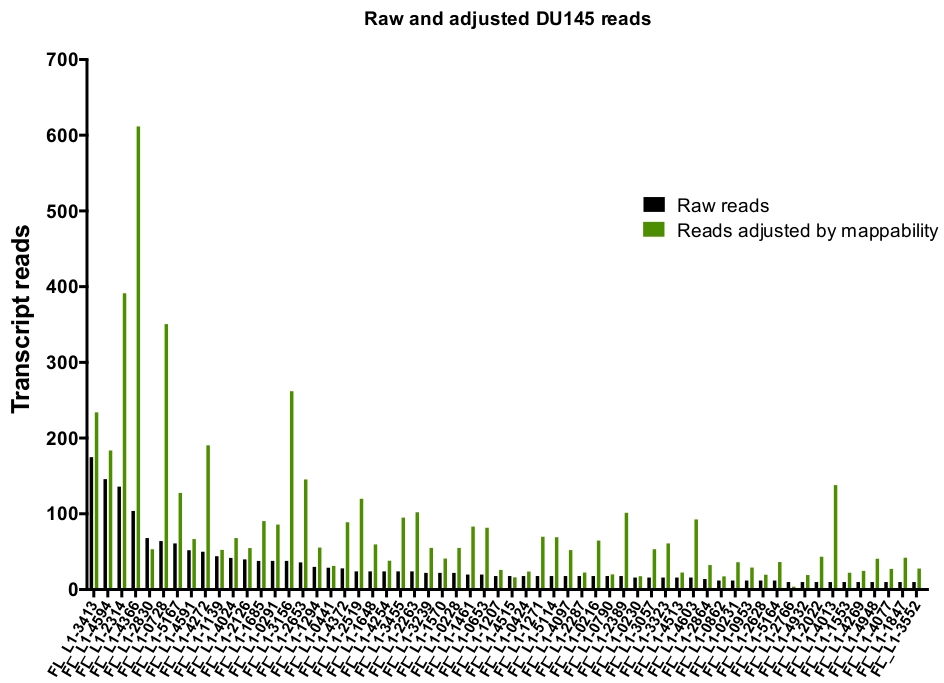
Após a Curação manual, o número de leituras que mapeiam exclusivamente para o loci L1 específico autenticamente expresso em DU145 variam de 175 leituras a um corte mínimo arbitrariamente escolhido de 10 leituras (Figura 5). Essa abordagem de identificar as leituras de transcrição mapeadas exclusivamente para L1s limita a capacidade de quantificar a expressão com precisão. Para isso, foi criado um fator de correção para cada Locus baseado em sua mappability. Para criar esse fator de correção, os primeiros bedtools foram usados para extrair o número de leituras mapeadas exclusivamente do arquivo de Bam genômica HeLa que alinhou a todos os Locos L1 de comprimento total e grafou aqueles loci das leituras de transcrição mapeadas mais altas para as mais baixas (suplementar Figura 1). Foi designado arbitrariamente que L1s com 400 leituras tiveram o mappability cheio da cobertura. O número de leituras capazes de mapear para um locus L1 na amostra de sequenciamento genômica de HeLa foi dimensionado em relação a 400 leituras e que o número em escala foi então multiplicada para o número de leituras que mapearam para cada loci L1 autenticamente expresso em DU145 (tabela suplementar 2) . Como esperado, os elementos L1 que apresentaram maiores escores de correção para a mappability vieram de subfamílias mais jovens como L1PA2 (tabela suplementar 2). Uma vez que as leituras foram ajustadas para escores de mappability em cada locus, a quantitação para a expressão para a maioria dos Locos aumentou (Figura 6). O número de leituras que mapearam exclusivamente para o loci L1 específico autenticamente expresso com correções de mappability em DU145 variou de 612 a 4 leituras e houve uma re-ordenação de loci expressando o mais alto para o mais baixo (Figura 6).

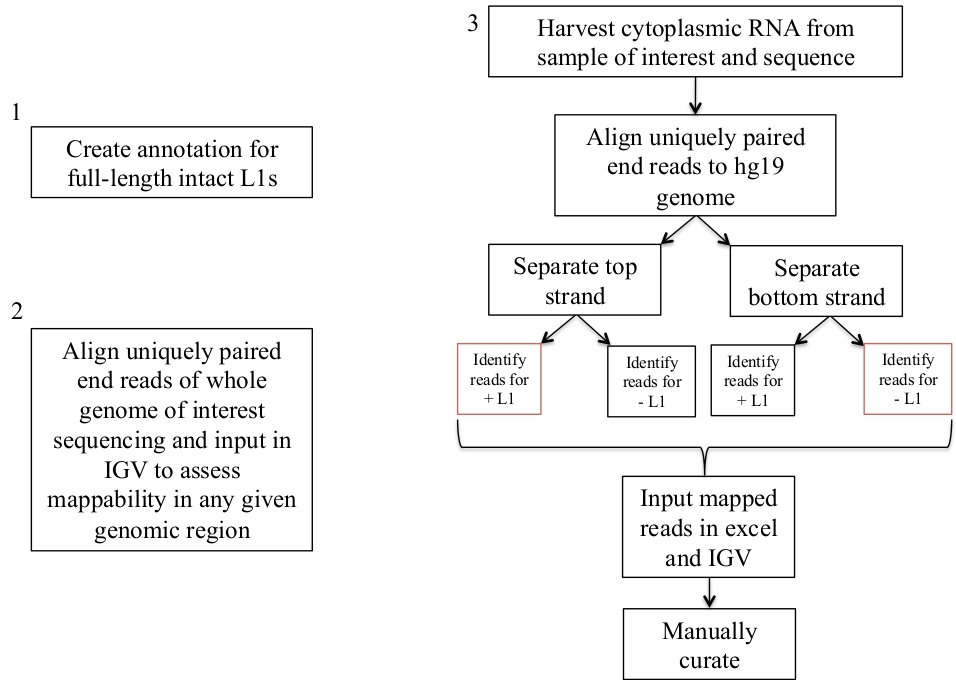
Figura 1: esquema de fluxo de trabalho.
Graficamente descritos são as etapas para identificar L1s expressas em uma amostra humana. Observe que as etapas 1 e 2 não precisam ser repetidas se os arquivos apropriados já estão disponíveis. Estes arquivos apropriados podem ser baixados de suplemento arquivo 1a-b e suplemento arquivo 2. As caixas em vermelho indicam as etapas em que o programa de cobertura de bedtools é usado para contar o número de leituras mapeando para L1s na mesma direção de sentido. Estes loci com sentido orientado mapeamento leituras são o L1s que deve ser manualmente curadoria. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

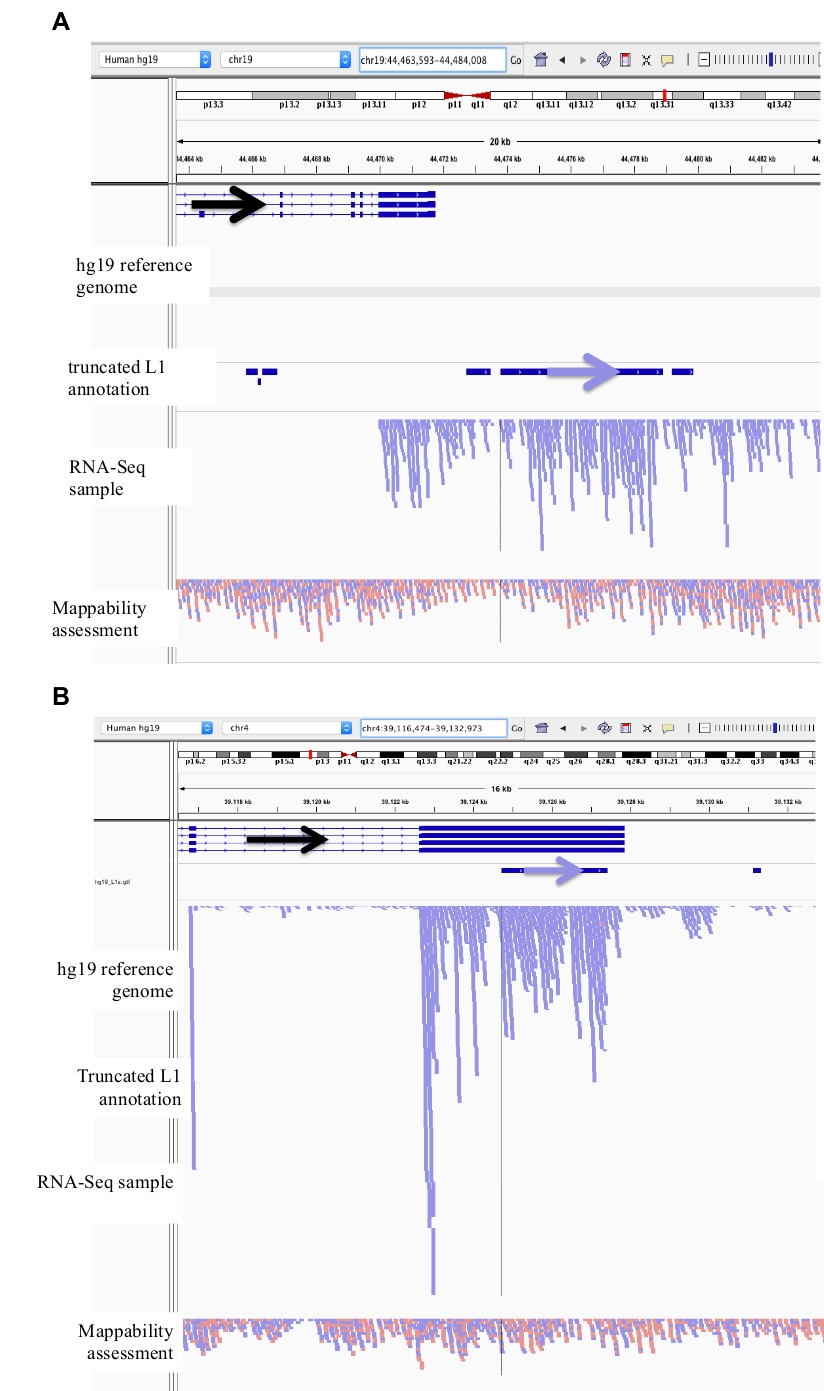
Figura 2: exemplos de Locos L1 com curadoria em DU145.
Carregado em IGV são o genoma de referência, o arquivo de anotação de comprimento total L1 GFF correspondente a versão do genoma de referência (suplemento arquivo 1), o arquivo DU145 Bam e, por último, o arquivo genômica Hela Bam para avaliar a mappability, que estão todos disponíveis em cima do autor Solicitação. As setas foram adicionadas para auxiliar na visualização da direção do L1 anotado. Setas e leituras em vermelho são orientadas em seqüência da direita para a esquerda. Setas e leituras em azul são orientadas em seqüência da esquerda para a direita. a) em IGV, este Locus L1 parece ser expressa fora de seu próprio promotor, pois não há leituras a montante do L1 na orientação sentido para mais de 5 KB. Este L1 tem baixa mappability, não está em um gene, e tem a evidência da atividade esperada do promotor do antisense26. b) em IGV, este Locus L1 parece ser expressa fora de seu próprio promotor como não há leituras a montante do L1 na orientação sentido para mais de 5 KB. Este L1 tem baixa mappability e está dentro de um gene de direção oposta. c) em IGV, este Locus L1 foi rejeitado como um L1 expresso porque há leituras ascendentes na mesma orientação dentro de 5 KB. Este L1 está dentro de um gene do mesmo sentido assim que o Transcript lê é originando o mais provável do promotor do gene expressado. d) em IGV, este Locus L1 foi rejeitado como um L1 expresso porque há leituras ascendentes na mesma orientação dentro de 5 KB. Este L1 é a jusante de um gene altamente expressado no mesmo sentido assim que o Transcript lê é originando o mais provável do promotor daquele gene expresso e estendendo além do terminador normal do gene. e) em IGV, este Locus L1 foi rejeitado como um L1 expresso porque há leituras ascendentes na mesma orientação dentro de 5 KB. Este L1 não está dentro ou perto de um gene anotado no gene de referência assim que a origem destas transcrições dentro e a montante do elemento L1 sugerem um promotor un-anotated. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: o ruído de fundo origina do L1s truncado também.
Nossa anotação L1 não inclui truncado L1s como eles são uma fonte principal de ruído de fundo. As setas foram adicionadas para auxiliar na visualização da direção do L1 anotado. Setas e leituras em azul são orientadas em seqüência da esquerda para a direita. a) demonstrado é um exemplo de um L1 truncado na SUFAMÍLIA L1MB5 que é 2706 bps. Em IGV é aparente que as leituras originam da extensão a jusante de um gene expressado. b) mostrado é outro exemplo de um L1 truncado. Este L1 é um L1PA11 que é 4767 bps de comprimento. Em IGV é evidente que as leituras que mapeam excepcionalmente ao L1 originam do exon expressado, que o L1 está dentro. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: o transcrito lê esse mapa excepcionalmente a todo o L1s intacto completo no genoma humano expressado na linha de pilha do tumor da próstata DU145.
No preto estão os Locus específicos a ser identificados como autenticamente expressos depois que o curadoria manual e no vermelho é o loci específico a ser rejeitado como leituras autenticamente expressas após o curadoria manual. Em cinza são loci com menos de dez leituras de mapeamento para cada um. Como esses loci representam uma pequena fração de leituras de transcrição, eles não foram manualmente curate. As marcas de escala do eixo x denotam cada 100 de comprimento total, L1s intacto. aproximadamente 4.500 loci não são mostrados graficamente como eles tinham zero mapeado leituras. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: transcrição lê que o mapa exclusivamente para autenticamente expressos full-length L1s intacta na linha celular tumor de próstata DU145.
São mostrados os números de leituras de transcrição que mapeiam para Locos específicos em células DU145 após a Curação manual. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: lê o mapeamento para o L1 autenticamente expresso quando ajustado por mappability.
São mostrados os números de leituras de transcrição ajustadas por pontuações de mappability específicas do loci que mapeiam para Locos L1 manualmente curados em células DU145. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Arquivo suplementar 1: anotações para o corpo inteiro, L1s humano intacto de acordo com a orientação. a) FL-L1-BLAST_RM_minus. GFF. b) FL-L1-BLAST_RM_plus. GFF. Por favor, clique aqui para baixar este arquivo.
Arquivo suplementar 2: scripts de supercomputador usados para automatizar o pipeline de Bioinformática detalhado na seção 4. Por favor, clique aqui para baixar este arquivo.
Figura suplementar 1: amostra de DNA genômica usada para determinar a mappability L1.
São mostrados o número de leituras de transcrição genômica da amostra de linha de célula HeLa que mapeiam exclusivamente para todos os 5.000 locos de comprimento total L1 no genoma. Foi designado que um L1 tem o mappability cheio da cobertura quando 400 lê o mapa ao L1. Por favor clique aqui para baixar esta figura.
Tabela suplementar 1: Curação manual de L1s em DU145. Por favor clique aqui para baixar esta tabela.
Tabela suplementar 2: L1s curadoria em DU145 com ajuste de mappability. Por favor clique aqui para baixar esta tabela.
Discussão
A atividade L1 tem demonstrado causar dano genético e instabilidade contribuindo para a doença27,28,29. Das aproximadamente 5.000 cópias L1 full-length, somente algumas dúzias evolutionariamente jovens L1s respondem para a maioria da atividade do retrotransposição2. Entretanto, há uma evidência que mesmo alguns mais velhos, retrotransposicionalmente-incompentent L1s são ainda capazes de produzir proteínas prejudiciais do ADN30. Para apreciar inteiramente o papel de L1s na instabilidade e na doença genomic, a expressão L1 a nível locus-específico deve ser compreendida. Entretanto, o fundo elevado de seqüências L1-relacionadas incorporados em outros RNAs não relacionados à retrotransposição L1 levanta um desafio significativo em interpretar a expressão L1 autêntica. Outro desafio na identificação e, portanto, compreensão de padrões de expressão de loci L1 individuais ocorre por causa de sua natureza repetitiva que não permite que muitas seqüências de leitura curtas para mapear para um único Locus único. Para superar esses desafios, desenvolvemos a abordagem descrita acima na identificação da expressão de Locos L1 individuais usando dados de RNA-Seq.
Nossa abordagem filtra o alto nível (mais de 99%) de ruído transcricional gerado a partir de sequências L1 que não estão relacionadas com a retrotransposição L1, tomando uma série de passos. O primeiro passo envolve a preparação do RNA citoplasmático. Selecionando para o RNA cytoplasmic, as leituras L1-relacionadas encontradas dentro do mRNA intronic expressado no núcleo são esgotadas significativamente. Na preparação da biblioteca de sequenciamento, outra etapa tomada para reduzir o ruído transcricional não relacionada ao L1s incluem a seleção de transcrições poliadeniladas. Isso remove o ruído de transcrição relacionado ao L1 encontrado em espécies não mRNA. Uma outra etapa inclui arranjar em seqüência vertente-específica a fim identificar e eliminar transcritos L1-relacionados do antisense. O uso de uma anotação para o L1s completo com regiões de promotor funcionais ao identificar o número de transcritos RNA-Seq que mapeiam para L1s também elimina o ruído de fundo que de outra forma se origina de L1s truncado. Finalmente, a última etapa crítica na eliminação do ruído transcricional das sequências L1 não relacionadas à retrotransposição L1 é a Curação manual de L1s de comprimento total identificada para ter mapeado transcrições de RNA-Seq. A Curação manual envolve a visualização de cada lócus L1 bioinformaticamente identificado-a-ser-expresso no contexto de seu ambiente genómico circunvizinho para confirmar que a expressão se origina do promotor L1. Esta aproximação foi aplicada a DU145, uma linha celular do tumor da próstata. Mesmo com todas as etapas relacionadas à preparação adotadas para reduzir o ruído de fundo, aproximadamente 50% dos Locos L1 identificados bioinformaticamente em DU145 foram rejeitados como ruído de fundo L1 proveniente de outras fontes transcricional (Figura 4), enfatizando o rigor exigido para produzir resultados confiáveis. Essa abordagem usando a Curação manual é trabalhosa, mas necessária no desenvolvimento deste pipeline para avaliar e compreender o ambiente genómico em torno de um L1 de comprimento total. Os próximos passos incluem a redução da quantidade de curadoria manual necessária automatizando algumas das regras de curadoria, embora devido à natureza ainda não completamente conhecida da expressão genômica, fontes de expressão não anotadas no genoma de referência, regiões de baixa mappability, e mesmo complicando fatores envolvidos com a construção de um genoma da referência não é ser possível automatizar inteiramente a Curação L1 neste tempo.
O segundo desafio na identificação da expressão de Locos L1 individuais com sequenciamento relaciona-se com o mapeamento de transcrições L1 repetitivas. Nesta estratégia de alinhamento, é necessário que uma transcrição deve alinhar de forma única e colinearmente ao genoma de referência para ser mapeado. Ao selecionar para sequências de extremidade emparelhada que mapeiam concoremente, a quantidade de transcrições que se alinham exclusivamente a Locos L1 encontrados no genoma de referência aumenta. Essa estratégia de mapeamento exclusivo fornece confiança no chamado de mapeamento de leituras especificamente para um único Locus L1, embora potencialmente subestimar a quantidade de expressão de cada L1 repetitivo identificado para ser autenticamente expresso. Para aproximadamente corrigir essa subestimação, um escore de "mappability" para cada Locus L1 baseado em sua mappability foi desenvolvido e aplicado ao número de leituras de transcrição exclusivamente mapeadas (Figura 6). É de notar que, idealmente, a mappability deve ser pontuada para a cobertura completa lê através do comprimento total L1 de acordo com a amostra WGS correspondente. Aqui, nós usamos WGS de pilhas de HeLa para determinar contagens do mappability de cada Locus L1 a fim inflar ou esvaziar leituras que mapeiam ao loci L1 em linhas de pilha do tumor da próstata DU145. Este cálculo do mappability é uma contagem bruta da correção, mas o ' mappability completo da cobertura escolhida de 400 leituras foi determinado com a natureza dinâmica de linhas de pilha do tumor na mente. Pode ser observado na Figura 1 suplementar, que há alguns Locos L1 com HELA WGS com número extremamente alto de leituras mapeadas. Estes provavelmente vêm das seqüências duplicadas do cromossoma dentro de HeLa que não estão dentro do genoma da referência, que é porque aqueles loci não foram escolhidos para ser representativos da cobertura completa do mappability. Em vez disso, foi determinado que a média de 100% de cobertura de leitura ocorre em torno de 400 leituras de acordo com a Figura 1 suplementar e, em seguida, foi assumido que esta média se aplica à linha de células da próstata tumor DU145 também.
Esta estratégia do alinhamento com 100-200 BP lê da tecnologia do RNA-Seq seleciona preferencialmente também para evolutionarmente mais velho L1s dentro do genoma da referência como L1s mais velhos acumularam sobre mutações originais do tempo que os fazem mais mappable. Esta abordagem, portanto, tem sensibilidade limitada quando se trata de identificar o mais jovem de L1s, bem como não-referência, L1s polimóica. Para identificar o mais novo de L1s, sugerimos o uso de 5 ' RACE seleção de transcritos L1 e tecnologia de sequenciamento como PacBio que fazem uso de leituras mais21. Isto permite o mapeamento mais original e conseqüentemente a identificação confiável do expressado, jovem L1s. o uso de RNA-Seq e PacBio abordagens em conjunto pode levar a uma lista mais abrangente de L1s autenticamente expressa. Para identificar os L1s polimoróricos autenticamente expressos, os primeiros passos seguintes incluem a construção e inserção de sequências popoliméricos no genoma de referência.
Os desafios biológicos e técnicos no estudo de sequências de repetição são grandes, embora com o procedimento rigoroso acima para remover o ruído transcricional das sequências L1 não relacionadas ao retrotransposição usando a tecnologia de sequenciamento de RNA, começamos a peneirar os grandes níveis de ruído de fundo transcricional e ser para identificar com confiança e de forma estrita os padrões de expressão L1 e a quantidade no nível do locus individual.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Gostaríamos de agradecer ao Dr. Yan Dong pelas células tumorais da próstata DU145. Gostaríamos de agradecer ao Dr. Nathan Ungerleider por sua orientação e aconselhamento na criação de scripts de supercomputador. Parte deste trabalho foi financiado pela NIH Grants R01 GM121812 para PD, R01 AG057597 para VPB, e 5TL1TR001418 para TK. Também gostaríamos de reconhecer o apoio do cancer Crusaders e do centro de bioinformática do Tulane Cancer Center.
Materiais
| Name | Company | Catalog Number | Comments |
| 1 M HEPES | Affymetrix | AAJ16924AE | |
| 5 M NaCl | Invitrogen | AM9760G | |
| Agilent bioanalyzer 2100 | Agilent technologies | ||
| Agilent RNA 6000 Nano Kit | Agilent technologies | 5067-1511 | |
| bedtools.26.0 | https://bedtools.readthedocs.io/en/latest/content/installation.html | ||
| bowtie-0.12.8 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.8/ | ||
| Cell scraper | Olympus plastics | 25-270 | |
| Chloroform | Fisher | C298-500 | |
| Digitonin | Research Products International Corp | 50-488-644 | |
| Ethanol | Fisher | A4094 | |
| Gibco (Phosphate Buffered Saline) | Invitrogen | 10-010-049 | |
| Homogenizer | Thomas Scientific | BBI-8541906 | |
| IGV 2.4 | https://software.broadinstitute.org/software/igv/download | ||
| Isopropanol | Fisher | A416-500 | |
| mac2unix | https://sourceforge.net/projects/cs-cmdtools/files/mac2unix/ | ||
| Q-tips | Fisher | 23-400-122 | |
| RNAse later solution | Invitrogen | AM7022 | |
| RNaseZap RNase Decontamination Solution | Invitrogen | AM9780 | |
| samtools-1.3 | https://sourceforge.net/projects/samtools/files/ | ||
| sratoolkit.2.9.2 | https://github.com/ncbi/sra-tools/wiki/Downloads | ||
| SUPERase·In RNase Inhibitor | Invitrogen | AM2694 | |
| Trizol | Invitrogen | 15-596-018 | |
| Water (DNASE, RNASE free) | Fisher | BP2484100 |
Referências
- International Human Genome Sequencing. Initial sequencing and analysis of the human genome. Nature. 409, 860 (2001).
- Brouha, B., et al. Hot L1s account for the bulk of retrotransposition in the human population. Proceedings of the National Academy of Sciences of the United States of America. 100 (9), 5280-5285 (2003).
- Dombroski, B. A., Mathias, S. L., Nanthakumar, E., Scott, A. F., Kazazian, H. H. Isolation of an active human transposable element. Science. 254 (5039), 1805 (1991).
- Swergold, G. D. Identification, characterization, and cell specificity of a human LINE-1 promoter. Molecular and Cellular Biology. 10 (12), 6718-6729 (1990).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular and Cellular Biology. 21 (6), 1973-1985 (2001).
- Deininger, L., Batzer, M. A., Hutchison, C. A., Edgell, M. H. Master genes in mammalian repetitive DNA amplification. Trends in Genetics. 8 (9), 307-311 (1992).
- Boissinot, S., Chevret, P., Furano, A. L1 (LINE-1) Retrotransposon Evolution and Amplification in Recent Human History. Molecular Biology and Evolution. 17 (6), 915-918 (2000).
- Khazina, E., Weichenrieder, O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proceedings of the National Academy of Sciences of the United States of America. 106 (3), 731-736 (2009).
- Martin, S. L., Bushman, F. D. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Molecular and Cellular Biology. 21 (2), 467-475 (2001).
- Feng, Q., Moran, M. H., Kazazian, H. H., Boeke, J. D. Human L1 Retrotransposon Encodes a Conserved Endonuclease Required for Retrotransposition. Cell. 87 (5), 905-916 (1996).
- Mathias, S. L., Scott, A. F., Kazazian, H. H., Boeke, J. D., Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science. 254 (5039), 1808 (1991).
- Luan, D. D., Korman, M. H., Jakubczak, J. L., Eickbush, T. H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 72 (4), 595-605 (1993).
- van den Hurk, J. A. J. M., et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Human Genetics. 113 (3), 268-275 (2003).
- Miné, M., et al. A large genomic deletion in the PDHX gene caused by the retrotranspositional insertion of a full-length LINE-1 element. Human Mutation. 28 (2), 137-142 (2007).
- Solyom, S., et al. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Human Mutation. 33 (2), 369-371 (2012).
- Hancks, D. C., Kazazian, H. H. Roles for retrotransposon insertions in human disease. Mobile DNA. Mobile DNA. 7, 9-9 (2016).
- Tubio, J. M. C., et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345 (6196), 1251343-1251343 (2014).
- Ewing, A. D., et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Research. 25 (10), 1536-1545 (2015).
- Beck, C. R., Garcia-Perez, J. L., Badge, R. M., Moran, J. V. LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics. 12, 187-215 (2011).
- Philippe, C., et al. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife. 5, e13926 (2016).
- Deininger, P., et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Research. 45 (5), e31-e31 (2017).
- Jin, Y., Tam, O. H., Paniagua, E., Hammell, M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 31 (22), 3593-3599 (2015).
- . . Agilent RNA 6000 Nano Kit Guide. , (2017).
- Mueller, O. L., Schroeder, A. . RNA Integrity Number (RIN) –Standardization of RNA Quality Control. , (2016).
- Robinson, J. T., et al. Integrative genomics viewer. Nature Biotechnology. 29, 24 (2011).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular Cellular Biology. 21 (6), 1973-1985 (2001).
- Belancio, V. P., Deininger, L., Roy-Engel, A. M. LINE dancing in the human genome: transposable elements and disease. Genome Medicine. 1 (10), 97-97 (2009).
- Iskow, R. C., et al. Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell. 141 (7), 1253-1261 (2010).
- Scott, E. C., et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Research. 26 (6), 745-755 (2016).
- Kines, K. J., Sokolowski, M., deHaro, D. L., Christian, C. M., Belancio, V. P. Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci. Nucleic Acids Research. 42 (16), 10488-10502 (2014).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados