
Method Article
Transcrição iniciar o mapeamento do site usando super-baixa entrada Carrier-CAGE
Neste Artigo
Resumo
A análise do tampão da expressão de gene (gaiola) é um método para o mapeamento quantitativo genoma-largo de mRNA 5 ' extremidades para capturar locais do começo da transcrição do RNA polymerase II em uma definição single-nucleotide. Este trabalho descreve um protocolo de baixa entrada (SLIC-CAGE) para a geração de bibliotecas de alta qualidade usando nanograma-quantidades de RNA total.
Resumo
A análise do tampão da expressão de gene (gaiola) é um método usado para a deteção da definição do único-nucleotide de locais do começo da transcrição do RNA polymerase II (TSSs). A deteção exata de TSSs realça a identificação e a descoberta de promotores principais. Além disso, potenciadores ativos podem ser detectados através de assinaturas de iniciação de transcrição bidirecional. É descrito aqui um protocolo para executar a portador-gaiola da entrada super-baixa (SLIC-gaiola). Esta adaptação de SLIC do protocolo da gaiola minimiza perdas do RNA artificialmente aumentando a quantidade do RNA com o uso de uma mistura in vitro transcrita do portador do RNA que seja adicionada à amostra do interesse, assim permitindo a preparação da biblioteca do nanogram-quantidades do total RNA (i.e., milhares de células). O portador imita a distribuição esperada do comprimento do fragmento da biblioteca do ADN, eliminando desse modo os vieses que poderiam ser causados pela abundância de um portador homogêneo. Nos últimos estágios do protocolo, o portador é removido com a degradação com endonucleases homing e a biblioteca do alvo é amplificada. A biblioteca-alvo da amostra é protegida da degradação, porque os locais de reconhecimento do endonuclease do homing são longos (entre 18 e 27 BP), fazendo a probabilidade de sua existência nos genomas eucarióticas muito baixos. O resultado final é uma biblioteca de DNA pronta para sequenciamento de próxima geração. Todas as etapas do protocolo, até o seqüenciamento, podem ser concluídas dentro de 6 dias. A preparação do transportador requer um dia de trabalho completo; no entanto, ele pode ser preparado em grandes quantidades e mantido congelado em-80 ° c. Uma vez sequenciado, as leituras podem ser processadas para obter a resolução de nucleotídeo genoma-Wide TSSs. TSSs pode ser usado para a descoberta principal do promotor ou do potenciador, fornecendo a introspecção no Regulamento do gene. Uma vez agregado aos promotores, os dados também podem ser usados para criação de perfil de expressão centrada em 5 '.
Introdução
A análise do tampão da expressão de gene (gaiola) é um método usado para o mapeamento genoma-largo da definição do único-nucleotide de locais do começo da transcrição do RNA polymerase II (TSSs)1. Sua natureza quantitativa também permite 5 '-End centric perfil de expressão. As regiões que cercam os Tsss (cerca de 40 BP a montante e a jusante) são promotores centrais e representam o local físico onde o RNA polimerase II e os fatores de transcrição geral se ligam (revistos anteriormente2,3). As informações sobre locais exatos de TSSs podem ser usadas para a descoberta do promotor principal e para monitorar a dinâmica do promotor. Além disso, como potenciadores ativos exibem assinaturas de transcrição bidirecional, os dados CAGE também podem ser usados para descoberta de potenciador e monitoramento da dinâmica do potenciador4. A metodologia CAGE tem aumentado recentemente em popularidade devido à sua ampla aplicação e uso em projetos de pesquisa de alto perfil, como ENCODE5, modencode6e Fantom Projects7. Além, a informação de TSS está provando igualmente ser importante para distinguir o tecido saudável e doente, porque os TSSs doença-específicos podem ser usados para finalidades diagnósticas8.
Mesmo que vários métodos para mapeamento TSS estão disponíveis (CAGE, RAMPAGE, STRT, nanoCAGE, nanoCAGE-XL, oligo-tampando), nós e outros têm mostrado recentemente que gaiola é o método mais imparcial para capturar TSSs verdadeiro com o menor número de falsos positivos9 , o protocolo 10. The recente da gaiola, NAnT-icage11, é o protocolo o mais imparcial para o perfilamento de TSS, porque evita cortar os fragmentos às etiquetas curtas usando enzimas da limitação e não usa a amplificação do PCR. Uma limitação do protocolo nAnT-iCAGE é a exigência de uma grande quantidade de material de partida (por exemplo, 5 μg de RNA total para cada amostra). Para responder a perguntas específicas, biologicamente relevantes, muitas vezes é impossível obter tais quantidades elevadas de material de partida (por exemplo, para células classificadas por FACS ou estágios embrionários precoces). Finalmente, se nAnT-iCAGE for bem-sucedido, apenas 1-2 ng de material de biblioteca de DNA está disponível a partir de cada amostra, limitando assim a profundidade de sequenciamento alcançável.
Para permitir o perfilamento de TSS usando somente nanogramas do RNA total, nós desenvolvemos recentemente o portador-gaiola10 da super-baixa entrada (SLIC-gaiola, Figura 1). O SLIC-CAGE requer apenas 10 ng de RNA total para obter bibliotecas de alta complexidade. Nosso protocolo confia no portador sintético com cuidado projetado do RNA adicionado ao RNA do interesse para conseguir um total de 5 μg do material do RNA. O portador sintético imita a biblioteca do ADN do alvo na distribuição do comprimento para evitar os vieses potenciais que poderiam ser causados por moléculas homogêneas no excesso. A seqüência do portador é baseada na seqüência do gene da sintetase de leucyl-tRNA de Escherichia coli (tabela 1) para duas razões. Primeiro, qualquer sobra do portador na biblioteca final, mesmo se sequenciado, não mapeará para um genoma eucariótico. Em segundo lugar, como E. coli é uma espécie mesófilos, seus genes do Housekeeping são otimizados para a escala de temperatura apropriada para SLIC-Cage. A seqüência transportadora também é incorporado com sites de reconhecimento de endonuclease homing para permitir a degradação específica do DNA derivado das moléculas de RNA transportadora. O alvo, a biblioteca derivada da amostra permanece intacto, como os locais de reconhecimento de endonuclease homing são longos (I-CeuI = 27 BP; I-SceI = 18 BP) e estatisticamente improvável de ser encontrado em genomas eucarióticos. Após a degradação específica do portador e a remoção dos fragmentos pela exclusão do tamanho, a biblioteca do alvo é PCR amplificada e pronta para arranjar em seqüência da próxima geração. Dependendo da quantidade inicial de RNA (1-100 ng), espera-se que entre 13-18 ciclos de amplificação de PCR sejam necessários. A quantidade final de DNA por cada amostra varia entre 5-50 ng, produzindo material suficiente para seqüenciamento muito profundo. Ao usar somente 1-2 ng do RNA total, os TSSs verdadeiros podem ser detectados; no entanto, espera-se que as bibliotecas sejam de menor complexidade. Por último, como SLIC-CAGE é baseado no protocolo nAnT-iCAGE11, ele permite a multiplexação de até oito amostras antes do sequenciamento.
Protocolo
1. preparação do transportador
-
Preparação de modelos de DNA para transcrição in vitro
- Prepare a mistura do PCR para cada molde do PCR combinando 41 μL da água, 20 μL do amortecedor de 5x HF, 8 μL de 2,5 milímetros dNTPs, 10 μL do primer dianteiro original de 10 μM (PCR_GN5_f1, tabela 2; os primers são dissolvidos e diluídos na água) , 10 μL de plasmídeo modelo de 2 ng/μL contendo o gene portador sintético e 1 μL de Phusion polymerase. Misture a mistura de PCR introduzindo pipetagem. Uma mistura mestra para todos os 10 modelos pode ser preparada de uma só vez (Prepare-se para 11 reações).
- Adicionar 90 μL da mistura de PCR a 10 μL de cada primer reverso de 10 μM (PCR_N6_r1-R10, tabela 2). Misture com pipetagem.
- A PCR amplifica os modelos utilizando o seguinte programa: 98 ° c para 60 s, (98 ° c para 10 s, 50 ° c para 30 s, 72 ° c para 30 s) 35 ciclos, 72 ° c por 10 min, segure a 4 ° c.
-
Purificação de gel de modelos de DNA amplificados por PCR
- Prepare um gel de agarose a 1% (o agarose de baixa fusão é recomendado).
- Para diminuir o volume, concentre as misturas de reacção de PCR de 100 μL a 20 μL de volume total utilizando o concentrador a vácuo a uma temperatura média baixa (30 – 40 ° c).
- Adicione 6 μL do corante de carregamento 6x, misture bem e carregue no gel. Funcione a electroforese por 30 minutos no amortecedor de 1x TAE na tensão apropriada para o tanque usado da electroforese (5 – 10 V/cm). Em paralelo executar uma 100 BP ou 1.000 BP escada de DNA.
- Usando um bisturi limpo, extirpar as fatias de gel contendo o produto alvo PCR. Evite o excesso de gel de agarose. Purify os produtos do PCR usando um jogo da extração do gel (de acordo com as instruções do fabricante).
Nota: A260/A230 rácios de DNA isolados de géis de agarose são tipicamente baixos (0,1 – 0,3). O produto-alvo esperado e os produtos laterais são mostrados na Figura 2a. Os rendimentos esperados de 100 μL de reações de PCR são 1,2 – 3 μg. as reações podem ser ampliadas para obter um rendimento mais elevado.
-
Transcrição in vitro de moléculas transportadoras
- Transcribe o portador RNA in vitro usando o T7 RNA polymerase de acordo com as instruções do fabricante. Configurar 10 – 20 μL de reações (o kit recomendado está na tabela de materiais).
- Purify o RNA transcrito in vitro usando um kit de purificação de RNA. Configurar a digestão do DNA em solução usando DNase eu seguindo as instruções padrão do fabricante, e eluir o RNA em 50 μL de água. Para aumentar o rendimento de eluição, deixe a água na coluna durante 5 minutos antes da centrifugação.
Nota: Tenha cuidado para não exceder a capacidade máxima de ligação das colunas (no kit mencionado na tabela de materiais, a capacidade é de até 100 μg). O rendimento esperado dos modelos de PCR 1 – 10 (1 KBP a 200 BP de comprimento) é de 25 – 50 μg de 10 μL de reações de transcrição in vitro. As reações podem ser ampliadas para obter um estoque maior de moléculas de portador.
-
Nivelamento de moléculas de RNA de portador transcritas in vitro
- Prepare a mistura de nivelamento combinando 2 μL de tampão tampando 10x, 1 μL de GTP de 10 milímetros, 1 μL de SAM de 2 milímetros (diluído recentemente), e 1 μL da enzima tampando de vaccinia por o RNA do portador.
- Misture até 10 μg de cada molécula transportadora em 15 μL de volume total e de desnaturamento durante 10 min a 65 ° c. Coloc no gelo imediatamente para impedir a formação secundária da estrutura.
- Misture o RNA portador desnaturado com 5 μL da mistura tampando e incubar por 1 h a 37 ° c.
- Purify moléculas de RNA tampadas usando um kit de purificação de RNA — siga o protocolo de limpeza do fabricante. RNA elute em 30 μL de água. Para aumentar o rendimento de eluição, deixe a água na coluna durante 5 minutos antes da centrifugação.
Nota: Meça a concentração usando o espectrofotômetro do microespectrômetro. A relação A260/A280 esperada é > 2 e A260/A230 é > 2. Note que para algumas amostras de RNA A260/A230 pode estar entre 1,3 – 2. O rendimento esperado ao utilizar 10 μg de RNA não tampado é de 9 – 10 μg de RNA tampado.
- Prepare a mistura do transportador tampado e sem tampos combinando os valores descritos na tabela 3. Misture bem sacudendo o tubo e meça a concentração usando o espectrofotômetro do microespectrômetro.
Nota: Se for necessária uma maior concentração do transportador para caber na reacção de transcrição inversa (ver abaixo), a mistura transportadora pode ser concentrada utilizando o concentrador a vácuo a baixa temperatura média (30-35 ° c) até atingir a concentração final desejada. As etapas 2 – 14 são modificadas a partir do protocolo padrão nAnT-iCAGE relatado por Murata et al.11
2. transcrição reversa
- Combine 1 μL do primer RT (2,5 mM TCT-N6 dissolvido em água, para a seqüência ver tabela complementar 1), 10 ng de RNA total de interesse e 4.990 ng de mistura transportadora (tabela 3) em 10 μL de volume total em uma placa de PCR de baixa ligação. Misture por sacudendo o tubo.
Nota: Se o RNA da amostra for demasiado diluído para transcrição inversa (ver abaixo), combine-o com a quantidade adequada da transportadora, concentre-se utilizando o concentrador de vácuo para 9 μL de volume total e adicione 1 μL do primer RT. Adicionando o Earl portador, para alcangar 5 μg do RNA no total impede a perda da amostra. - Aqueça a mistura da etapa 2,1 em 65 ° c por 5 minutos, e coloc no gelo imediatamente para impedir o renaturation.
-
Preparar a mistura de transcrição reversa (RT).
- Para cada amostra, combine 6,1 μL de água (RNase-e DNase-Free), 7,6 μL de tampão de primeira vertente 5x, 1,9 μL de 0,1 M DTT, 1 μL de dNTPs de 10 mM, 7,6 μL de mistura de trealose/sorbitol (ver receita em Murata et al.11) e 3,8 μl da transcriptase reversa recomendada (ver < c 6 > tabela de materiais). Misture bem, sacudendo o tubo.
- Adicionar 28 μL da mistura RT no tubo de PCR com 10 μL de RNA, transportadora e o primer RT (volume total 38 μL). Misture bem com pipetagem.
Nota: A mistura é altamente viscosa devido ao Trehalose/sorbitol. Misture até visivelmente homogêneo. - Incubar em um Thermal cycler usando o seguinte programa: 25 ° c para 30 s, 50 ° c para 60 minutos, e preensão em 4 ° c.
-
Purificação de cDNA: híbridos de RNA usando grânulos magnéticos SPRI
- Adicionar 68,4 μL dos grânulos RNAse e DNase-Free SPRI recomendados (ver tabela de materiais) a 38 μl da mistura RT (grânulos para a proporção da amostra 1.8:1). Misture bem introduzindo pipetagem e incubar durante 5 min à temperatura ambiente (RT).
- Separe os grânulos em um suporte magnético por 5 min. descarte o sobrenadante e lave as contas duas vezes com 200 μL de 70% de etanol (preparado na hora).
Nota: O etanol é adicionado aos grânulos sem misturar e enquanto o tubo está no suporte magnético. O etanol adicionado é imediatamente removido. Cuidado deve ser tomado para não perder qualquer miçangas durante as lavas, pois pode levar à perda da amostra. - Enquanto o tubo ainda está no suporte magnético, retire todos os vestígios de etanol. Gotículas de etanol podem ser removidas e empurradas para fora do tubo usando uma pipeta P10. Não deixe as contas secas.
- Adicione 42 μL de água pré-aquecida a 37 ° c aos grânulos e eluir a amostra introduzindo com pipting para cima e para baixo 60x.
Nota: Tenha cuidado para não causar a formação de espuma por pipetagem, pois pode causar perda de grânulos (ou seja, amostra ligada) na espuma. - Incubar em 37 ° c por 5 minutos sem a tampa para permitir a evaporação de quantidades de traço do etanol.
- Separe os grânulos em um suporte magnético por 5 min e transfira o sobrenadante para uma nova placa.
Nota: Tente recuperar todo o sobrenadante para impedir a perda da amostra ao evitar o Carryover do grânulo. Use a pipeta P10 para obter as últimas gotas de amostra.
3. oxidação
- Adicionar 2 μL de NaOAc de 1 M (pH 4,5) na reacção de RT purificada. Misturar com pipetagem, adicionar 2 μL de 250 mM NaIO4 e misturar novamente.
- Incubar no gelo por 45 min. Cubra a placa com a folha de alumínio para evitar a luz.
- Adicionar 16 μL de Tris-HCl (pH 8,5) na mistura de oxidação para neutralizar o pH.
- Purify o cDNA oxidado: híbrido do RNA usando grânulos magnéticos de SPRI. Adicione 108 μL de grânulos de SPRI a 60 μL da mistura da oxidação (1.8:1 grânulos à relação da amostra). Repita a purificação conforme descrito nas etapas 2.6.1 – 2.6.6. Elute usando 42 μL de água pré-aquecida a 37 ° c.
Nota: Prepare recentemente 250 mM NaIO4 adicionando 18,7 μL de água por 1 mg de Naio4. O NaIO4 é sensível à luz; Portanto, manter a solução em um tubo coberto com folha de alumínio ou em um tubo resistente à luz.
4. biotinilação
- Adicionar 4 μL de NaOAc de 1 M (pH 6,0) no tubo contendo a amostra oxidada purificada e misturar com pipetagem.
- Adicionar 4 μL de solução de biotina a 10 mM, misturar com pipetagem e incubar durante 2 h a 23 ° c num termociclador para evitar a luz.
Nota: Prepare a solução de biotina misturando 50 mg de biotina com 13,5 mL de DMSO. Faça alíquotas de uso único e congele em-80 ° c. - Purify cDNA biotinylated: híbridos do RNA usando grânulos magnéticos de SPRI. Adicionar 12 μL de 2-propanol e misturar com pipetagem. Adicione 108 μL de grânulos de SPRI (1.8:1 grânulos à relação da amostra) e repita a purificação como descrito nas etapas 2.6.1 – 2.6.6. Elute usando 42 μL de água pré-aquecida a 37 ° c.
Nota: O protocolo pode ser pausado aqui, e as amostras congeladas em-80 ° c.
5. RNase eu Ddigestão
- Prepare a mistura RNase I misturando 4,5 μL de tampão 10x RNase I com 0,5 μL de RNase I (10 U/μL) por cada amostra. Misture com pipetagem.
- Adicionar 5 μL da mistura a cada amostra purificada (45 μL no total). Misture com pipetagem e incubar durante 30 min a 37 ° c.
6. preparação de grânulos de streptavidin
- Para cada amostra, misture 30 μL da pasta de grânulos de estreptavidina com 0,38 μL de tRNA de 20 mg/mL. Incubar no gelo por 30 min e misture a cada 5 min, sacudendo o tubo.
Nota: Ressuscitar o estreptavidina grânulos chorume bem antes de Pipetar por flicking o frasco. A solução de tRNA deve ser preparada de acordo com Murata et al.11 - Separe os grânulos no suporte magnético por 2 – 3 min. Retire o sobrenadante.
- Lave as contas retendo em 15 μL de tampão a. Separe as esferas do suporte magnético durante 2 – 3 min e retire o sobrenadante. Repita a lavagem e retire o sobrenadante.
- Suspender os grânulos em 105 μL de tampão A e adicionar 0,19 μL de 20 mg/mL de tRNA. Misture bem com pipetagem.
Nota: Os grânulos devem ser preparados frescos antes do uso. Comece a preparação dos grânulos durante a digestão de RNase I. Para várias amostras preparar as contas em conjunto em um único tubo.
7. tampão-aprisionamento
-
Ligação de amostra
- Adicionar 105 μL de grânulos de streptavidina preparados a 45 μL da amostra tratada com RNase I. Misture bem introduzindo pipetagem e incubar a 37 ° c durante 30 min. Misture pipetando a cada 10 min.
- Separe os grânulos no suporte magnético por 2 – 3 min. Retire o sobrenadante.
-
Grânulos de lavagem
- Adicionar 150 μL de tampão de lavagem a e suspender as contas com pipetagem. Separe os grânulos no suporte magnético por 2 – 3 min e retire o sobrenadante.
- Adicionar 150 μL do tampão de lavagem B e suspender as contas com pipetagem. Separe os grânulos no suporte magnético por 2 – 3 min e retire o sobrenadante.
- Adicionar 150 μL do tampão de lavagem C e suspender as contas com pipetagem. Separe os grânulos no suporte magnético por 2 – 3 min e retire o sobrenadante.
Nota: Os amortecedores B e C devem ser pré-aquecidos a 37 ° c. As receitas para os buffers de lavagem A, B e C são descritas em Murata et al.11
-
lançamento do cDNA
- Prepare 1x RNase I tampão misturando 58,5 μL de água com 6,5 μL de 10x RNase I tampão.
- Resuspend as contas em 35 μL de 1x RNase I tampão. Incubar a 95 ° c por 5 min e transfira diretamente no gelo por 2 min para evitar a reassociação do cDNA. Segure as tampas durante a transferência para o gelo, pois eles podem pop-off devido à acumulação de pressão.
- Separe os grânulos por 2 – 3 min em um suporte magnético e transfira o sobrenadante para uma nova placa.
- Resuspend as contas em 30 μL de 1x RNase I tampão. Separe os grânulos no suporte magnético por 2 – 3 min e transfira o sobrenadante para o sobrenadante previamente coletado (o volume total de cDNA eluído deve ser de cerca de 65 μL).
8. remoção de RNA por RNase H e RNase I digestão
- Por amostra, combine 2,4 μL de água, 0,5 μL de tampão 10x RNase I, 0,1 μL de RNase H e 2 μL de RNase I.
- Adicionar 5 μL da mistura aos 65 μL da amostra de cDNA liberada e misturar com pipetagem. Incubar a 37 ° c durante 15 min e segure a 4 ° c.
- Purify o cDNA da mistura da digestão de RNase usando grânulos magnéticos de SPRI. Adicionar 126 μL de grânulos de SPRI a 70 μL de reacção de degradação e misturar com pipetagem. Siga etapas da purificação como descrito para a purificação dos grânulos de SPRI em 2.6.1-2.6.6. Elute usando 42 μL de água pré-aquecida a 37 ° c conforme descrito.
- Prepare RNase eu misturo combinando 4,5 μL de 10x RNase I buffer e 0,5 μL de RNase I.
- Adicionar 5 μL da mistura de RNase aos 40 μL da amostra de cDNA purificada. Misturar com pipetagem e incubar a 37 ° c durante 30 min. Segure a 4 ° c.
- Purify a amostra usando grânulos magnéticos de SPRI. Adicionar 81 μL de grânulos de SPRI a 45 μL de reacção de degradação e misturar com pipetagem. Siga etapas da purificação como descrito para a purificação dos grânulos de SPRI em 2.6.1-2.6.6. Elute usando 42 μL de água como descrito.
9. ligadura de 5 ' linker
- Concentre a amostra de cDNA purificada em 4 μL utilizando o concentrador de vácuo. Mantenha a temperatura a 30 – 35 ° c. Teste o volume usando uma pipeta. Se a amostra secar à completude, dissolva-se adicionando 4 μL de água.
Nota: É melhor evitar a secagem à integralidade para impedir a perda da amostra. - Incubar a amostra concentrada a 95 ° c por 5 min e imediatamente colocar no gelo por 2 min para evitar a renaturação. Segure as tampas durante a transferência dos tubos como as tampas podem pop-off devido à acumulação de pressão.
- Incubar 4 μL do linker 2,5 μM 5 ' a 55 ° c durante 5 min e coloque imediatamente no gelo durante 2 min para evitar a renaturação.
- Misture 4 μL do linker 2,5 μM 5 ' com 4 μL da amostra.
Nota: O vinculador 5 ' deve ser preparado de acordo com a tabela complementar 2, tabela complementar 3, tabela complementar 4, e tabela complementar 5. Diluir o linker de 10 μM 5 ' para uma concentração de 2,5 μM utilizando 100 mM de NaCl antes da utilização. - Adicione 16 μL da pré-mistura de ligadura (ver tabela de materiais) ao vinculador misto 5 ' e a amostra e misture bem com pipetagem. Incubar a 16 ° c por 16 h.
- Purify a mistura da ligadura usando grânulos magnéticos de SPRI. Adicione 43,2 μL de grânulos de SPRI e siga os passos 2.6.1 – 2.6.6. Elute como descrito utilizando 42 μL de água pré-aquecida a 37 ° c.
- Repita a purificação feita na etapa 9,6 adicionando 72 μL de grânulos de SPRI ao sobrenadante transferido (1.8:1 grânulos à relação da amostra).
Nota: 5 ' linkers contêm códigos de barras que permite a junção de até oito amostras antes do sequenciamento (oito códigos de barras de trinucleotídeo estão disponíveis, conforme descrito em Murata et al.11 e tabela complementar 1).
10. ligadura de 3 ' linker
- Concentre a amostra purificada em 4 μL utilizando o concentrador de vácuo conforme descrito no passo 9,1.
- Incubar a amostra concentrada a 95 ° c por 5 min e imediatamente colocar no gelo por 2 min para evitar a renaturação. Segure as tampas durante a transferência dos tubos, pois as tampas podem estourar devido à acumulação de pressão.
- Incubar 4 μL do linker 2,5 μM 3 ' a 65 ° c durante 5 min e coloque imediatamente no gelo durante 2 min para evitar a renaturação.
- Adicionar 4 μL do linker 2,5 μM 3 ' aos 4 μL da amostra concentrada.
- Adicionar 16 μL de pré-mistura de ligadura e misturar bem com pipetagem. Incubar a 16 ° c por 16 h.
- Purify a mistura da ligadura usando grânulos magnéticos de SPRI. Adicione 43,2 μL de grânulos de SPRI e siga os passos 2.6.1 – 2.6.6. Elute como descrito usando 42 μL de água pré-aquecida a 37 ° c.
Nota: O vinculador 3 ' deve ser preparado de acordo com as tabelas complementares 6 e tabela suplementar 7. Diluir o vinculador 10 μM 3 ' para uma concentração de 2,5 μM usando 100 mM NaCl.
11. dephosphorylation
- Prepare a mistura SAP combinando 4 μL de água, 5 μL de tampão SAP 10x e 1 μL de enzima SAP.
- Adicionar 10 μL de mistura de SAP à amostra ligada purificada (volume total 50 μL) e incubar no termociclador utilizando o seguinte programa: 37 ° c por 30 min, 65 ° c durante 15 min, e segure a 4 ° c.
12. degradação da vertente superior do linker de 3 ' usando a enzima específica da excisão do uracil
- Adicione 2 μL de enzima de excisão específica de uracil (ver tabela de materiais) à amostra dephosphorylated, misture introduzindo com pipting e incubar no termociclador usando o seguinte programa: 37 ° c por 30 minutos, 95 ° c por 5 minutos, e coloc imediatamente no gelo por 2 minutos a evitar recozimento da vertente superior fragmentada.
- Purifique a mistura de reacção adicionando 93,6 μL de grânulos magnéticos de SPRI à mistura de 52 μl e misture bem com pipetagem. Repita os passos de purificação 2.6.1 – 2.6.6. Elute com 42 μL de água pré-aquecida a 37 ° c, conforme descrito.
13. síntese de segunda vertente
- Prepare a segunda mistura da síntese da vertente (os volumes são expressos por a amostra) combinando 5 μL do amortecedor da reacção da polimerase do ADN 10x, 2 μL da água, 1 μL de dNTPs de 10 milímetros, 1 μL do primer da segunda vertente do nAnT-iCAGE de 50 μM (a seqüência está na tabela suplementar 1) e 1 μL de D Polimerase exonuclease-deficiente de ND (veja a polimerase recomendada na tabela de materiais).
- Adicionar 10 μL da mistura à amostra purificada e misturar bem com pipetagem (o volume total é de 50 μL). Incubar no termociclador usando o seguinte programa: 95 ° c por 5 min, 55 ° c por 5 min, 72 ° c por 30 min, e segure a 4 ° c.
14. degradação do primer de síntese de segunda vertente utilizando exonuclease I
- Adicionar 1 μL de exonuclease I à segunda mistura de síntese de vertente. Misture bem introduzindo pipetagem e incubar a 37 ° c durante 30 min, seguido de uma fixação a 4 ° c.
- Purify o ADN encalhado dobro adicionando 91,8 μL de grânulos magnéticos de SPRI a 51 μL da amostra exonuclease I-tratada. Repita as etapas de purificação descritas em 2.6.1 – 2.6.6. e eluir com 42 μL de água pré-aquecida a 37 ° c como descrito.
- Concentre a amostra utilizando o concentrador de vácuo a 15 μL, conforme descrito no passo 9,1.
15. qualidade e controle de quantidade
- Use 1 μL das amostras concentradas e execute um chip de DNA de alta sensibilidade em um analisador de qualidade de DNA. O perfil/quantidade esperados é apresentado na Figura 3.
16. primeira rodada de degradação do transportador
- Prepare o mix de degradação combinando 2 μL de água, 2 μL de tampão enzimático de restrição 10x, 1 μL de I-SceI e 1 μL de I-CeuI.
- Adicionar 6 μL da mistura de degradação a 14 μL da amostra concentrada e misturar com pipetagem. Incubar a 37 ° c por 3 h seguido de 20 min de desativação a 65 ° c e segure a 4 ° c.
- Purify a mistura da degradação usando grânulos magnéticos de SPRI. Adicione 5 μL de água para aumentar o volume da mistura de degradação e adicione 45 μL de grânulos de SPRI (1,8:1 grânulos à relação da amostra). Repita a purificação conforme descrito nas etapas 2.6.1 – 2.6.6. e eluir com 42 μL de água pré-aquecida a 37 ° c.
- Concentre a amostra eluída de 42 μL a 20 μL do volume total, conforme descrito no passo 9,1.
17. controle do nível de degradação e determinação do número de ciclos de amplificação de PCR
- Prepare a mistura de qPCR para amplificar bibliotecas inteiras (mistura do adaptador). Combine 3,8 μL de água, 5 μL de qPCR premix (2x), 0,1 μL de 10 μM adaptor_f1 Primer (5 '-AATGATACGGCGACCACCGA-3 ') e 0,1 μL de 10 μM adaptor_r1 Primer (5 '-CAAGCAGAAGACGGCATACGA-3 ') para cada amostra (ver tabela de materiais para a pré-mistura recomendada de qPCR).
- Combine 9 μL de mistura do adaptador qPCR com 1 μL de amostra do passo 16,4 e misture bem com pipetagem.
- Prepare a mistura de qPCR para amplificar o ADN derivado do portador (mistura do portador). Combine 3,8 μL de água, 5 μL de qPCR premix (2x), 0,1 μL de 10 μM carrier_f1 Primer (5 '-GCGGCAGCGTTCGCTATAAC-3 ') e 0,1 μL de 10 μM adaptor_r1 primer para cada amostra
- Combine 9 μL de mistura de portador de qPCR com 1 μL da amostra do passo 16,4 e misture bem com pipetagem.
- Programe o programa qPCR: 95 ° c por 3 min (95 ° c para 20 s, 60 ° c para 20 s, 72 ° c por 2 min) repetiu 40x, seguido por uma curva de desnaturação específica do instrumento (65 – 95 ° c) e segure a 4 ° c.
Nota: Prepare um controle negativo substituindo a amostra por água.
18. amplificação do PCR da biblioteca do alvo
- Prepare o mix de amplificação de PCR combinando 6 μL de água, 0,5 μL de primer 10 μM adaptor_f1, 0,5 μL de 10 μM adaptor_r1 primer e 25 μL de PCR premix (2x). Misture com pipetagem (ver tabela de materiais para a pré-mistura de PCR recomendada).
- Adicionar 32 μL da mistura de PCR a 18 μL da amostra a partir do passo 16,4. Misture cuidadosamente com pipetagem.
- Ajuste a amplificação do PCR: 95 ° c por 3 minutos, (98 ° c para 20 s, 60 ° c para 15 s, 72 ° c para 2 minutos) 12-18 ciclos, 72 ° c por 2 minutos e preensão em 4 ° c.
Nota: O número exato de ciclos de PCR é determinado pelos resultados da qPCR e corresponde ao valor de TC obtido com a mistura de primer do adaptador (o número de ciclos de PCR é igual ao valor do CT). - Purify a amostra amplificada adicionando 90 μL de grânulos magnéticos de SPRI a 50 μL da amostra amplificada e misture-a completamente introduzindo com pipting. Repita as etapas de purificação descritas nas etapas 2.6.1 – 2.6.6. e eluir a amostra usando 42 μL de água como descrito.
19. segunda rodada de degradação do portador
- Repita os passos 16.1 – 16.3.
- Purify a mistura da degradação usando grânulos magnéticos de SPRI. Adicionar 10 μL de água à amostra para aumentar o volume e misturar com 30 μL de grânulos de SPRI (1:1 grânulos para a razão da amostra). Repita a purificação conforme descrito nas etapas 2.6.1 – 2.6.6. e eluir com 42 μL de água pré-aquecida a 37 ° c, conforme descrito.
- Concentre a amostra eluída de 42 μL a 30 μL de volume total.
20. seleção de tamanho da biblioteca
- Misturar 24 μL de grânulos magnéticos SPRI com 30 μL da amostra a partir do passo 19,3. (0.8:1 grânulos à relação da amostra). Repita as etapas de purificação conforme descrito nas etapas 2.6.1 – 2.6.6. e eluir a amostra em 42 μL de água como descrito.
- Concentre a amostra em aproximadamente 14 μL, conforme descrito na etapa 9,1.
21. controle de qualidade
-
Avaliação da distribuição de tamanho
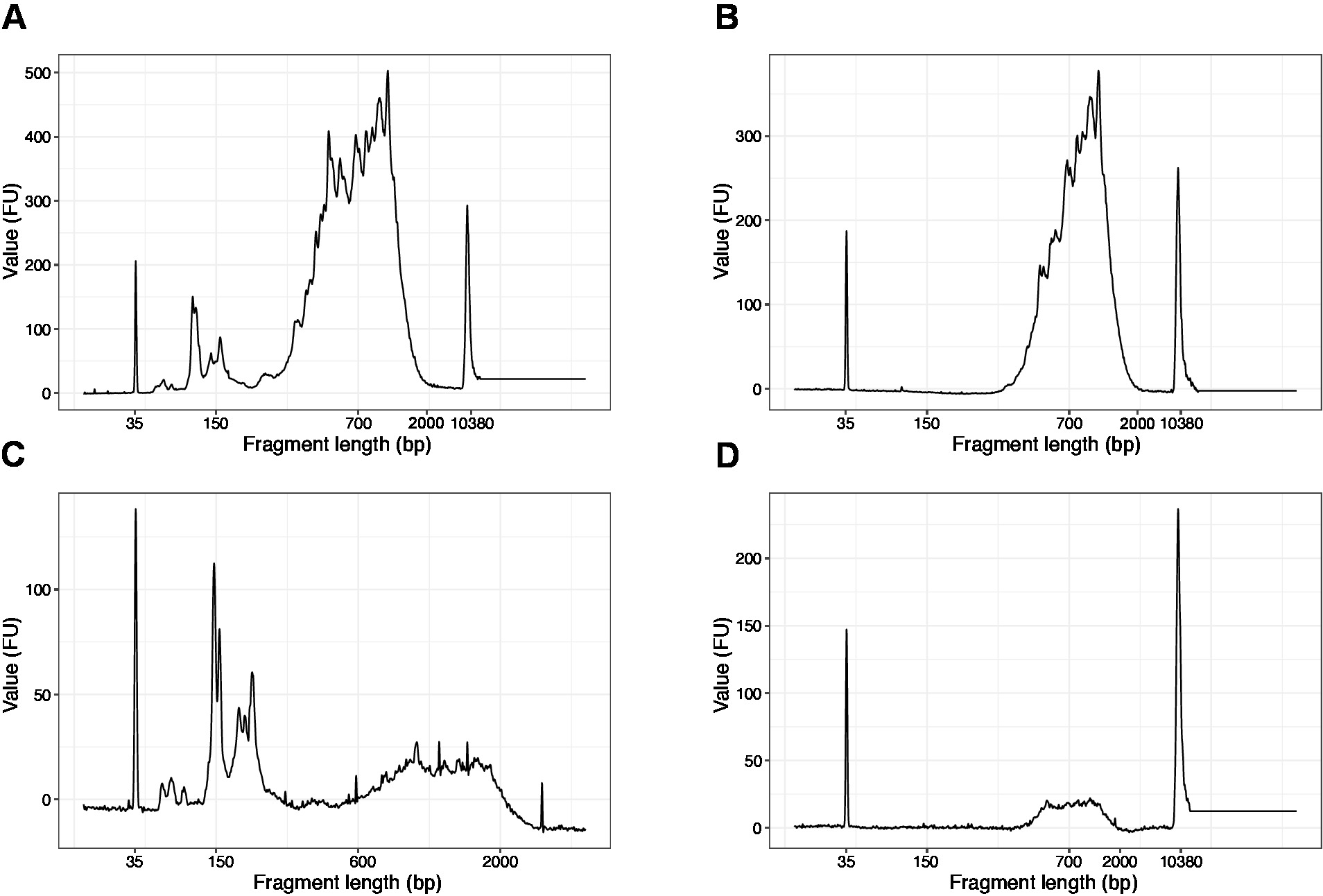
- Execute 1 μL da amostra no chip de DNA de alta sensibilidade. Os resultados esperados são apresentados na Figura 4.
Nota: Se os fragmentos inferiores a 200 BP estiverem visíveis (ver exemplo na figura 4a, C), a seleção de tamanho (etapas 20,1 – 20,2) deve ser repetida até que os fragmentos curtos sejam removidos (Figura 4B, D). Geralmente uma rodada adicional de seleção de tamanho é suficiente. Se a quantidade de fragmentos curtos for grave (como na Figura 4C), a proporção de grânulos para amostra deve ser diminuída para 0,6:1.
- Execute 1 μL da amostra no chip de DNA de alta sensibilidade. Os resultados esperados são apresentados na Figura 4.
-
Controle de qualidade da degradação do portador
- Repita os passos 17.1 – 17.5.
Nota: Dependendo da concentração das bibliotecas estimadas na execução de chip de DNA HS (análise de região), as amostras precisam ser diluídas antes da qPCR. Utilizar 0,5 μL da amostra para evitar a perda da amostra e diluir 100 – 500x em água (diluir para 1 – 20 PG/μL de concentração final). A diferença esperada entre os valores de TC obtidos com o adaptador e a mistura transportadora é de 5 – 10.
- Repita os passos 17.1 – 17.5.
-
Quantificação da biblioteca
- Prepare a diluição de trabalho do padrão lambda de DNA misturando 20 μL de 100 mg/mL de DNA lambda padrão com 980 μL de 1x TE (Prepare-se diluindo 20x TE fornecido no kit de quantificação de DNA). A diluição do DNA lambda pode ser armazenada a-20 ° c.
- Prepare as diluições seriais padrão lambda DNA misturando o padrão lambda diluído e 1x TE de acordo com a tabela complementar 8.
Nota: Para maior precisão, recomenda-se adicionar 100 μL de tampão TE 1x a todos os tubos e remover o volume 1x TE como exigido por volume do lambda diluído a ser adicionado. Não utilize mais de 1 μL da biblioteca; recomenda-se a utilização de 384 placas de poços para esta medição.
Resultados
Este relato descreve o protocolo completo de SLIC-CAGE para a obtenção de bibliotecas prontas para sequenciamento a partir de nanogramas de material de RNA total inicial (Figura 1). Para obter a mistura sintética do portador do RNA, primeiramente, os moldes do portador do PCR precisam de ser preparados e gel-purified para eliminar produtos laterais do PCR (Figura 2a). Cada molde do PCR (dez no total) é produzido usando um avanço comum, mas uma primeira demão reversa diferente (tabela 2), conduzindo aos comprimentos diferentes do molde do PCR para permitir a variabilidade do tamanho de portadores sintéticos do RNA. Uma vez purificada, os modelos de PCR são utilizados para a transcrição in vitro das moléculas transportadoras. Um único produto portador do RNA é esperado se os moldes são gel-purified (veja a gel-análise representativa na Figura 2b). A preparação do transportador pode ser dimensionada dependendo da necessidade, e quando preparada, misturada e congelada em-80 ° c para uso futuro.
Usando a quantidade mínima recomendada de RNA total da amostra (10 NG) combinada com 16-18 ciclos da amplificação do PCR, as bibliotecas de alta complexidade de SLIC-CAGE podem ser conseguidas. O número de ciclos de PCR necessários para amplificar a biblioteca final depende muito da quantidade de RNA de entrada total utilizada (o número esperado de ciclos é apresentado na tabela 4).
Após a primeira rodada de degradação, nos resultados da qPCR (etapa 17), a diferença esperada entre os valores de TC obtidos com o primer adaptor_f1 ou carrier_f1 é de 1-2, com valores de TC obtidos com adaptor_f1 menor do que com carrier_f1.
A distribuição dos comprimentos dos fragmentos na biblioteca final está entre 200-2.000 BP com o tamanho médio do fragmento de 700-900 BP (baseado na análise da região usando o software de Bioanalyzer, Figura 4B, D). Fragmentos mais curtos, conforme apresentado na figura 4a, C, têm que ser removidos por rodadas adicionais de tamanho-exclusão (etapas 20-21). Estes fragmentos curtos são artefactos da amplificação do PCR e não a biblioteca do alvo. Observe que fragmentos mais curtos se aglomeram melhor nas células de fluxo de seqüenciamento e podem causar problemas de seqüenciamento.
A quantidade esperada de material de biblioteca obtida por amostra é entre 5-50 ng. Quantidades significativamente inferiores são indicativas de perda de amostra durante o protocolo. Se a quantidade baixa obtida for suficiente para seqüenciamento (2-3 ng das bibliotecas agrupadas é necessária), as bibliotecas podem ser de menor complexidade (veja abaixo).
Dependendo da máquina de sequenciamento, a quantidade da biblioteca carregada na célula de fluxo pode precisar ser otimizada. Usando um Illumina HiSeq 2500, carregando 8-12 pM SLIC-CAGE bibliotecas dá em média 150-200 milhões de leituras, com > 80% de leituras passando Pontuação de qualidade p30 como limiar.
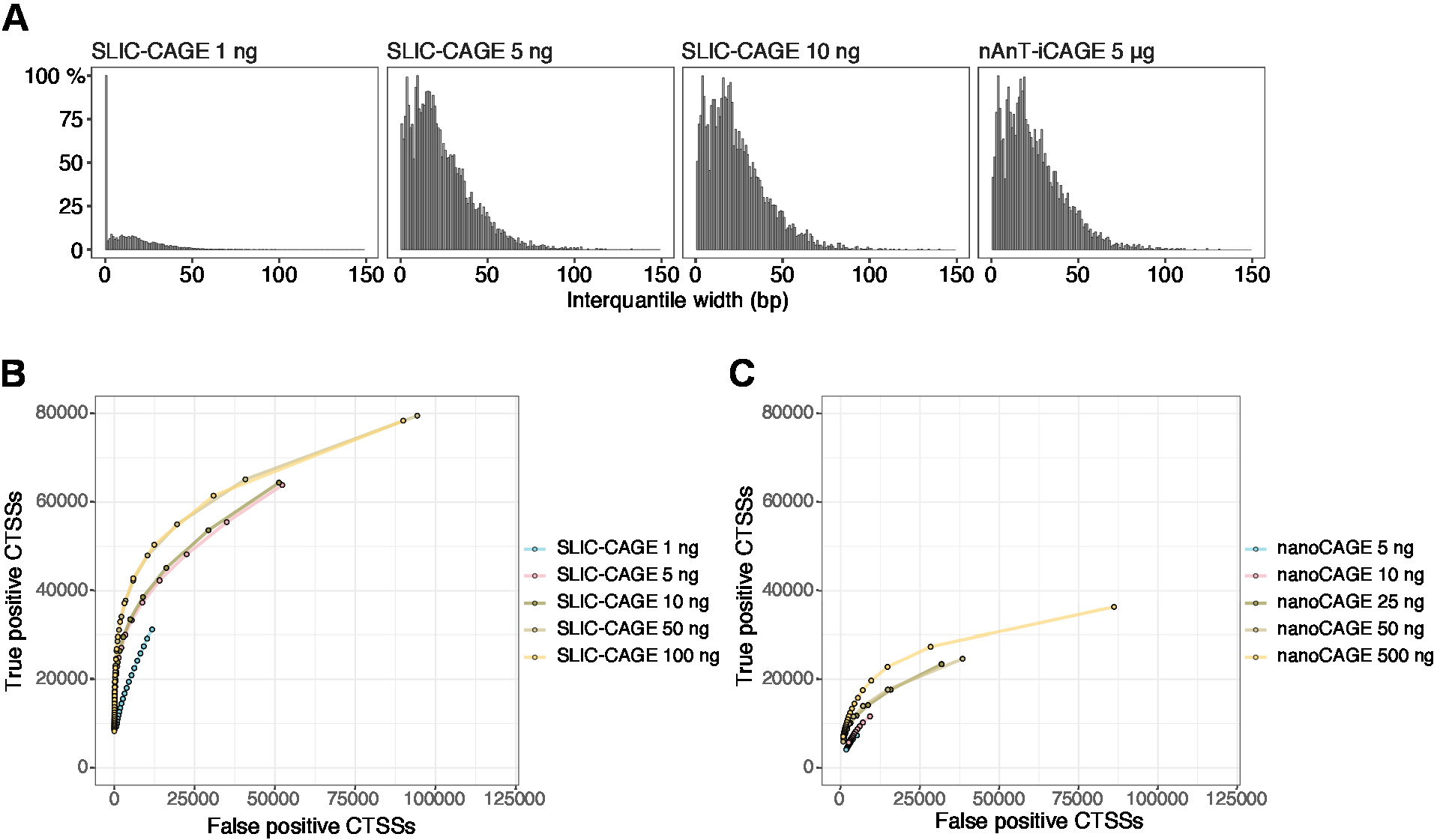
As leituras obtidas são então mapeadas para o genoma de referência [para leituras de 50 BP, Bowtie212 podem ser usadas com parâmetros padrão que permitem desencontros zero por sequência de semente (22 BP)]. As eficiências de mapeamento esperadas dependem da quantidade total de entrada de RNA e são apresentadas na tabela 5. As leituras mapeadas exclusivamente podem ser carregadas no ambiente de computação gráfica e estatística R13 e processadas usando Cager (pacote BioConductor14). A vinheta de pacote é fácil de seguir e explica o fluxo de trabalho e o processamento dos dados mapeados em detalhes. Um controle visual fácil da complexidade da biblioteca é a distribuição da largura do promotor, já que as bibliotecas de baixa complexidade terão promotores artificialmente estreitos (Figura 5a, biblioteca SLIC-Cage derivada de 1 ng do RNA total, para detalhes ver Publicação10). No entanto, mesmo as bibliotecas SLIC-CAGE de baixa complexidade permitem a identificação de CTSSs verdadeiros, com maior precisão do que métodos alternativos para mapeamento de TSS de baixa/média entrada (Figura 5b, C).

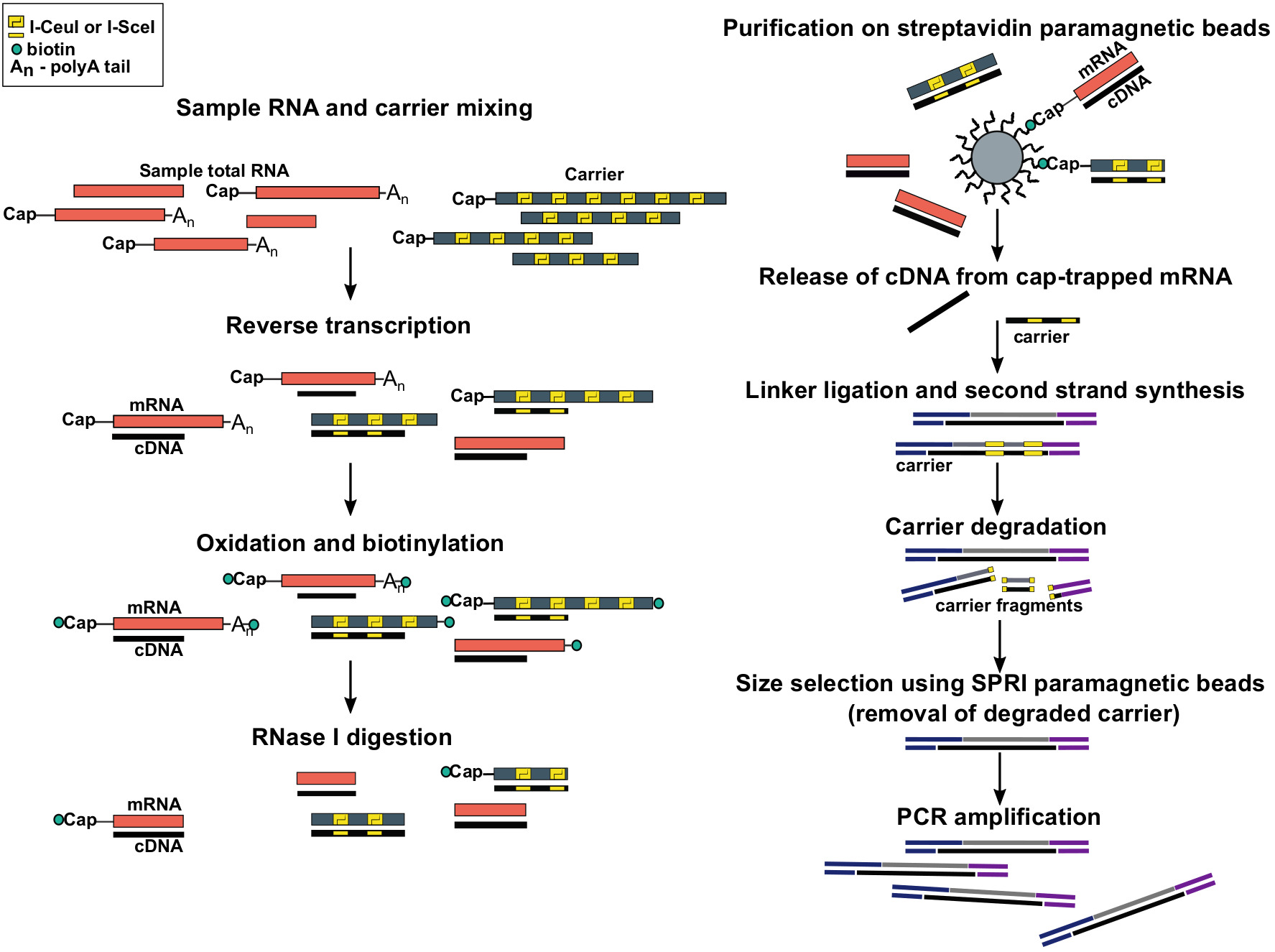
Figura 1: Passos no protocolo SLIC-Cage. O RNA da amostra é misturado com a mistura do portador do RNA para conseguir 5 μg do material total do RNA. o cDNA é sintetizado através da transcrição reversa e o tampão é oxidado usando o periodate do sódio. A oxidação permite o acessório da biotina à tampa usando a hidrazida da biotina. A biotina fica presa à extremidade 3 ′ do mRNA, pois também é oxidada usando periodato de sódio. Para eliminar a biotina de mRNA: híbrido de cDNA com o cDNA incompletamente sintetizado e das extremidades 3 do ′ de mRNA, as amostras são tratadas com o cDNA de RNase I. que alcangou a extremidade 5 do ′ de mRNA é selecionada então pela purificação da afinidade em grânulos magnéticos do estreptavidina ( Cap-trapping). Após a liberação de cDNA, 5 ′-e 3 '-linkers são ligated. As moléculas da biblioteca que se originam do portador são degradadas usando I-SceI e I-CeuI homing endonucleases e os fragmentos são removidos usando grânulos magnéticos de SPRI. A biblioteca é então PCR amplificada. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: gel representativo-análise de moldes do PCR do portador e de transcritos in vitro do portador. (A) moldes do PCR do portador antes da purificação do gel: o primeiro poço contem o marcador de 1 KBP, seguido pelos moldes do PCR do portador 1, 1-10. B) transcrições in vitro do transportador: o primeiro poço contém o marcador de 1 KBP, seguido das transcrições do transportador 1-10. Os transcritos do portador foram desnaturados pelo aquecimento por 5 minutos em 95 ° c antes do carregamento. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: traço de qualidade de DNA representativo (chip de DNA de alta sensibilidade) de SLIC-Cage antes da primeira rodada de degradação do portador. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: traços de qualidade de DNA representativo (chip de DNA de alta sensibilidade) de bibliotecas de SLIC-Cage após amplificação de PCR. (A) biblioteca SLIC-Cage que requer seleção de tamanho adicional para remoção de fragmentos curtos. (B) biblioteca da SLIC-gaiola após o tamanho-seleção usando grânulos de 0.6 x SPRI à relação da amostra. (C) SLIC-Cage biblioteca de menor quantidade de saída que requer tamanho-seleção para a remoção de fragmento curto. (D) SLIC-Cage biblioteca de menor quantidade de saída após o tamanho-seleção usando 0.6:1 SPRI contas para a razão da amostra. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Validação de bibliotecas SLIC-Cage. (A) distribuição de larguras de interquantil de agrupamento de Tags em bibliotecas de SLIC-Cage preparadas a partir de 1, 5, ou 10 ng de RNA total de s. cerevisiae e na biblioteca NAnT-icage preparada a partir de 5 μg de RNA total de s. cerevisiae . Uma grande quantidade de clusters de tag estreitos na biblioteca 1 ng SLIC-CAGE indica sua baixa complexidade. (B) curvas ROC para identificação de CTSS em bibliotecas de S. cerevisiae SLIC-Cage. Todos os CTSSs do nAnT-iCAGE de S. cerevisiae foram usados como um jogo verdadeiro. (C) curvas ROC para identificação de CTSS em bibliotecas nanocagem de S. cerevisiae . Todos os CTSSs do nAnT-iCAGE de S. cerevisiae foram usados como um jogo verdadeiro. A comparação de curvas de ROC mostra que SLIC-CAGE supera fortemente nanoCAGE na identificação de CTSS. Foram utilizados dados de ArrayExpress E-MTAB-6519. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Tabela 1: sequência do gene sintético portador. Sites i-SCEI são ousados e em itálico em roxo, e i-ceui reconhecimentos locais são verdes. Por favor, clique aqui para baixar este arquivo.
| Transportadora | primer reverso 5 '-3 ' | Comprimento do produto do PCR/BP | |
| 1 | PCR_N6_r1: NNNNNNCTACGTGTCGCAGACGAATT | 1034 | |
| 2 | PCR_N6_r2: NNNNNNTATCCAGATCGTTGAGCTGC | 966 | |
| 3 | PCR_N6_r3: NNNNNNCACTGCGGGATCTCTTTACG | 889 | |
| 4 | PCR_N6_r4: NNNNNNGCCGTCGATAACTTGTTCGT | 821 | |
| 5 | PCR_N6_r5: NNNNNNAGTTGACCGCAGAAGTCTTC | 744 | |
| 6 | PCR_N6_r6: NNNNNNGTGAAGAATTTCTGTTCCCA | 676 | |
| 7 | PCR_N6_r7: NNNNNNCTCGCGGCTCCAGTCATAAC | 599 | |
| 8 | PCR_N6_r8: NNNNNNTATACGCGATGTTGTCGTAC | 531 | |
| 9 | PCR_N6_r9: NNNNNNACCGCCGCGCCTTCCGCAGG | 454 | |
| 10 | PCR_N6_r10: NNNNNNCAGGACGTTTTTGCCCAGCA | 386 | |
| * Primer forward é o mesmo para todos os modelos de transportadora. Sublinhado é a sequência de promotor T7. PCR_GN5_f1: TAATACGACTCACTATAGNNNNNCAGCGTTCGCTA | |||
Tabela 2: primers para amplificação do modelo do portador. Primer forward é o mesmo para todos os modelos de transportadora. Sublinhado é a sequência de promotor T7. PCR_GN5_f1: Taatacgactcactatagnnnnncagcgttcgcta. Usando os primers reversos de deferimento, os moldes do PCR e daqui o portador RNAs do comprimento de deferimento são produzidos.
| Transportadora | Comprimento | Não tampado/μg | tampado/μg |
| 1 | 1034 | 3,96 | 0,45 |
| 2 | 966 | 8,36 | 0,95 |
| 3 | 889 | 4,4 | 0,5 |
| 4 | 821 | 6,6 | 0,75 |
| 5 | 744 | 4,4 | 0,5 |
| 6 | 676 | 3, 8 | 0,35 |
| 7 | 599 | 4,4 | 0,5 |
| 8 | 531 | 3,96 | 0,45 |
| 9 | 454 | 2,64 | 0,3 |
| 10 | 386 | 2,2 | 0,25 |
Tabela 3: mistura do portador do RNA. No total 49 μg da mistura transportadora 0,3-1 KBP: sem tampos = 44 μg, tampado = 5 μg.
| Entrada total do RNA/ng | Ciclos de PCR |
| 1 ng | 18 |
| 2 ng | 17 |
| 5 ng | 16 |
| 10 ng | 15-16 |
| 25 ng | 14-15 |
| 50 ng | 13-15 |
| 100 ng | 12-14 |
Tabela 4: Número esperado de ciclos do PCR na dependência da entrada total do RNA da amostra. O número aproximado de ciclos é baseado em experimentos realizados com Saccharomyces cerevisiae, Drosophila melanogaster e RNA total de Mus musculus .
| Entrada de RNA total/ng | % geral mapeado | % mapeados exclusivamente | % transportadora |
| 1 ng | 30 | 20-30 | 30 |
| 2 ng | 60 | 20-50 | 10 |
| 5 ng | 60-70 | 40-60 | 5-10 |
| 10 ng | 60-70 | 40-60 | 5-10 |
| 25 ng | 65-80 | 40-70 | 0-5 |
| 50 ng | 65-80 | 40-70 | 0-3 |
| 100 ng | 70-85 | 40-70 | 0-2 |
Tabela 5: eficiência de mapeamento esperada e na dependência da quantidade total de entrada de RNA. Os números aproximados são apresentados e baseados em experimentos realizados com Saccharomyces cerevisiae e Mus musculus RNA total.
Discussão
Para preparações bem sucedidas da biblioteca de SLIC-CAGE, é crítico usar pontas e tubos Low-Binding para impedir a perda da amostra devido à adsorção da amostra. Em todas as etapas que envolvem a recuperação do sobrenadante, recomenda-se recuperar o volume de entiresample. Como o protocolo tem várias etapas, a perda de amostra contínua levará a bibliotecas sem êxito.
Se CAGE (nAnT-iCAGE) não tiver sido realizado rotineiramente, é melhor testar SLIC-CAGE com diferentes quantidades de entrada (10 ng, 20 ng, 50 ng, 100 ng, 200 ng) da mesma amostra total de RNA e comparar com as bibliotecas nAnT-iCAGE que são preparadas usando 5 μg de RNA total. Se a biblioteca nAnT-iCAGE não for bem-sucedida (menos de 0,5-1 ng da biblioteca de DNA obtida por amostra), o SLIC-CAGE dificilmente funcionará e a perda da amostra deverá ser minimizada.
Um passo crítico para garantir que bibliotecas de alta qualidade desprovidas de RNA ou rRNA degradado não tampado é a captura de tampa descrita na seção 7. É altamente importante que os grânulos de estreptavidina estejam completamente resrepended em tampões de lavagem e que os bufferes da lavagem estejam removidos antes de continuar à etapa seguinte da lavagem ou à eluição do cDNA.
Se os resultados do qPCR após a primeira rodada de degradação do portador não mostrarem diferença entre o uso de primers adaptor_f1 e carrier_f1, continuar o protocolo ainda é recomendado. Se após a segunda rodada de degradação do portador, a diferença nos valores de TC for inferior a cinco, recomenda-se uma terceira rodada de degradação do transportador. Nós nunca encontramos uma terceira rodada de degradação necessária, e se isso ocorrer, recomenda-se substituir os estoques de endonuclease homing.
Os círculos adicionais da amplificação do PCR podem ser adicionados ao protocolo se a quantidade final da biblioteca obtida não é bastante para arranjar em seqüência. A amplificação do PCR pode então ser ajustada com número mínimo de ciclos da amplificação necessários para render bastante material para arranjar em seqüência, tendo em consideração a perda da amostra que não pode ser evitada na seleção do tamanho. A purificação ou a seleção do tamanho usando grânulos magnéticos de SPRI devem então ser executadas até que todos os fragmentos pequenos (< 200 BP) sejam removidos (se necessário, usam 0.6:1 grânulos à relação da amostra), e a biblioteca deve ser quantificada usando Picogreen.
As bibliotecas podem ser sequenciadas em modo single-end ou emparelhado. Usando o sequenciamento de fim emparelhado, informações sobre isoformas de transcrição podem ser obtidas. Além, porque a transcrição reversa é executada usando um primer aleatório (TCT-N6, n6 que é um hexamer aleatório), a informação do 3 '-fim seqüenciado pode ser usada como identificadores MOLECULARS originais (Umi) para recolher duplicatas do PCR. Como um número moderado de ciclos da amplificação do PCR é usado (até 18), o uso de UMIs foi encontrado previamente para ser desnecessário.
Como o núcleo do protocolo depende nAnT-iCAGE11, SLIC-Cage usa oito códigos de barras. Portanto, não há suporte para multiplexação de mais de oito amostras no momento. Além disso, tanto o SLIC-CAGE quanto o nAnT-iCAGE não são adequados para capturar RNAs menores que 200 BP, pois os protocolos são projetados para remover linkers e artefatos de PCR por meio da exclusão de tamanho com grânulos AMPure XP.
SLIC-CAGE é o único método de resolução de baixo-nucleotídeo de baixa entrada imparcial para mapear sites de início de iniciação de transcrição usando nanogramas de material de RNA total. Os métodos alternativos confiam na atividade de switching do molde da transcriptase reversa ao RNA tampado código de barras em vez do tampão-aprisionamento (por exemplo, NanoCAGE15 e nanopare16). Devido à alternância de modelos, esses métodos exibem vieses específicos de sequência na detecção de Tsss, levando a um número aumentado de Tsss falsos positivos e a números diminuídos de Tsss verdadeiros9,10.
Divulgações
Uma patente para o RNA/ADN degradável do portador foi enchida.
Agradecimentos
Este trabalho foi apoiado pelo Wellcome Trust Grant (106954) concedido ao B. L. e ao financiamento do núcleo do Conselho de pesquisa médica (MRC) (MC-A652-5QA10). O N. C. foi apoiado pela bolsa de longa duração EMBO (EMBO ALTF 1279-2016); E. P. foi apoiado pelo Conselho de pesquisa médica do Reino Unido; B. L. foi apoiado pelo Conselho de pesquisa médica do Reino Unido (MC UP 1102/1).
Materiais
| Name | Company | Catalog Number | Comments |
| 2-propanol, Bioultra, for molecular biology, ≥99.5% | Sigma-Aldrich | 59304-100ML-F | Used in RNAclean XP purification. |
| 3' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 3'linkers is described in the supplementary of this protocol. | ||
| 5' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 5'linkers is described in the supplementary of this protocol. | ||
| Agencourt AMPure XP, 60 mL | Beckman Coulter | A63881 | Purification of DNA |
| Agencourt RNAClean XP Kit | Beckman Coulter | A63987 | Purification of RNA and RNA:cDNA hybrids in CAGE steps. |
| Axygen 0.2 mL Polypropylene PCR Tube Strips and Domed Cap Strips | Axygen (available through Corning) | PCR-0208-CP-C | Or any 8-tube PCR strips (used only for water and mixes). |
| Axygen 1 x 8 strip domed PCR caps | Axygen (available through Corning) | PCR-02CP-C | Caps for PCR plates. |
| Axygen 1.5 mL Maxymum Recovery Snaplock Microcentrifuge Tube | Axygen (available through Corning) | MCT-150-L-C | Low-binding 1.5 mL tubes, used for enzyme mixes or sample concentration. |
| Axygen 96 well no skirt PCR microplate | Axygen (available through Corning) | PCR-96-C | Low-binding PCR plates - have to be used for all steps in the protocol. Note that plates should be cut to contain 2 x 8 wells for easier visibility of the samples |
| Bioanalyzer (or Tapestation): RNA nano and HS DNA kits | Agilent | To determine quality of RNA, efficient size selection and final quality of the library (Tapestation can also be used) | |
| Biotin (Long Arm) Hydrazide | Vector laboratories | SP-1100 | Biotinylation/tagging |
| Cutsmart buffer | NEB | Restriction enzyme buffer | |
| Deep Vent (exo-) DNA Polymerase | NEB | M0259S | Second strand synthesis |
| DNA Ligation Kit, Mighty Mix | Takara | 6023 | Used for 5' and 3'-linker ligation |
| dNTP mix (10 mM each) | ThermoFisher Scientific | 18427013 | dNTP mix for production of carrier templates (or any dNTPs suitable for PCR) |
| Dynabeads M-270 Streptavidin | Invitrogen | 65305 | Cap-trapping. Do not use other beads as these are optimised with the buffers used. |
| DynaMag-2 Magnet | ThermoFisher Scientific | 12321D | Magnetic stand for 1.5 mL tubes - used to prepare Streptavidin beads. |
| DynaMag-96 Side Skirted Magnet | ThermoFisher Scientific | 12027 | Magnetic stand for PCR plates (96 well-plates) - used with cut plates to contain 2 x 8 wells. |
| Ethanol, BioUltra, for molecular biology, ≥99.8% | Sigma-Aldrich | 51976-500ML-F | Used in AMPure washes. Any molecular biology suitable ethanol can be used. |
| Exonuclease I (E. coli) | NEB | M0293S | Leftover primer degradation |
| Gel Loading Dye, Purple (6x), no SDS | NEB | B7025S | agarose gel loading dye |
| HiScribe T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S | Kit for carrier in vitro transcription |
| Horizontal electrophoresis apparatus | purification of carrier DNA templates from agarose gels | ||
| I-Ceu | NEB | R0699S | Homing endonuclease used for carrier degradation. |
| I-SceI | NEB | R0694S | Homing endonuclease used for carrier degradation. |
| KAPA HiFi HS ReadyMix (2x) | Kapa Biosystems (Supplied by Roche) | KK2601 | PCR mix for target library amplification |
| KAPA SYBR FAST qPCR kit (Universal) 2x | Kapa Biosystems (Supplied by Roche) | KK4600 | qPCR mix to assess degradation efficiency and requiered number of PCR amplification cycles |
| Micropipettes and multichannel micropipettes (0.1-10 µL, 1-20 µL, 20-200 µL) | Gilson | Use of Gilson with the low-binding Sorenson tips is recommended. Other micropippetes might not be compatible. Different brand low-binding tips may not be of equal quality and may increase sample loss. | |
| Microplate reader | For Picogreen concentration measurement of the final library. Microplates are used to allow small volume measurement and reduce sample waste. | ||
| nuclease free water | ThermoFisher Scientific | AM9937 | Or any nuclease (DNase and RNase) free water |
| PCR thermal cycler | incubation steps and PCR amplficication | ||
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific | F530S | DNA polymerase for amplification of carrier templates (or any high fidelity polymerase) |
| QIAquick Gel Extraction Kit (50) | Qiagen | 28704 | Purification of carrier PCR templates from agarose gels. |
| qPCR machine | determining PCR amplification cyle number and degree of carrier degradation | ||
| Quant-iT PicoGreen dsDNA Reagent | ThermoFisher Scientific | P11495 | Used to measure final library concentration - recommended as, in our hands, it is more accurate and reproducible than Qubit. |
| Quick-Load Purple 100 bp DNA Ladder | NEB | N0551S | DNA ladder |
| Quick-Load Purple 1 kb Plus DNA Ladder | NEB | N0550S | DNA ladder |
| Ribonuclease H | Takara | 2150A | Digestion of RNA after cap-trapping. |
| RNase ONE Ribonuclease | Promega | M4261 | Degradation of single stranded RNA not protected by cDNA. |
| RNase-Free DNase Set | Qiagen | 79254 | Removal of carrier DNA templates after in vitro transcription. |
| RNeasy Mini Kit | Qiagen | 74104 | For cleanup of carrier RNA from in vitro transcription or capping |
| Sodium acetate, 1 M, aq.soln, pH 4.5 RNAse free | VWR | AAJ63669-AK | Or any nuclease (DNase and RNase) free solution |
| Sodium acetate, 1 M, aq.soln, pH 6.0 RNAse free | Or any nuclease (DNase and RNase) free solution | ||
| Sodium periodate | Sigma-Aldrich | 311448-100G | Oxidation of vicinal diols |
| Sorenson low binding aerosol barrier tips, MicroReach Guard, volume range 10 μL, Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719390-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 1,000 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719463-1000EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 20 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719412-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 200 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719447-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| SpeedVac Vacuum Concentrator | concentrating samples in various steps to lower volume | ||
| SuperScript III Reverse Transcriptase | ThermoFisher Scientific | 18080044 | Used for reverse transcription (1st CAGE step) |
| Trehalose/sorbitol solution | Preparation is described in Murata et al. 2014. | ||
| Tris-HCl, 1 M aq.soln, pH 8.5 | 1 M solution, DNase and RNase free | ||
| tRNA (20 mg/mL) | tRNA solution. Preparation is described in Murata et al. 2014. | ||
| UltraPure Low Melting Point Agarose | ThermoFisher Scientific | 16520050 | Or any suitable pure low-melt agarose. |
| USB Shrimp Alkaline Phosphatase (SAP) | Applied Biosystems (Provided by ThermoFisher Scientific) | 78390500UN | |
| USER Enzyme | NEB | M5505S | Degradation of 3'linker's upper strand, Uracil Specific Excision Reagent/Enzyme |
| Vaccinia Capping System | NEB | M2080S | Enzymatic kit for in vitro capping of carrier molecules |
| Wash buffer A | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer B | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer C | Cap trapping washes. Preparation is described in Murata et al. 2014. |
Referências
- Shiraki, T., et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proceedings of the National Academy of Sciences of the United States of America. 100 (26), 15776-15781 (2003).
- Haberle, V., Lenhard, B. Promoter architectures and developmental gene regulation. Seminars in Cell and Developmental Biology. 57, 11-23 (2016).
- Haberle, V., Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nature Reviews Molecular Cell Biology. 19 (10), 621-637 (2018).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Celniker, S. E., et al. Unlocking the secrets of the genome. Nature. 459 (7249), 927-930 (2009).
- Consortium, F., et al. A promoter-level mammalian expression atlas. Nature. 507 (7493), 462-470 (2014).
- Boyd, M., et al. Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies. Nature Communications. 9 (1), 1661 (2018).
- Adiconis, X., et al. Comprehensive comparative analysis of 5'-end RNA-sequencing methods. Nature Methods. , (2018).
- Cvetesic, N., et al. SLIC-CAGE: high-resolution transcription start site mapping using nanogram-levels of total RNA. Genome Research. 28 (12), 1943-1956 (2018).
- Murata, M., et al. Detecting expressed genes using CAGE. Methods in Molecular Biology. 1164, 67-85 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9, 357 (2012).
- A language and environment for statistical computing. Available from: https://www.R-project.org/ (2017)
- Haberle, V., Forrest, A. R., Hayashizaki, Y., Carninci, P., Lenhard, B. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Research. 43 (8), e51 (2015).
- Poulain, S., et al. NanoCAGE: A Method for the Analysis of Coding and Noncoding 5'-Capped Transcriptomes. Methods in Molecular Biology. 1543, 57-109 (2017).
- Schon, M. A., Kellner, M. J., Plotnikova, A., Hofmann, F., Nodine, M. D. NanoPARE: parallel analysis of RNA 5' ends from low-input RNA. Genome Research. 28 (12), 1931-1942 (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados