
Method Article
Aplicações de hibridização in situ de RNA/DNA viral de próxima geração na pesquisa do vírus da imunodeficiência humana/vírus da imunodeficiência símia
Neste Artigo
Resumo
Aqui, apresentamos um ensaio de hibridização in situ de última geração para identificar sequências específicas de RNA ou DNA viral em tecidos embebidos em parafina fixada em formalina (FFPE). Essa abordagem permite a visualização de cópias baixas de RNA e DNA em menos de 24 h com sensibilidade e especificidade muito altas.
Resumo
A hibridização in situ é uma técnica poderosa para identificar sequências específicas de RNA ou DNA dentro de células individuais em seções de tecido, fornecendo informações importantes sobre processos fisiológicos e patogênese de doenças. A hibridização in situ (ISH) tem sido usada há muitos anos para avaliar a localização de células infectadas por vírus, mas recentemente uma abordagem ISH de última geração foi desenvolvida com uma estratégia exclusiva de design de sonda que permite amplificação simultânea de sinal e supressão de fundo para obter visualização de molécula única, preservando a morfologia do tecido. Este ISH de próxima geração é baseado em uma abordagem como PCR ramificada, mas realizada in situ e é mais fácil, sensível e reprodutível do que os métodos clássicos de ISH ou abordagens de PCR in situ na detecção rotineira de RNA ou DNA em tecidos embebidos em parafina fixada em formalina (FFPE). Nos últimos anos, nosso laboratório tem aplicado esta plataforma ISH para a detecção de células positivas para imunodeficiência humana (HIV) e imunodeficiência símia (SIV), RNA viral (vRNA) e/ou DNA viral (vDNA) em uma infinidade de tecidos FFPE. Com este manuscrito técnico detalhado, gostaríamos de compartilhar nosso conhecimento e conselhos com todos os indivíduos interessados em usar a próxima geração de ISH em suas pesquisas.
Introdução
ISH é a abordagem experimental usada para direcionar e visualizar fitas complementares de DNA, RNA ou ácido nucleico modificado (ou seja, sondas) para sequências específicas de DNA ou RNA dentro de uma célula ou seção de tecido. O ISH permite a localização e visualização específicas de sequências nucléicas específicas em tecidos, importante para entender o nível de expressão, organização, distribuição e interações entre o alvo e seu ambiente celular, o que é uma informação valiosa que não pode ser obtida com o uso de outras técnicas populares, como qPCR. Até recentemente, a ISH era comumente realizada com um DNA complementar marcado ou um RNA complementar (ribossonda). Essas sondas foram diretamente conjugadas com bases marcadas com rádio, fluorescência ou antígeno (por exemplo, 35S, FITC e digoxigenina) e, em seguida, localizadas e quantificadas no tecido usando abordagens de autorradiografia, microscopia de fluorescência ou detecção imuno-histoquímica, respectivamente. Embora essas tecnologias in situ continuem a ser abordagens valiosas, há amplo espaço para melhorias no desenvolvimento de abordagens menos trabalhosas, mais simples, mais rápidas, sensíveis e específicas.
Uma abordagem ISH comercial alternativa de próxima geração (por exemplo, ensaio RNAscope), descrita pela primeira vez em 2012, para a detecção de RNA mensageiro do hospedeiro (mRNA) é baseada em PCR ramificado. A detecção de mRNA é realizada em células e tecidos FFPE, com uma sensibilidade que se aproxima da visualização de uma molécula de RNA único em células individuais1. A especificidade dessa abordagem é alcançada sob a condição única de que duas sondas alvo duplo Z se liguem contíguas às suas respectivas sequências complementares de RNA (ou DNA) para que um pré-amplificador de sinal se ligue sequencialmentea 1. Isso permite o início de uma cascata de amplificação de sinal por meio de etapas de hibridização subsequentes semelhantes ao DNA ramificado (bDNA) 1 , 2 . Além disso, essa abordagem é notavelmente rápida e fácil, com resultados obtidos em apenas 1 dia (<8 h), uma vantagem significativa em comparação com até 4 semanas com técnicas alternativas, incluindo Radio-ISH 1,2. Esta ISH de próxima geração abriu novas perspectivas e oportunidades para a pesquisa de HIV / SIV. Os principais obstáculos para a cura do HIV são os reservatórios celulares e teciduais que se estabelecem durante os estágios iniciais da doença 3,4. O objetivo geral desta técnica é identificar, localizar e, finalmente, entender os principais compartimentos de tecido que atuam como um reservatório viral e são persistentes dentro de um hospedeiro infectado. Isso, por sua vez, ajudará no desenvolvimento de estratégias eficazes de cura contra o HIV.
Neste manuscrito, explicamos nosso protocolo ISH multiplex de RNA/DNA duplex de próxima geração (por exemplo, RNAscope/DNAscope) em detalhes e explicamos como modificamos o protocolo RNA ISH existente para otimizar o ISH de próxima geração para nossas amostras e alvos específicos. Este protocolo permite a visualização, localização e quantificação do RNA viral do HIV/SIV e do DNA viral em seções de tecido de 5 μm. A visualização simultânea de vRNA e vDNA é realizada combinando dois conjuntos de sondas personalizadas: um sentido, visando a fita codificadora de vDNA (sonda C1 SIVmac239 Gag-Pol-Sense [416141-C1]) e um anti-sense, visando transcritos de vRNA (sonda C2 SIVmac239 Vif-Env-Nef-Tar-Anti-Sense [416131-C2]) cobrindo diferentes regiões do genoma viral (Tabela 1), utilizando dois canais de visualização diferentes, C1 e C2. Neste protocolo, os canais C1 e C2 nos permitem visualizar sinais em cores diferentes (ou seja, AP em vermelho e HRP em marrom) e detectar as sondas com diferentes abordagens. Excluindo o processamento e corte da fixação do tecido, este ensaio leva 2 dias. Apresentado aqui é o protocolo de hibridização in situ de vRNA e vDNA duplex que pode ser realizado em pellets de células ou seções de tecido.
Protocolo
1. Preparações de corte e lâmina
- Apare os blocos de parafina e use um micrótomo para cortar seções de 5 +/- 1 μm. Monte as seções ou pellets de células em lâminas de microscópio carregadas em um banho-maria livre de RNase a 40-45 °C. Secar as lâminas ao ar durante a noite a 37 °C ou RT.
NOTA: As lâminas podem ser armazenadas por até 3 meses em temperatura ambiente (RT) e 6 meses a 4 °C. - Desparafinizar slides FFPE.
- Asse as lâminas em forno seco por 1 h a 60 °C.
- Em uma capela de exaustão, encha duas placas de coloração de lâminas com ~ 200 mL de xileno fresco e duas placas de coloração adicionais com ~ 200 mL de etanol 100% fresco. Cubra os recipientes com tampas.
- Coloque as lâminas em uma prateleira e mergulhe no primeiro prato contendo xileno. Incubar durante 5-10 min em RT com agitação.
- Coloque as lâminas no segundo prato contendo xileno e incube por 5-10 min em RT com agitação.
- Coloque imediatamente as lâminas no prato contendo 100% de etanol. Incube as lâminas por 5-10 min em RT com agitação.
- Coloque imediatamente as lâminas no segundo prato contendo 100% de etanol e incube por 5-10 min em RT com agitação.
- Remova o rack do etanol, bata suavemente na lateral do rack para remover o excesso de etanol e enxágue em água sem RNase por 5-15 min.
2. Preparação do forno
- Ligue o forno de hibridização e ajuste a temperatura para 40 °C.
- Coloque um pano ou toalha de papel absorvente resistente em uma bandeja e molhe completamente com água bidestilada para permitir o controle da umidade.
- Insira a bandeja coberta no forno e feche a porta do forno. Aqueça a bandeja por pelo menos 30 min a 40 °C antes de usar. Mantenha a bandeja no forno quando não estiver em uso.
3. Recuperação de epítopos induzida por calor
- Prepare o tampão de recuperação do alvo de hibridização ISH à base de citrato 0,5x (10 nmol/L, pH = 6, consulte a Tabela de Materiais). Leve para ferver em um copo na placa de aquecimento.
- Execute a recuperação de epítopos induzida por calor colocando as lâminas no tampão de recuperação do alvo de ebulição por 30 min.
- Remova as lâminas do tampão de recuperação alvo e lave imediatamente em água bidestilada Desidrate em etanol 100% por 5 min antes de secar ao ar.
- Depois que as lâminas secarem ao ar, aplique caneta de barreira hidrofóbica para circundar a seção de tecido na lâmina. Certifique-se de permitir que a barreira hidrofóbica seque completamente ao ar.
4. Pré-tratamento de protease
- Coloque as lâminas secas em um suporte de lâminas com trava e, em seguida, prepare os reagentes de pré-tratamento de protease (solução de digestão de protease, 2,5 μg / mL) diluindo com PBS estéril e frio na proporção de 1:5. Misture bem.
NOTA: Três reagentes de protease diferentes com concentrações diferentes são fornecidos no kit disponível comercialmente. Protease III (padrão), Protease IV (forte) e Protease Plus (leve). Teste empiricamente o tempo de digestão da protease e a diluição antes da implementação em um estudo, pois as condições ideais variam de acordo com o tipo de tecido, fixação e espessura (ver Discussão). - Dispense a solução de protease diluída em lâminas para cobrir completamente as seções de tecido. Incube imediatamente as lâminas por 20 min a 40 °C em um forno (preparado na etapa 1.4), garantindo que as lâminas sejam seladas na bandeja de hibridização úmida. Não deixe as seções de tecido secarem pelo restante do protocolo.
- Enxágue imediatamente 3x submergindo o rack deslizante de travamento em uma bandeja de lavagem cheia de água bidestilada
- Execute o bloqueio da peroxidase endógena deixando cair a solução de peroxidase em cada seção do tecido para cobri-la completamente. Incube as lâminas por 10 min em RT. Uma vez feito isso, enxágue as seções 3x em água bidestilada
5. Hibridização da sonda e amplificação de sinal
NOTA: Para evitar a evaporação, certifique-se de que a bandeja com controle de umidade vede adequadamente para que os tecidos não sequem durante as etapas de incubação. Coloque a câmara de umidade de volta no forno durante a etapa de lavagem para garantir que permaneça a 40 °C.
- Misture a sonda C2 e a sonda C1 na proporção de 1:50 pipetando 1 volume de sonda C2 para 50 volumes de sonda C1 em um tubo, conforme sugerido pelo fabricante. Inverta o tubo várias vezes. Pré-aqueça a mistura da sonda alvo em forno a 40 °C por ~10 min para dissolver qualquer precipitação antes do uso.
NOTA: As sondas de alvo misto podem ser armazenadas a 4 °C por até 6 meses. - Remova os slides da água. Enxágue e bata ou sacuda as lâminas para remover o excesso de água das seções de tecido. Dispense imediatamente a sonda nas lâminas, garantindo que cada seção de tecido esteja completamente coberta sem bolhas de ar. Incubar a mistura da sonda na câmara de humidade durante a noite a 40 °C.
- No dia seguinte, retire as lâminas do forno e coloque-as na bandeja de lavagem contendo 0.5x tampão de lavagem por 5 min em RT. Repita a etapa de lavagem mais uma vez.
- Remova as lâminas do tampão de lavagem. Enxágue e bata ou sacuda as lâminas para remover o excesso de tampão de lavagem das seções de tecido.
- Dispense comercialmente o reagente AMP 1 pronto para uso (2 nmol/L) em tampão de hibridização B (20% formamida, 5x SSC, 0,3% sulfato de lítiododecil, 10% sulfato de dextrana, reagentes bloqueadores) nas lâminas, garantindo cobertura completa da seção de tecido sem bolhas de ar. Incubar durante 30 min a 40 °C na câmara de humidade. Repita as etapas 1.6.3-1.6.4, realizando a lavagem por 2 min cada.
- Dispense AMP 2 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 15 min a 40 °C. Repita as etapas 5.3-5.4, realizando a lavagem por 2 min cada.
- Dispense AMP 3 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 30 min a 40 °C. Repita as etapas 5.3-5.4, realizando a lavagem por 2 min cada.
- Dispense AMP 4 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 15 min a 40 °C. Repita as etapas 5.3-5.4, realizando a lavagem por 2 min cada.
- Dispense AMP 5 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 30 min a 40 °C. Repita os passos 5.3-5.4, realizando a lavagem por 2 min cada.
- Dispense AMP 6 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 15 min a 40 °C. Repita os passos 5.3-5.4, realizando a lavagem por 2 min cada.
- Antes da detecção, enxágue as lâminas uma vez em 1x TBS-Tween 20 (0.05% v/v). Remova as lâminas do tampão de lavagem, enxágue e bata ou sacuda as lâminas para remover o excesso de tampão de lavagem das seções de tecido. Coloque imediatamente na bandeja de lavagem cheia de 1x tampão TBS-Tween.
6. Detecção de sinal alvo do canal 1 (C1)
NOTA: Isso é realizado usando a Fosfatase Alcalina Vermelha e a Amplificação Rápida de Cromogênio Vermelho 6 de kits de detecção 2-plex (consulte a Tabela de Materiais) contendo rótulos de fosfatase alcalina e detecção cromogênica. Ele usa vermelho rápido como substrato para gerar um sinal vermelho.
- Prepare a solução de trabalho vermelha rápida (FR) usando uma diluição de 1:60 de Fast RED-B para Fast RED-A. Misture bem. Para reduzir o precipitado e obter um sinal mais limpo, filtre a solução de cromogênio através de uma membrana MCE de 0,45 μm usando uma seringa.
NOTA: Use a solução Fast RED-B dentro de 5 min. Não exponha à luz solar direta ou luz ultravioleta. - Remova as lâminas da TBS-Tween, enxágue e bata ou mova as lâminas para remover o excesso de tampão das seções de tecido.
- Dispense a solução FR misturada e filtrada em cada seção de tecido, certificando-se de que cada seção esteja completamente coberta. Incubar em RT por 6-8 min. Observe ao microscópio.
- Enxágue as lâminas em tampão de lavagem 0.5x 2x. Remova as lâminas do tampão de lavagem, enxágue e bata ou sacuda as lâminas para remover o excesso de tampão de lavagem das seções de tecido.
- Dispense AMP 7 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 10 min a 40 °C. Repita os passos 5.3-5.4, realizando a lavagem por 2 min cada.
- Dispense AMP 8 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 15 min a 40 °C. Repita os passos 5.3-5.4, realizando a lavagem por 2 min cada.
- Dispense AMP 9 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 30 min a 40 °C. Repita os passos 5.3-5.4, realizando a lavagem por 2 min cada.
- Dispense AMP 10 disponível comercialmente. Certifique-se de que a seção de tecido esteja completamente coberta sem bolhas de ar. Incubar as lâminas na câmara de humidade durante 30 min a 40 °C. Repita os passos 5.3-5.4, realizando a lavagem por 2 min cada.
7. Detecção de sinal alvo do canal 2 (C2)
NOTA: Isso é feito usando kits de cromogênio Brown HRP e DAB disponíveis comercialmente (consulte a Tabela de Materiais). A amplificação 10 da detecção 2-plex contém marcadores de peroxidase de rábano e a detecção cromogênica é realizada usando DAB para gerar um sinal marrom.
- Para uma detecção ideal do sinal DAB, use kits disponíveis comercialmente e siga as instruções do fabricante (consulte a Tabela de Materiais). Observe ao microscópio.
CUIDADO: DAB é tóxico. Siga as precauções e diretrizes de segurança apropriadas ao manusear e descartar este produto químico.
8. Contracoloração e montagem
- Contra-core as lâminas com hematoxilina.
- Contra-core as lâminas por 30 s com 50% de hematoxilina filtrada fresca enquanto agita as lâminas. Os slides aparecerão em roxo. Enxágue imediatamente em água corrente enquanto agita as lâminas para cima e para baixo até que a água esteja limpa. As seções de tecido permanecerão roxas.
- Para obter um melhor contraste, coloque as lâminas contracoradas em água destilada saturada com carbonato de lítio por 1 min. Enxágue bem em água corrente pelo menos 3x enquanto agita as lâminas. Use água bidestilada para o enxágue final.
- Como o FR é sensível a solventes orgânicos, as lâminas coradas com FR precisarão ser cobertas com meio de montagem à base de água e secas durante a noite em RT.
- Monte os slides.
- Certifique-se de que as seções de tecido cobertas com meio de montagem à base de água estejam secas.
- Mergulhe as lâminas em xileno antes de deslizar a tampa usando o reagente de montagem. Certifique-se de evitar ou remover quaisquer bolhas de ar entre a lamínula e a seção de tecido e deixe secar por 16 horas em RT.
9. Protocolo de análise quantitativa de imagens para RNAscope usando CellProfiler5
- Resumidamente, certifique-se de que o software separará as colorações de hematoxilina e FR em imagens separadas. Identifique e meça os objetos de interesse: núcleos, vírions, células positivas e coloração FR positiva agregada. Armazene as medidas em um arquivo CSV e salve a imagem analisada.
- Selecione a opção "Desmisturar cores", que separa as manchas, dividindo a região de interesse (ROI) original em imagens separadas de hematoxilina e FR.
- Se a hemossiderina, tatuagem ou características semelhantes interferirem na análise, adicione uma segunda etapa "Unmix" com hematoxilina, FR e DAB. Use a segunda imagem FR para encontrar os pixels FR mais intensos. Esta segunda imagem será usada como uma máscara para a coloração FR verdadeira.
- Opcionalmente, suavize as imagens manchadas antes de limitá-las. Esta é uma decisão empírica e nem sempre necessária. Limite as imagens manchadas individuais usando "IdentifyPrimaryObjects" para selecionar pixels positivos.
- Identifique os três tipos diferentes de objetos (vírion, agregados de vírion, célula produtiva).
- Certifique-se de que os núcleos sejam objetos de 4 a 100 pixels (px) de diâmetro corados com hematoxilina. Desgrude e preencha os buracos após o limiar. Os objetos "IntenseFastRed" têm 4-100 px de diâmetro e incluem vírions, células positivas e coloração positiva agregada, como visto em células dendríticas foliculares (FDCs) em folículos de células B (BCF). Essa imagem é usada para filtrar falsos positivos (por exemplo, hemossiderina).
- Certifique-se de que os pequenos positivos FR sejam objetos de 2 a 12 px de diâmetro. Esta medição inclui vírions e células vDNA+. Descarte todos os objetos fora desse intervalo. Desgrude e preencha os buracos após o limiar.
- Certifique-se de que FR grandes positivos: objetos de 9 a 100 px de diâmetro. Esta medição inclui células positivas de vRNA+ e coloração positiva agregada. Diminuir após o limiar. O tamanho de objetos FR positivos pequenos e grandes pode se sobrepor. Eles serão separados em uma etapa posterior.
NOTA: O software (por exemplo, Cellprofiler) usa pixels para tamanhos de objetos, e os usados aqui são derivados de lâminas digitalizadas a 0,2510 μm/pixel (40x).
- Defina e extraia resultados.
- Depois que qualquer objeto for identificado, defina e extraia os resultados para o número de vírions, células infectadas produtivas e núcleos.
- Identifique vírions mascarando FastRedSmallPositives com o objeto IntenseFastRed definido para remover falsos positivos (por exemplo, hemossiderina).
- Em seguida, identifique as células positivas e agregue a coloração FR positiva. Remova falsos positivos e vírions de FastRedLargePositives mascarando-os com IntenseFastRed e com o conjunto de objetos vírion.
- Extraia células positivas dos FastRedLargePositives refinados mascarando novamente com Nuclei. Divida os objetos que não estão mais se tocando e filtre os resultados por área do objeto, removendo objetos pequenos (≤6 px). Isso remove as manchas criadas pelo mascaramento da sobreposição dos núcleos. O resultado são as células positivas.
- Por fim, defina os positivos agregados de FR. Esta etapa usa IdentifyTertiaryObjects, localizando objetos contidos em um objeto pai maior. Nesse caso, o conjunto de objetos FastRedLargePositives refinado é o pai e as células positivas são subtraídas.
- Conte o número de vírions e células positivas.
- Meça a área positiva agregada e converta-a de pixels para mm2. Opcionalmente, registar a área ocupada pelos vírions e células positivas em mm2 se a análise exigir a utilização de uma célula padrão e tamanhos de viriões em vez de contagens diretas.
- Sobreponha os objetos positivos na imagem original e salve o resultado.
NOTA: Os dados de ISH são relatados pelo número de vírions por 106 núcleos (células) e pelo número de células vRNA+ produtivamente infectadas por 106 núcleos (células) para uma melhor compreensão e para facilitar a comparação com os dados de qPCR, mas os resultados também podem ser relatados por área de tecido em mm2.
Resultados
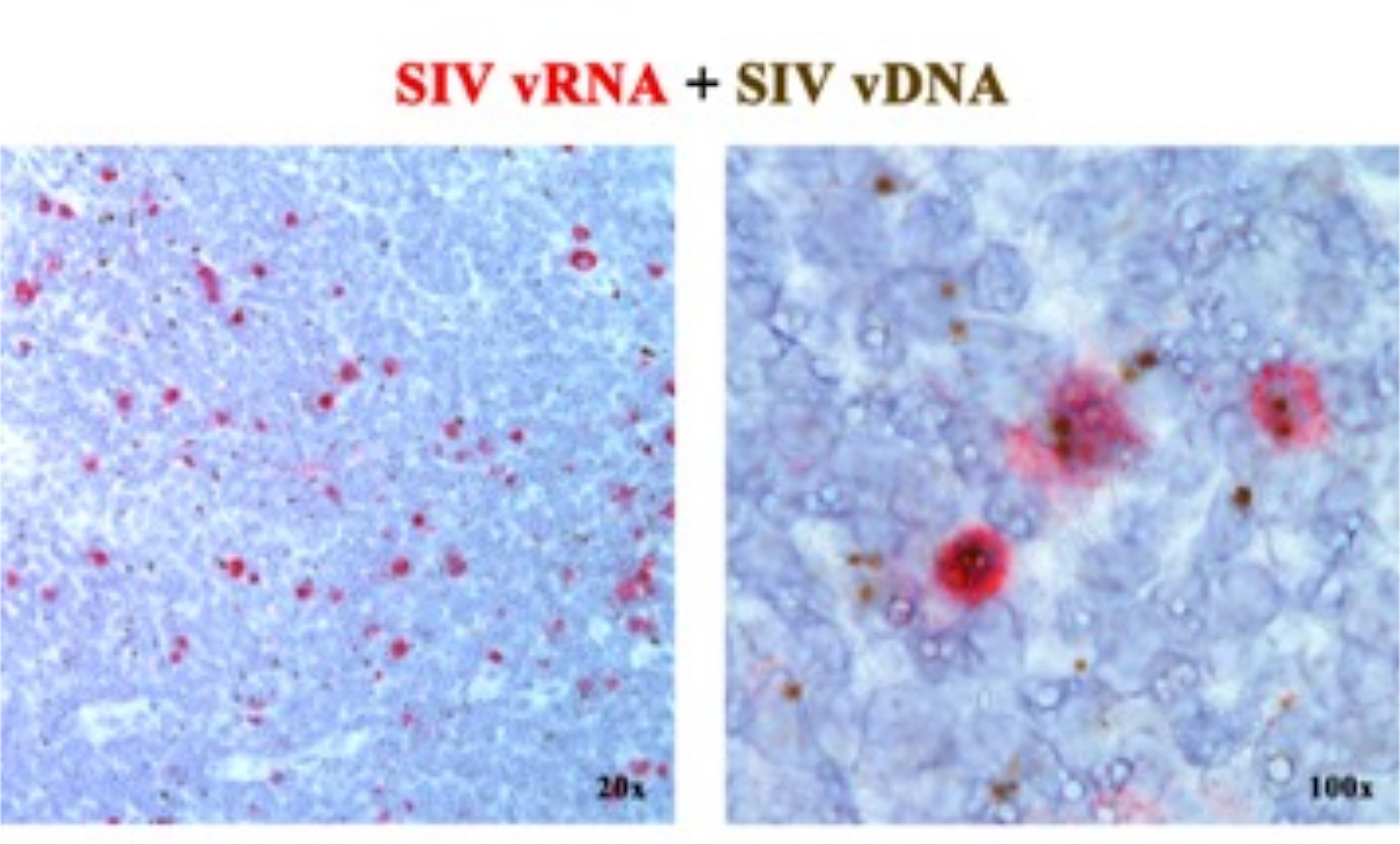
Em um manuscrito anterior 2,6,7,8,9,10,11, relatamos que as plataformas ISH de próxima geração que detectam vRNA ou vDNA podem ser combinadas, usando sondas sensoriais (vDNA) direcionadas à porção 5 'gag-pol do genoma SIV / HIV e sondas antisense (vRNA) direcionadas a genes na metade 3' do genoma (vif, vpx, vpr, tat, env e nef), bem como o elemento TAR no genoma 5' (Tabela 1). Essa abordagem distingue células transcricionalmente ativas (vRNA+, vDNA+) de células infectadas transcricionalmente inativas (supostamente latentes) ou células que abrigam provírus transcricionalmente incompetentes (vRNA-, vDNA+) na mesma seção de tecido2 (Figura 1).

Figura 1: Detecção de RNA viral e vDNA na mesma seção de tecido. Combinação de ensaio de hibridização de RNA (vermelho) e hibridização de DNA (marrom) em um linfonodo RM agudamente infectado por SIV, demonstrando a capacidade de detectar vRNA e vDNA na mesma seção de tecido e fornecendo uma abordagem poderosa para identificar células vDNA+ vRNA- transcricionalmente silenciosas in situ. Esta figura foi modificada de Deleage C. et al.2. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Conjuntos de sondas de RNA/DNAscope de plexo único | |||
| Nome | Catálogo ACD # | Número de ZZ | Descrição |
| SIVmac239 (Anti-Sense) | 312811 | 83 | Direcionamento de sonda anti-sentido dentro de 1251-9420bp de D01065.1 (gag, pol, vif, vpx, vpr, tat, env e nef) |
| SIVmac239 (Sentido) | 314071 | 83 | Sonda de detecção direcionada à fita reversa dentro de 1251-9420bp de D01065.1 (gag, pol, vif, vpx, vpr, tat, env e nef) |
| V-HIV1-Clado A (Anti-Sense) | 416101 | 80 | Direcionamento de sonda anti-sentido dentro de 879-7629bp do Consenso do Clado A do HIV-1 (gag, pol, vif, vpr, tat, rev, vpu, env e nef) |
| V-HIV1-Clado A (Sentido) | 426341 | 80 | Sonda de detecção direcionada à fita reversa dentro de 879-7629bp do Consenso do Clado A do HIV-1 (gag, pol, vif, vpr, tat, rev, vpu, env e nef) |
| V-HIV1-Clado B (Anti-Sense) | 416111 | 78 | Direcionamento de sonda anti-sentido dentro de 854-8291bp de AF324493.2, HIV-1 Clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env e nef) |
| V-HIV1-Clado B (Sentido) | 425531 | 78 | Sonda de detecção direcionada à fita reversa dentro de 854-8291bp de AF324493.2, HIV-1 Clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env e nef) |
| V-HIV1-Clado D (Anti-Sense) | 416121 | 76 | Direcionamento de sonda anti-sentido dentro de 894-7697bp do Consenso do Clado D do HIV-1 (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| V-HIV1-Clado D (Sentido) | 426351 | 76 | Sonda de detecção direcionada à fita reversa dentro de 894-7697bp do consenso do clado D do HIV-1 (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| Conjuntos de sondas multiplex de RNA/DNAscope | |||
| Nome | Catálogo ACD # | Número de ZZ | Descrição |
| V-SIVmac239-gag-pol-Sense-C1 | Rolamento 416141-C1 | 40 | Sonda de detecção direcionada à fita reversa dentro de 1251-4093bp de D01065.1 (gag e pol) |
| V-SIVmac239-vif-env-nef-tar-C2 (Anti-sentido) | 416131-C2 | 47 | Segmentação de sonda antidetecção dentro de 5381-10257bp de D01065.1 (vif, vpx, vpr, tat, env, nef e o elemento TAR) |
| V-HIV1-Clade_B-gag-pol-sense-C1 | Rolamento 444051-C1 | 40 | Sonda de detecção direcionada à fita reversa dentro de 854-3940bp de AF324493.2, HIV-1 Clade B NL4-3 (gag e pol) |
| V-HIV1-Clade_B-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Anti-sentido) | 444061-C2 | 40 | Direcionamento de sonda anti-sentido dentro de 5042-9673bp de AF324493.2,, HIV-1 Clade B NL4-3 (vif, vpr, tat, env, nef e o elemento TAR) |
| V-HIV1-Clade_C-gag-pol-sense-C1 | Rolamento 444021-C1 | 48 | Sonda de detecção direcionada à fita reversa dentro de 888-5032bp da sequência de consenso do Clado C do HIV-1 (gag e pol) |
| V-HIV1-Clade_C-vif-vpr- rev-vpu-env-nef-tar-C2 (Anti-sentido) | 444041-C2 | 49 | Direcionamento de sonda anti-sentido dentro de 5078-9698bp da sequência de consenso do Clado C do HIV-1 (vif, vpr, tat, env, nef e o elemento TAR) |
| V-HIV1-Clade_AE-gag-pol-sense-C1 | Rolamento 444011-C1 | 55 | Sonda de detecção direcionada à fita reversa dentro de 890-4812bp de AF259954.1, HIV-1 Clade AE (gag e pol) |
| V-HIV1-Clade_AE-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Anti-sentido) | Rolamento 444031-C2 | 57 | Direcionamento de sonda anti-sentido dentro de 5052-9694bp de AF259954.1, HIV-1 Clade AE (vif, vpr, tat, env, nef e o elemento TAR) |
Tabela 1: Lista de sondas para direcionar vRNA e vDNA de HIV-1 e SIV.
Discussão
A hibridização in situ é um ensaio meticuloso que requer rigor e conhecimento básico de química de ácidos nucleicos, biologia celular e histologia para ser capaz de adaptar cada etapa crítica para localizar um alvo em um ambiente bem conservado. Nesta discussão, gostaríamos de destacar as etapas críticas em que a solução de problemas é crucial para obter resultados precisos e interpretáveis.
A fixação e o processamento dos tecidos são críticos e devem ser abordados antecipadamente para garantir que o ensaio possa produzir os melhores resultados. O fixador PFA tamponado neutro (4% recém-preparado) é ideal para o ensaio duplex. No entanto, o ensaio também pode ser realizado em tecidos congelados (OCT) com as condições adequadas de fixação pós-criosecção.
O pré-tratamento das seções de tecido é uma etapa crucial. Existem duas etapas de pré-tratamento neste ensaio: a primeira é uma recuperação de epítopos induzida por calor (HIER). Esta etapa é importante para a reversão das ligações cruzadas da ponte de metileno e restauração das estruturas proteicas, o que é necessário em tecidos fixos. A eficiência deste tratamento depende do tempo, temperatura, tipo de tampão de recuperação e pH. O segundo pré-tratamento é uma recuperação de epítopo induzida por protease (PIER). Esta etapa cliva peptídeos, expondo o antígeno ou nucleotídeos, e usa enzimas, incluindo proteinase K, tripsina e pepsina. Esta é uma etapa extremamente sensível que pode danificar a morfologia do tecido e o alvo de interesse. A concentração da enzima, bem como o tempo e a temperatura de incubação são críticos neste processo. A superdigestão leva a uma má demarcação dos núcleos e dificuldade nas etapas de quantificação. É fundamental encontrar um equilíbrio entre o acesso ideal ao alvo de RNA/DNA e as condições de pré-tratamento que não danifiquem o tecido ou o alvo de interesse. Cada tipo de tecido tem um nível diferente de sensibilidade a cada um desses pré-tratamentos e cada parâmetro (concentração enzimática, tempo, temperatura) deve ser testado empiricamente.
O rigor do tampão de lavagem é baseado em três parâmetros principais: temperatura, concentração de sais e detergente e tempo. O tampão de lavagem é um tampão citrato de sódio salino (SSC) e a concentração de sal dentro do tampão controla o rigor durante as etapas de lavagem. Em seu protocolo, a ACD aconselha o uso do tampão de lavagem a uma concentração final de 0,1x SSC, 0,03% de dodecil sulfato de lítio. Ao trabalhar no DNAscope e na otimização multiplex, determinamos que o uso do tampão de lavagem em uma concentração final de 0,05x SSC nos deu melhores resultados para visualizar o sinal de DNA e ajudou consideravelmente a reduzir a hibridização fora do alvo inespecífica resultante da incubação noturna da sonda sensorial.
A escolha da abordagem de detecção, cromogênio (vermelho ou marrom) versus fluorescência precisa ser pensada com base no tipo de tecido e no objetivo antes de iniciar o ensaio. A abordagem cromogênica vermelha dará um bom contraste, porque o vermelho não é encontrado naturalmente nos tecidos. O cromógeno marrom dará resultados semelhantes ao cromogênio vermelho. No entanto, é importante ter em mente que alguns produtos de degradação do sangue presentes no tecido têm cor semelhante, e a tinta da tatuagem será difícil de separar do sinal marrom durante a quantificação. Uma abordagem de detecção de fluorescência permitirá uma distinção clara de diferentes marcadores celulares e a multiplexação oferecerá um ensaio perfeito para fenotipar as células que abrigam vRNA e/ou vDNA.
Vários controles são necessários para garantir a especificidade das sondas e a qualidade do ensaio. Cada sonda recém-projetada deve ser testada em tecidos de controle positivo e negativo conhecidos ou pellets de células. Freqüentemente, geramos plasmídeos contendo nossa sequência alvo e realizamos a transfecção em linhagens celulares para gerar controles positivos. Para cada execução, adicionamos um tecido negativo conhecido (HIV ou SIV negativo), um controle sem sonda contendo apenas o diluente da sonda e um controle tratado com RNase para garantir a qualidade e especificidade do ensaio.
A quantificação é uma etapa extremamente importante e deve ser realizada usando as ferramentas e algoritmos apropriados com base na pergunta feita. Neste manuscrito, apresentamos um software de análise de imagens (por exemplo, Cellprofiler), que escolhemos após avaliação de várias opções. Estimamos que este software era o melhor software para nossas necessidades, mas existem vários programas de software de análise de imagem que podem ser usados.
Divulgações
Os autores não têm nada a divulgar.
Agradecimentos
Este projeto foi financiado integralmente com fundos federais do Instituto Nacional do Câncer, Institutos Nacionais de Saúde, sob o Contrato nº. HHSN261200800001E e pelo Oregon National Primate Research Center NIH grant award P51OD011092 (JDE). O conteúdo desta publicação não reflete necessariamente as opiniões ou políticas do Departamento de Saúde e Serviços Humanos, nem a menção de nomes comerciais, produtos comerciais ou organizações implica endosso do governo dos EUA. O duplex foi desenvolvido com a ajuda do Advanced Cell Diagnostics.
Materiais
| Name | Company | Catalog Number | Comments |
| ACD HybEZII Hybridization system (110V) with ACD EZ-Batch Slides system | ACD | 321710 | Hybridization oven |
| CAT Hematoxylin | Biocare medical | CATHE-GAL | colorstain |
| Clear-Mount | ELECTRON MICROSCOPY SCIENCES | 17985-15 | mounting reagent for red chromogen |
| Immpact DAB Peroxidase Kit | Vector | SK-4105 | Used to reveal HRP - DAB (Brown) to replace the DAB coming in the ACD kit |
| lithium carbonate | Fisher chemical | L119-500 | bluing solution |
| paraformaldehyde | ELECTRON MICROSCOPY SCIENCES | 15714-S | for tissue fixation (4%) |
| PBS | life technology | 14190-136 | |
| Permount Mounting Medium | ThermoFisher Scientific | SP15-100 | mounting regaent for brown chromogen |
| Prolong Gold | ThermoFisher Scientific | P36930 | mounting regaent for fluorescence |
| ribonucleases A | ThermoFisher Scientific | 12091039 | for RNAse treatment in DNAscope protocol |
| ribonucleases T1 | Roche | R1003 | for RNAse treatment in DNAscope protocol |
| RNAscope 2.5, 2-plex detection reagent | ACD | 322430 | Brown and red kit chromogen detection |
| RNAscope Target Retrieval Reagents | ACD | 322000 | retrieval buffer |
| SuperFrost Plus Glass Slides | ThermoFisher Scientific | 12-550-17 | |
| TBS | BOSTON BIOPRODUCTS | BM-301-4L | for washes |
| TSA Plus Fluorescence palette kit (Cy3, Cy5, TMR, Fluorescein) | Perkin elmer | NEL760001KT | HRP Fluorescence detection |
| Tween 20 | SIGMA | P1379-1L | for washes |
| XYLENE 20LT | ThermoFisher Scientific | AC422680200 |
Referências
- Wang, F., et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnosis. 14 (1), 22-29 (2012).
- Deleage, C., et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathogens and Immunity. 1 (1), 68-106 (2016).
- Sengupta, S., Siliciano, R. F. Targeting the Latent Reservoir for HIV-1. Immunity. 48 (5), 872-895 (2018).
- Churchill, M. J., Deeks, S. G., Margolis, D. M., Siliciano, R. F., Swanstrom, R. HIV reservoirs: what, where and how to target them. Nature Reviews Microbiology. 14 (1), 55-60 (2016).
- McQuin, C., et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biology. 16 (7), e2005970 (2018).
- Deleage, C., Chan, C. N., Busman-Sahay, K., Estes, J. D. Next-generation in situ hybridization approaches to define and quantify HIV and SIV reservoirs in tissue microenvironments. Retrovirology. 15 (1), 4 (2018).
- Deleage, C., Turkbey, B., Estes, J. D. Imaging lymphoid tissues in nonhuman primates to understand SIV pathogenesis and persistence. Current Opinion in Virology. 19, 77-84 (2016).
- Estes, J. D., et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nature Medicine. 23 (11), 1271-1276 (2017).
- Mavigner, M., et al. Simian Immunodeficiency Virus Persistence in Cellular and Anatomic Reservoirs in Antiretroviral Therapy-Suppressed Infant Rhesus Macaques. Journal of Virology. 92 (18), (2018).
- Peterson, C. W., et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathogen. 14 (4), e1006956 (2018).
- Deleage, C., et al. Impact of early cART in the gut during acute HIV infection. Journal of Clinical Investigation Insight. 1 (10), e87065 (2016).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados