
Method Article
Preparação da amostra TMT para envio de instalações proteômicas e análise de dados subsequentes
Neste Artigo
Resumo
Apresentamos um protocolo de rotulagem de etiquetagem tandem (TMT) otimizado que inclui informações detalhadas para cada uma das seguintes etapas: extração de proteínas, quantificação, precipitação, digestão, rotulagem, submissão a uma instalação de proteômica e análises de dados.
Resumo
As tecnologias proteômicas são metodologias poderosas que podem ajudar a entender nossos mecanismos de ação em sistemas biológicos, fornecendo uma visão global do impacto de uma doença, tratamento ou outra condição no proteome como um todo. Este relatório fornece um protocolo detalhado para a extração, quantificação, precipitação, digestão, rotulagem e análise subsequente de dados de amostras de proteínas. Nosso protocolo de rotulagem TMT otimizado requer uma menor concentração de etiquetas e consegue dados consistentemente confiáveis. Usamos este protocolo para avaliar perfis de expressão proteica em uma variedade de tecidos de camundongos (ou seja, coração, músculo esquelético e cérebro) bem como células cultivadas in vitro. Além disso, demonstramos como avaliar milhares de proteínas a partir do conjunto de dados resultante.
Introdução
O termo "proteômica" foi definido pela primeira vez como a caracterização em larga escala de todo o complemento proteico de uma célula, tecido ou organismo1. As análises proteômicas permitem a investigação de mecanismos e processos celulares envolvidos no desenvolvimento de doenças, vias terapêuticas e sistemas saudáveis utilizando técnicas para realizar a quantificação relativa dos níveis de expressão proteica2. As descrições iniciais de tais estudos foram publicadas em 1975 e demonstraram o uso de eletroforese de gel poliacrilamida bidimensional (2D-PAGE) para este fim1,,3. O método 2D separa proteínas baseadas na carga (focalização isoelétrica, IEF) e massa molecular (eletroforese de gel de poliacrilamida de sulfato de sódio, ou SDS-PAGE)4. Durante anos, a combinação de espectrometria de massa 2D-PAGE e subsequente em tandem realizada em cada componente gel foi a técnica de análise de expressão proteica não alvo mais comum realizada e identificou numerosos perfis de expressão proteica até então desconhecidos5,,6. Desvantagens gerais para a abordagem 2D-PAGE são que é demorado, não funciona bem para proteínas hidrofóbicas, e há limitações no número total de proteínas avaliadas devido à baixa sensibilidade7,,8.
A rotulagem estável de isótopos por aminoácidos no método de cultura celular (SILAC) tornou-se a próxima abordagem popular para identificar e quantificar a abundância de proteínas nas amostras9. Consiste na rotulagem metabólica de células incubadas em meio sem aminoácido essencial padrão e suplementadas com uma versão rotulada por isótopo desse aminoácido específico10. A vantagem dessa técnica é sua eficiência e rotulagem precisa9. A principal limitação à abordagem do SILAC é principalmente a redução da taxa de crescimento celular causada pela incorporação de rótulos de isótopos, o que pode ser particularmente desafiador em linhas celulares relativamente sensíveis modelando doenças humanas11.
Em 2003, uma nova e robusta técnica de proteômica envolvendo etiquetas isobáricas tandem mass tag (TMT) foi introduzida no campo12. A rotulagem TMT é um método poderoso devido à sua sensibilidade aumentada para detectar níveis relativos de expressão proteica e modificações pós-transacionais13. A partir desta data de publicação, foram desenvolvidos kits TMT que podem rotular simultaneamente 6, 10, 11 ou 16 amostras. Como resultado, é possível medir a abundância de peptídeos em múltiplas condições com réplicas biológicas ao mesmo tempo14,15,16. Recentemente, utilizamos TMT para caracterizar o perfil proteômico cardíaco de um modelo de camundongo da Síndrome de Barth (BTHS)17. Ao fazê-lo, pudemos demonstrar melhora generalizada nos perfis cardíacos de camundongos BTHS tratados com terapia genética e identificar novas proteínas impactadas pelo BTHS que revelaram novas vias terapêuticas envolvidas em cardiomiopatias.
Aqui, descrevemos um método detalhado para realizar análises de proteômica quantitativa multiplex TMT usando amostras de tecido ou pelotas de células. Pode ser benéfico realizar a preparação e rotulagem da amostra antes da submissão a um núcleo porque os peptídeos tripptic rotulados são mais estáveis do que amostras congeladas cruas, nem todos os núcleos têm experiência em manusear todos os tipos de amostras, e preparar amostras em laboratório pode economizar tempo para núcleos, que muitas vezes têm longas pendências. Para obter descrições detalhadas da porção de espectroscopia de massa deste processo, consulte Kirshenbaum et al. e Perumal et al.18,19.
O protocolo de preparação da amostra consiste nas seguintes etapas principais: extração, quantificação, precipitação, digestão e rotulagem. Os principais benefícios deste protocolo otimizado são que ele reduz os custos de rotulagem, melhora a extração de proteínas e gera consistentemente dados de alta qualidade. Além disso, descrevemos como analisar dados TMT para rastrear milhares de proteínas em um curto espaço de tempo. Esperamos que este protocolo incentive outros grupos de pesquisa a considerar a incorporação dessa metodologia poderosa em seus estudos.
Protocolo
O Comitê Institucional de Cuidados e Uso de Animais da Universidade da Flórida aprovou todos os estudos em animais.
1. Preparação de reagentes
- Prepare chaps lysis buffer (150 mM KCl, 50 mM HEPES pH = 7,4, 0,1% CHAPS e 1 comprimido de coquetel inibidor de protease por 50 mL de tampão). Tampão sem inibidores de protease pode ser armazenado a 4 °C por até 6 meses ou tampão com inibidor de protease armazenado a -20 °C até 1 ano.
- Prepare 100 mM trietilamônio bicarbonato (TEAB): Adicione 500 μL de 1 M TEAB a 4,5 mL de água ultrapura.
- Prepare 200 mM tris (2 carboxyethyl) cloridrato de fosfina (TCEP): Adicione 70 μL de 0,5 M TCEP, um reagente desnaturing, a 70 μL de água ultrapura. Em seguida, adicione 35 μL do TEAB de 1 M.
- Prepare 5% de hidroxilamina: Adicione 50 μL de 50% de hidroxilamina a 450 μL de 100 mM TEAB.
2. Extração de proteína
- Isole o músculo quadríceps de um rato eutanizado de acordo com um protocolo aprovado pela IACUC. Congele e mantenha a -80 °C ou continue com o protocolo para uso imediato.
- Corte para isolar aproximadamente 10 mg de tecido de rato quadríceps fresco ou congelado. Separe fibras usando pinças ao trabalhar com músculo esquelético. Alternativamente, se trabalhar com culturas celulares, resuspenda ~3 x 106 células em 300 μL de tampão de lise CHAPS e pule para o passo 2.4.
- Homogeneize o tecido utilizando um disruptor de contas usando tubos de 2 mL preenchidos com aproximadamente 200 μL de 1 mm de contas de zircônia/sílica e 500 μL de tampão de lise CHAPS. Dimensione para cima ou para baixo conforme apropriado (por exemplo, 5 mg de tecido em 250 μL de tampão de lise CHAPS).
- Realize a sonicação (10x para 10 s cada com 50% de amplitude e intervalos de 30 s no gelo) para liberar proteína ligada ao DNA. Os mesmos resultados de degradação do DNA podem ser obtidos com a seringa lysis passando o 10x de lysato através de uma agulha de 21 G presa a uma seringa de 1 mL, ou por incubação de benzonase (44 U/mL) a 37 °C por 30 min.
- Centrifugar o lysate a 16.000 x g por 10 min a 4 °C e transferir o supernasal para um novo tubo de centrífuga.
3. Medição de proteínas
- Determine a concentração proteica do supernacido utilizando protocolos estabelecidos (ver Tabela de Materiais).
NOTA: É melhor usar amostras a ≥2 μg/μL, mas também podem ser utilizadas amostras menos concentradas. Se for utilizada uma amostra menos concentrada, será necessário ajustar adequadamente os volumes dos reagentes de redução/alquilação na etapa 5.1. - Prepare uma diluição de curva padrão BSA usando tampão de lise CHAPS.
- Siga as instruções do fabricante e, após 15 min, leia a absorvância a 750 nm.
4. Redução/alquilação do tratamento de reagente
- Transfira 200 μg de proteína por condição para um novo tubo de centrífuga e ajuste para um volume final de 100 μL usando tampão de lise CHAPS. É possível escalar até 200 μL quando a concentração de proteína é muito baixa, mas não se esqueça de ajustar adequadamente o volume de reagente redutor/alquilante.
- Adicione 5 μL do TCEP de 200 mM e incubar amostras a 55 °C por 1h.
- Imediatamente antes do uso, prepare 375 mM iodoacetamida dissolvendo um tubo de iodoacetamida (i.e., 9 mgs) em 132 μL de 100 mM TEAB. Proteja esta solução da luz.
- Adicione 5 μL de iodoacetamida de 375 mM à amostra e incubar por 30 minutos à temperatura ambiente (RT) protegido da luz.
5. Precipitação metanol/clorofórmio20
- Adicione 400 μL de metanol a cada 100 μL de proteínas e brevemente amostras de vórtice.
- Centrifugar a 9.000 x g para 10 s na RT. Isto é para incorporar líquidos depositados nas laterais do tubo de amostra.
- Adicione 100 μL de clorofórmio à mistura e brevemente vórtice. Use 200 μL de clorofórmio se a amostra tiver uma alta concentração de fosfolipídios.
- Centrifugar a 9.000 x g para 10 s na RT. Isto é para incorporar líquidos depositados nas laterais do tubo de amostra.
- Adicione 300 μL de água e vórtice vigorosamente. É importante obter uma solução homogênea.
- Centrifugar a 9.000 x g por 1 min na RT. Tenha muito cuidado para evitar perturbar as camadas ao transferir o tubo para um rack.
NOTA: O tubo deve conter agora três fases: 1) Camada superior (ou seja, supernante), uma mistura de água e metanol; 2) Camada média (ou seja, interfase), proteína branca precipitada; e 3) Camada inferior (ou seja, fase inferior), clorofórmio. - Remova cuidadosamente o supernatante.
- Adicione 300 μL de metanol à fase interfase e inferior restante. Vórtice vigorosamente.
- Centrifugar a 9.000 x g por 2 min no RT. Tenha muito cuidado para evitar perturbar as camadas ao transferir o tubo para um rack.
- Remova cuidadosamente o supernatante.
- Aspire suavemente o máximo de líquido possível sob um fluxo de ar (por exemplo, usando um concentrador de vácuo) no RT até que a pelota esteja apenas um pouco úmida (~10 min). Como o tempo necessário pode ser diferente para cada amostra, verifique a cada 2 minutos para avaliar. Armazene a pelota a -80 °C até que seja mais processamento.
6. Digestão de proteínas
- Resuspend a pelota de proteína precipitada em 100 μL de tampão de lise TEAB.
NOTA: É opcional medir a concentração de proteína nesta etapa. - Imediatamente antes do uso, prepare 1 μg/μL trypsin adicionando 100 μL da solução de armazenamento de trippsina (ácido acético de 50 mM) ao fundo do frasco de vidro trypsin de 100 μg e incubar por 5 minutos na RT. Armazene o reagente restante em doses de uso único a -80 °C.
- Adicione 2,5 μL de trippsina por 100 μg de proteína. Digerir a amostra durante a noite a 37 °C. Esta etapa é crucial para a solubilização completa da proteína; não modifique essas condições. Após a digestão, é opcional medir a concentração de proteínas usando ensaios proteicos padrão.
7. Rotulagem de peptídeos
- Imediatamente antes do uso, equilibre os reagentes do kit de etiqueta TMT para RT.
- Dissolva cada um dos frascos de etiqueta TMT de 0,8 mg através da adição de 41 μL de acetonitrilo anidro a cada tubo. Incubar o reagente por 5 min no RT com vórtice ocasional. Centrifugar brevemente os tubos.
NOTA: Uma concentração de 0,8 mg de tag TMT geralmente é suficiente para rotular dois conjuntos. Outros pesquisadores, no entanto, demonstraram que essa concentração pode ser reduzida ainda mais e ainda produzir dados confiáveis15. - Adicione cuidadosamente 41 μL do reagente de etiqueta TMT a cada amostra de 100 μL.
- Incubar a reação por 1 h no RT.
- Adicione 8 μL de 5% de hidroxilamina à amostra e incubar por 15 minutos para saciar a reação.
- Divida as amostras em quantidades iguais em um novo tubo de centrífuga e armazene a -80 °C.
NOTA: Nesta etapa as amostras são estáveis e podem ser submetidas para espectroscopia em massa. É opcional medir a concentração neste momento usando ensaios proteicos padrão.
8. Espectroscopia em massa
- Submeter as amostras a uma instalação de proteômica (este estudo utilizou a instalação uf ICBR Proteomics Core), onde todas as amostras são combinadas e purificadas usando colunas de spin C18.
NOTA: Discuta com a instalação central como enviar as amostras antes de prepará-las para confirmar os passos exatos que preferem para submissões. - Solicite os seguintes procedimentos por amostra de multiplex combinada: extração de fase sólida, HPLC (SCX, SE), zip tip e LC-MS/MS (gradiente de 2 h para ID de proteína, se >10QE Plus).
- Uma vez que os dados são coletados, a instalação principal processará arquivos RAW usando software fornecido pelo fornecedor para identificação de proteínas.
9. Análise de dados
- Os dados são normalmente fornecidos do núcleo de volta para o usuário no formato 7z, que pode exigir aproximadamente 16 GB de espaço em disco por cada conjunto de dados (neste caso 11 amostras). Para o processamento de dados, certifique-se de que um computador esteja disponível com pelo menos 3,4 GHz.
- Extrair arquivos usando o Gerenciador de Arquivos 7-Zip. Esses arquivos extraídos contêm dados RAW, arquivo de formato pdStudy e arquivo de formato pdResultVer. Guarde todos os arquivos para mais análises.
- Abra o arquivo usando o Software Proteome Discovery 2.2.
NOTA: O formato do arquivo é "Nome do arquivo.pdStudy". Se o "Nome do arquivo.pdResultView" for aberto, não será possível selecionar a amostra de controle. - Selecione amostras de controle no painel "Samples".
- Abra o Resultado selecionando iD no painel "Resultados de Análise".
- Exportar para o software de planilha.
- Salvar dados brutos (todas as proteínas identificadas).
- Abra o arquivo de software de planilha. Isso conterá todas as proteínas que foram identificadas.
- No arquivo de software de planilha use a função "Filter" para tela "Protein FDR Confidence: Combined" em alta (coluna B), " #UniquePeptídeos" superior a 2 (coluna K), e qualquer um de " AbundanceRatio" em branco exclusivamente(coluna S até W).
- Insira uma coluna para o cálculo" p-value" com a função
=TTEST (grupo de controle, grupo experimental, caudas, tipo) - Insira uma coluna para "Significância Estatística" com a função
=IF(p-value<0,05, "Significância", "NS") - Use a função "Filter" para exibir "Statistical Meaning" mostrando "Significado". O resultado mostra as proteínas analisadas com significância estatística no grupo controle e no grupo experimental.
- Determine significativamente maior ou menor abundância de expressão proteica no grupo experimental em comparação com o grupo controle, insira uma coluna para "Regulação" com a função
=IF (AVERAGE(controlgroup)>AVERAGE (experimentalgroup),"Upregulated","Downregulated")
10. Métodos para avaliar hits significativos
- Para identificar as interações proteína-proteína entre os impactos significativos identificados nos estudos de TMT, use a Ferramenta de Pesquisa para a Recuperação de Genes/Proteínas Interativas (STRING) versão 11.021: https://string-db.org/
- Para classificar por grupos (ou seja, função molecular, processos biológicos e classes proteicas) use análise de proteínas através do software de classificação de ontologia de Relações Evolutivas (PANTHER)22: http://www.pantherdb.org/
- Para identificar interações proteicas em uma variedade de caminhos, use o software de análise de vias23.
11. Upload de dados proteômicos em um banco de repositório
- Para enviar dados proteômicos ao Banco de Dados de Proteómicos (PRIDE) ou ao Mass Spectrometry Interactive Virtual Environment (MassIVE) incluem as seguintes informações: os arquivos de lista de pico (arquivos de espectro de massa processados em um formato padrão, como mzXML, mzML, ou MGF), arquivos de resultados (identificações de espectro em um formato padrão, como mzIdentML ou mzTab), e arquivos de espectro bruto (arquivos de espectro de massa bruta em um formato não padrão ou específico de instrumentos, como . Arquivos RAW ou . Arquivos WIFF).
- Para enviar, crie uma conta e inclua informações como afiliação e detalhes do projeto. Em seguida, selecione os arquivos listados na etapa 11.1 e faça upload deles.
- Para criar um conjunto de dados oficial, execute um fluxo de trabalho de envio nesses arquivos carregados.
NOTA: Após a apresentação, o conjunto de dados será privado no banco de repositórios. Com a opção privada, os dados só estão disponíveis para usuários autorizados. Existem duas opções adicionais: 1) Conjunto de dados compartilhado, que dá acesso a revisores de revistas e colaboradores; ou 2) Conjunto de dados público, que aparecerá em pesquisas públicas de conjunto de dados. Outra característica importante desses repositórios é a capacidade de atualizar os dados carregados e associar publicações subsequentes com o conjunto de dados existente.
Resultados
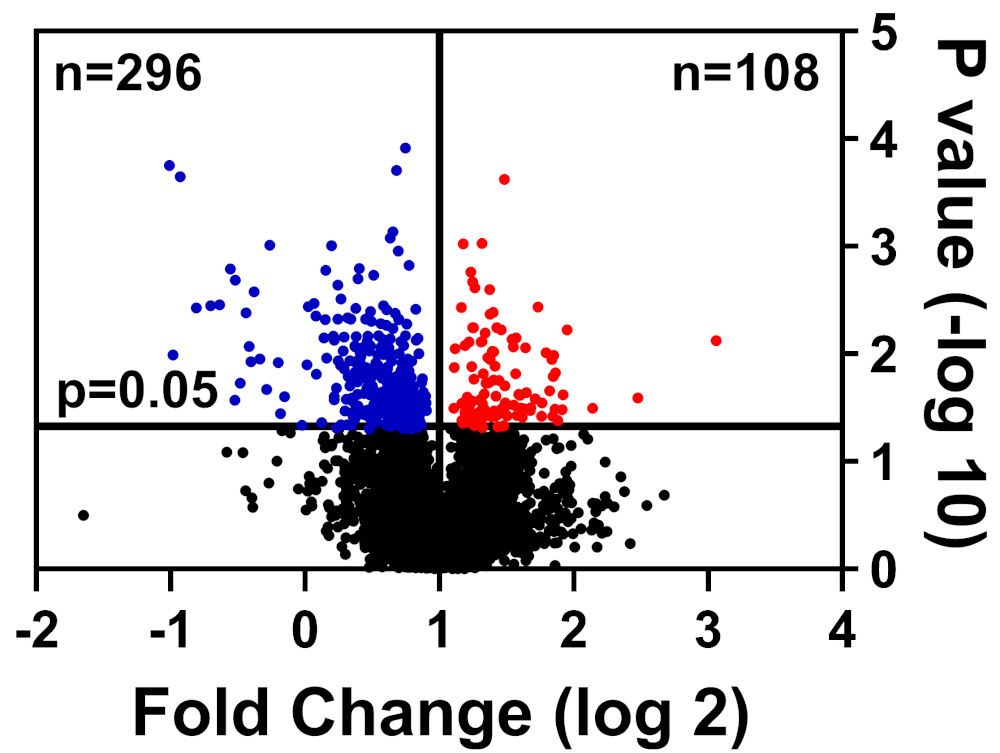
As células saudáveis e doentes foram límpidas no tampão CHAPS, preparadas como detalhadas em nosso método de rotulagem TMT, e submetidas ao Centro Interdisciplinar de Pesquisa em Biotecnologia da Universidade da Flórida (UF-ICBR) Núcleo de Proteômica para Cromatografia Líquida com espectrometria de massa tandem. Após a aquisição e entrega de dados do núcleo, o conjunto de dados foi aberto em software fornecido pelo fornecedor e foram aplicados os seguintes filtros de corte: ≥2 peptídeos únicos, íons repórteres para cada amostra de proteína presente em todos os canais, e incluem apenas proteínas significativamente alteradas (p ≤ 0,05). A Tabela 1 resume os dados: 39.653 peptídeos totais, dos quais 7.211 têm igual ou superior a dois peptídeos únicos, e 3.829 incluem íons repórteres para todos os canais. Os valores p para estes 3.829 peptídeos foram calculados pelo teste t do Aluno e p ≤ 0,05 foi considerado significativo. Além disso, foi utilizado um corte de dobra para determinar a distribuição relativa de proteínas de doentes em comparação com células saudáveis: downregulated (azul) ou upregulated (vermelho)(Figura 1).
A lista de expressão proteica significativamente desregulada foi avaliada utilizando-se o sistema de classificação ontologia PANTHER e as análises string. As análises das panteras mostraram uma lista categorizada de proteínas baseadas em significativamente menor(Figura 2A) ou maior abundância em células doentes baseadas na função molecular(Figura 2B). As análises de cordas de proteínas de abundância significativamente menor(Figura 2C) e maior(Figura 2D) identificaram múltiplas interações e fortes associações entre proteínas.

Figura 1: Enredo vulcânico exibindo proteínas cuja abundância não foi significativamente alterada (preta), significativamente reduzida (azul) ou significativamente aumentada (vermelho) em células de controle doentes versus saudáveis. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Avaliações representativas de hits significativamente desregulados identificados por PANTHER (A, B) e String (C, D) de proteínas de abundância significativamente menores ou maiores. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Peptídeos totais | Total identificado | ≥ 2 peptídeos únicos | Proteínas quantificadas | Proteínas significativamente alteradas | |
| Baixo | Alta | ||||
| 39653 | 7211 | 4457 | 3829 | 296 | 108 |
Tabela 1: Tabela representativa de proteínas quantificadas por análise de conjunto de dados.
Discussão
Para preparar com sucesso amostras para análise proteômica usando metodologias de rotulagem isótopos isobáricas baseadas em TMT, é crucial realizar extrações proteicas com muito cuidado a 4 °C e usar um tampão de lise que contenha um coquetel inibidor de protease24,25. O coquetel inibidor de protease é um reagente crucial para evitar a degradação inesperada da proteína durante a digestão proteica. Uma diferença fundamental entre nosso protocolo e o atual fornecido pelo fornecedor é que recomendamos fortemente o uso de tampão de lise CHAPS com base em nossa experiência com células e tecidos mamíferos. Também sugerimos o uso de uma abordagem de precipitação de proteína de metanol/clorofórmio para pelotas e tecidos celulares.
Idealmente, extração de proteínas, medição, tratamentos de reagente de redução/alquilação, e precipitações de metanol/clorofórmio são todas realizadas no mesmo dia. Seguindo esta recomendação resultará em concentrações proteicas mais precisas para rotulagem subsequente. O passo de precipitação proteica é importante para a remoção de reagentes que interferirão na espectrometria de massa tandem. A inclusão da etapa de precipitação aumenta significativamente a resolução do TMT26. Em suma, as principais vantagens do nosso protocolo TMT são a alta eficiência de rotulagem para diferentes tipos de amostras, sua reprodutibilidade e os dados confiáveis adquiridos.
À medida que a natureza multiplex desta estratégia de proteômica não alvo TMT continua a se expandir, ela aumentará progressivamente a capacidade dos pesquisadores em uma ampla variedade de campos para fazer novas descobertas. Especificamente no campo biomédico, nós e outros temos encontrado essa tecnologia cada vez mais informativa em estudos explorando novos mecanismos de ação em doenças e impactos relativos de diversas terapêuticas. Por todas essas razões, essa poderosa tecnologia complementa o repertório de outras abordagens de OMICS utilizadas em estudos de pesquisa modernos e fornece informações fundamentais que podem orientar o desenvolvimento terapêutico.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Gostaríamos de reconhecer a instalação de proteômica uf-ICBR para o processamento de nossas amostras. Este trabalho foi apoiado em parte pelos Institutos Nacionais de Saúde R01 HL136759-01A1 (CAP).
Materiais
| Name | Company | Catalog Number | Comments |
| 1 M Triethylammonium bicarbonate (TEAB), 50 mL | Thermo Fisher | 90114 | Reagent for protein labeling |
| 50% Hydroxylamine, 5 mL | Thermo Fisher | 90115 | Reagent for protein labeling |
| Acetic acid | Sigma | A6283 | Reagent for protein digestion |
| Anhydrous acetonitrile, LC-MS Grade | Thermo Fisher | 51101 | Reagent for protein labeling |
| Benzonaze nuclease | Sigma-Aldrich | E1014 | DNA shearing |
| Bond-Breaker TCEP solution, 5 mL | Thermo Fisher | 77720 | Reagent for protein labeling |
| BSA standard | Thermo | 23209 | Reagent for protein measurement |
| CHAPS | Thermo Fisher | 28300 | Reagent for protein extraction |
| Chloroform | Fisher | BP1145-1 | Reagent for protein precipitation |
| cOmplete, EDTA-free Protease Inhibitor Cocktail Tablet | Roche | 4693132001 | Reagent for protein extraction |
| DC Protein Assay | BioRad | 500-0116 | Reagent for protein measurement |
| Excel | Microsoft Office | Software for data analyses | |
| Heat block | VWR analog | 12621-104 | Equipment for protein digestion incubation |
| HEPES | Sigma | RDD002 | Reagent for protein extraction |
| Methanol | Fisher | A452-4 | Reagent for protein precipitation |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher | 90058 | Reagent for protein digestion |
| Potassium chloride | Sigma | 46436 | Reagent for protein extraction |
| Sigma Plot 14.0 | Sigma Plot 14.0 | Software for data analyses | |
| Sonicator | Fisher Scientific | FB120 | DNA shearing |
| Spectra Max i3x Multi-Mode Detection Platform | Molecular Devices | Plate reader for protein measurement | |
| Thermo Scientific Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher | 23275 | Reagent for protein measurement |
| Thermo Scientific Pierce Quantitative Fluorescent Peptide Assay | Thermo Fisher | 23290 | Reagent for protein measurement |
| Thermo Scientific Proteome Discoverer Software | Thermo Fisher | OPTON-30945 | Software for data analyses |
| TMT 10plex Isobaric Label Reagent Set 0.8 mg, sufficient reagents for one 10plex isobaric experiment | Thermo Fisher | 90110 | Reagent for protein labeling |
| TMT11-131C Label Reagent 5 mg | Thermo Fisher | A34807 | Reagent for protein labeling |
| Water, LC-MS Grade | Thermo Fisher | 51140 | Reagent for protein extraction |
Referências
- Graves, P. R., Haystead, T. A. Molecular biologist's guide to proteomics. Microbiology and Molecular Biology Reviews. 66 (1), 39-63 (2002).
- Erdjument-Bromage, H., Huang, F. K., Neubert, T. A. Sample Preparation for Relative Quantitation of Proteins Using Tandem Mass Tags (TMT) and Mass Spectrometry (MS). Methods in Molecular Biology. 1741, 135-149 (2018).
- O'Farrell, P. H. High resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry. 250 (10), 4007-4021 (1975).
- Rabilloud, T., Lelong, C. Two-dimensional gel electrophoresis in proteomics: a tutorial. Journal of Proteomics. 74 (10), 1829-1841 (2011).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics. 1 (5), 376-386 (2002).
- Anderson, N. G., Anderson, N. L. Twenty years of two-dimensional electrophoresis: Past, present and future. Electrophoresis. 17 (3), 443-453 (1996).
- Haynes, P. A., Yates, J. R. Proteome profiling-pitfalls and progress. Yeast. 17 (2), 81-87 (2000).
- Bunai, K., Yamane, K. Effectiveness and limitation of two-dimensional gel electrophoresis in bacterial membrane protein proteomics and perspectives. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 815 (1-2), 227-236 (2005).
- Sury, M. D., Chen, J. X., Selbach, M. The SILAC fly allows for accurate protein quantification in vivo. Molecular & Cellular Proteomics. 9 (10), 2173-2183 (2010).
- Zhang, G., Neubert, T. A. Use of stable isotope labeling by amino acids in cell culture (SILAC) for phosphotyrosine protein identification and quantitation. Methods in Molecular Biology. 527, 79-92 (2009).
- Wang, X., et al. SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Scientific Reports. 8 (1), 8441 (2018).
- Thompson, A., et al. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry. 75, 1895-1904 (2003).
- Cheng, L., Pisitkun, T., Knepper, M. A., Hoffert, J. D. Peptide Labeling Using Isobaric Tagging Reagents for Quantitative Phosphoproteomics. Methods in Molecular Biology. 1355, 53-70 (2016).
- Navarrete-Perea, J., Yu, Q., Gygi, S. P., Paulo, J. A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research. 17 (6), 2226-2236 (2018).
- Zecha, J., et al. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Molecular & Cellular Proteomics. 18 (7), 1468-1478 (2019).
- Bachor, R., Waliczek, M., Stefanowicz, P., Szewczuk, Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules. 24 (4), E701 (2019).
- Suzuki-Hatano, S., et al. AAV9-TAZ Gene Replacement Ameliorates Cardiac TMT Proteomic Profiles in a Mouse Model of Barth Syndrome. Molecular Therapy - Methods & Clinical Development. 13, 167-179 (2019).
- Kirshenbaum, N., Michaelevski, I., Sharon, M. Analyzing large protein complexes by structural mass spectrometry. Journal of Visualized Experiments. (40), e1954 (2010).
- Perumal, N., et al. Sample Preparation for Mass-spectrometry-based Proteomics Analysis of Ocular Microvessels. Journal of Visualized Experiments. (144), e59140 (2019).
- Wessel, D., Flügge, U. I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical Biochemistry. 138, 141-143 (1984).
- Jensen, L. J., et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, D412-D416 (2009).
- Mi, H., Muruganujan, A., Casagrande, J. T., Thomas, P. D. Large-scale gene function analysis with the PANTHER classification system. Nature Protocols. 8 (8), 1551-1566 (2013).
- Cirillo, E., Parnell, L. D., Evelo, C. T. A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Frontiers in Genetics. 8, 174 (2017).
- Plaxton, W. C. Avoiding Proteolysis during the Extraction and Purification of Active Plant Enzymes. Plant and Cell Physiology. 60 (4), 715-724 (2019).
- Ryan, B. J., Henehan, G. T., Walls, D., Loughran, S. T. . Protein Chromatography: Methods and Protocols. , 53-69 (2017).
- Fic, E., Kedracka-Krok, S., Jankowska, U., Pirog, A., Dziedzicka-Wasylewska, M. Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis. 31 (21), 3573-3579 (2010).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados