
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Dna-Tethered RNA Polymerase para Transcrição In vitro Programável e Computação Molecular
Neste Artigo
Resumo
Descrevemos a engenharia de uma nova polimerase T7 RNA com DNA para regular reações de transcrição in vitro. Discutimos os passos para a síntese e caracterização proteica, validamos a regulação transcricional de prova de conceito e discutimos suas aplicações em computação molecular, diagnóstico e processamento de informações moleculares.
Resumo
A nanotecnologia de DNA permite a automontagem programável de ácidos nucleicos em formas e dinâmicas prescritas pelo usuário para diversas aplicações. Este trabalho demonstra que conceitos da nanotecnologia de DNA podem ser usados para programar a atividade enzimática da polimerase T7 RNA (RNAP) derivada de phage e construir redes de regulação genética sintética escalável. Primeiro, um T7 RNAP com oligonucleotídeo é projetado através da expressão de um RNAP com marca SNAP n-terminal e posterior acoplamento químico da tag SNAP com um oligonucleotídeo modificado por benziloguanina (BG). Em seguida, o deslocamento da cadeia nucleico-ácido é usado para programar transcrição de polimerase sob demanda. Além disso, conjuntos auxiliares de ácido nucleico podem ser usados como "fatores de transcrição artificial" para regular as interações entre o T7 RNAP programado pelo DNA com seus modelos de DNA. Este mecanismo de regulação de transcrição in vitro pode implementar uma variedade de comportamentos de circuito, como lógica digital, feedback, cascata e multiplexing. A composabilidade dessa arquitetura normativa genética facilita a abstração, padronização e escala do design. Esses recursos permitirão a prototipagem rápida de dispositivos genéticos in vitro para aplicações como biosensação, detecção de doenças e armazenamento de dados.
Introdução
A computação em DNA usa um conjunto de oligonucleotídeos projetados como meio de computação. Esses oligonucleotídeos são programados com sequências para serem montados dinamicamente de acordo com a lógica especificada pelo usuário e responder a entradas específicas de ácido nucleico. Em estudos de prova de conceito, a saída da computação normalmente consiste em um conjunto de oligonucleotídeos fluorescentes rotulados que podem ser detectados através de leitores de eletroforese de gel ou fluorescência. Ao longo dos últimos 30 anos, foram demonstrados circuitos computacionais de DNA cada vez mais complexos, como várias cascatas lógicas digitais, redes de reação química e redes neurais1,2,3. Para auxiliar na preparação desses circuitos de DNA, modelos matemáticos têm sido usados para prever a funcionalidade dos circuitos genéticos sintéticos4,5, e ferramentas computacionais foram desenvolvidas para o projeto de sequência de DNA ortogonal6,7,8,9,10 . Em comparação com computadores baseados em silício, as vantagens dos computadores de DNA incluem sua capacidade de interagir diretamente com biomoléculas, operar em solução na ausência de uma fonte de alimentação, bem como sua compactação e estabilidade globais. Com o advento do sequenciamento da próxima geração, o custo de sintetizar computadores de DNA vem diminuindo nas últimas duas décadas a uma taxa mais rápida do que a Lei11de Moore. As aplicações desses computadores baseados em DNA estão começando a surgir, como para o diagnóstico de doenças12,13, para alimentar a biofísica molecular14, e como plataformas de armazenamento de dados15.

Figura 1: Mecanismo de deslocamento da cadeia de DNA mediado por toehold. O toehold, δ, é uma sequência livre e desvinculada em um duplex parcial. Quando um domínio complementar (δ*) é introduzido em uma segunda vertente, o domínio δ livre serve como um toehold para hibridização, permitindo que o resto da cadeia (ônica*) desloque lentamente seu concorrente através de uma reação reversível zipping/unzipping conhecida como migração de fios. À medida que o comprimento do δ aumenta, o ΔG para a reação para a frente diminui, e o deslocamento acontece mais facilmente. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Até o momento, a maioria dos computadores de DNA utilizam um motivo bem estabelecido no campo da nanotecnologia dinâmica de DNA conhecida como deslocamento de fios de DNA mediados por toehold (TMDSD, Figura 1)16. Este motivo consiste em um duplex de DNA parcialmente duplo (dsDNA) exibindo saliências curtas de "toehold" (ou seja, 7 a 10 nucleotídeos (nt)). Os fios de "entrada" de ácido nucleico podem interagir com os duplexes parciais através do dedo do dedo do sol. Isso leva ao deslocamento de um dos fios do duplex parcial, e este fio liberado pode então servir como entrada para duplexes parciais a jusante. Assim, o TMDSD permite a cascata de sinais e o processamento de informações. Em princípio, os motivos ortogonais de TMDSD podem operar de forma independente na solução, permitindo o processamento paralelo de informações. Houve uma série de variações na reação TMDSD, como a troca de fios de DNA mediada por toehold (TMDSE)17, toeholds "sem vazamento" com domínios de dois comprimentos18, dedos incompatíveis de sequência19e deslocamento de fios mediados por "handhold"20. Esses princípios inovadores de design permitem energias tmdsd mais finamente ajustadas e dinâmicas para melhorar o desempenho da computação de DNA.
Circuitos genéticos sintéticos, como circuitos genéticos transcricionais, também são capazes de calcular21,22,23. Esses circuitos são regulados por fatores de transcrição proteica, que ativam ou reprimem transcrição de um gene ligando-se a elementos específicos de DNA regulatório. Comparados com circuitos baseados em DNA, circuitos transcricionais têm várias vantagens. Em primeiro lugar, a transcrição enzimática tem uma taxa de rotatividade muito maior do que os circuitos de DNA catalíticos existentes, gerando assim mais cópias de saída por cópia única de entrada e fornecendo um meio mais eficiente de amplificação de sinal. Além disso, circuitos transcricionais podem produzir diferentes moléculas funcionais, como aptamers ou codificação de RNA (mRNA) mensageiro para proteínas terapêuticas, como saídas de computação, que podem ser exploradas para diferentes aplicações. No entanto, uma grande limitação dos circuitos transcricionais atuais é a falta de escalabilidade. Isso ocorre porque há um conjunto muito limitado de fatores de transcrição ortogonais baseados em proteínas, e o novo desenho de novos fatores de transcrição proteica permanece tecnicamente desafiador e demorado.

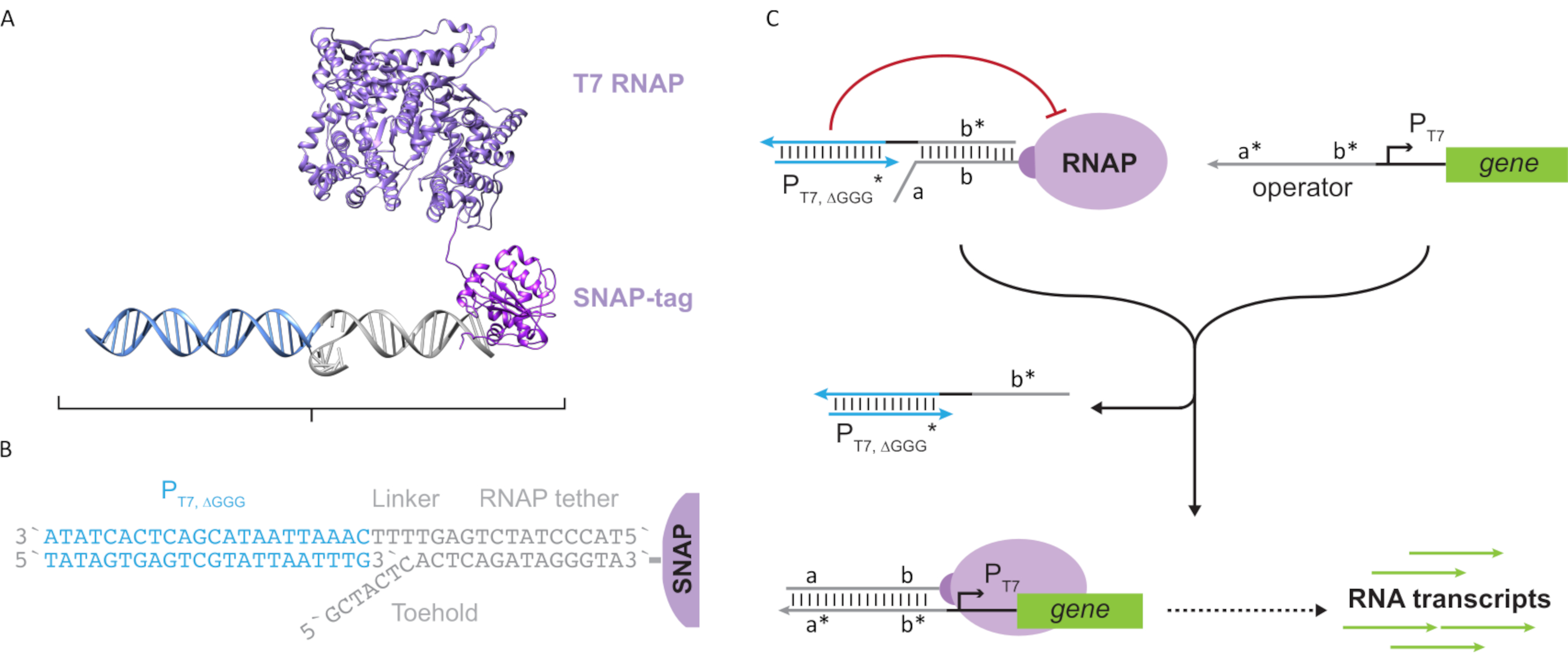
Figura 2: Abstração e mecanismo do complexo de polimerase "tether" e "cage" ( A e B) Uma corda oligonucleotídeo é enzimáticamente rotulada para uma polimerase T7 através da reação snap-tag. Uma gaiola composta por um promotor T7 "falso" com uma saliência complementar de tether permite hibridizar a atividade transcricional de corda e bloco. (C) Quando o operador(a*b*) está presente, ele se liga ao toehold sobre a corda oligonucleotídeo(ab)e desloca a região b* da gaiola, permitindo que a transcrição ocorra. Este número foi modificado de Chou e Shih27. Abreviaturas: RNAP = RNA polymerase. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Este artigo introduz um novo bloco de construção para computação molecular que combina as funcionalidades dos circuitos transcricionais com a escalabilidade dos circuitos baseados em DNA. Este bloco de construção é um T7 RNAP covalentemente anexado com uma corda de DNA de um único fio(Figura 2A). Para sintetizar este T7 RNAP ligado ao DNA, a polimerase foi fundida a um SNAP-tag24 n-terminal e recombinantemente expressa em Escherichia coli. A tag SNAP foi então reagida com um oligonucleotídeo funcionalizado com o substrato BG. A corda oligonucleotídeo permite o posicionamento de hóspedes moleculares nas proximidades da polimerase através da hibridização do DNA. Um desses convidados foi um bloqueador transcricional competitivo referido como uma "gaiola", que consiste em um duplex de DNA "falso" do promotor T7 sem gene a jusante(Figura 2B). Quando vinculada ao RNAP através de sua corda oligonucleotídeo, a gaiola paralisa a atividade de polimerase, superando outros modelos de DNA para a vinculação RNAP, tornando o RNAP em um estado "OFF"(Figura 2C).
Para ativar a polimerase a um estado "ON", foram projetados modelos de DNA T7 com domínios "operadores" de uma única linha a montante do promotor T7 do gene. O domínio do operador (ou seja, domínio a*b* Figura 2C) pode ser projetado para deslocar a gaiola do RNAP via TMDSD e posicionar o proximal RNAP ao promotor T7 do gene, iniciando assim a transcrição. Alternativamente, foram projetados modelos de DNA onde a sequência do operador era complementar aos fios auxiliares de ácido nucleico que são chamados de "fatores de transcrição artificial" (ou seja, fios TFA e TFB na Figura 3A). Quando ambas as vertentes forem introduzidas na reação, elas se reunirão no site do operador, criando um novo domínio pseudo-contíguo a*b*. Este domínio pode então deslocar a gaiola via TMDSD para iniciar a transcrição(Figura 3B). Estes fios podem ser fornecidos exogenously ou produzidos.

Figura 3: Programação seletiva da atividade de polimerase através de um ativador de interruptor de três componentes. (A) Quando os fatores de transcrição (TFA e TFB) estão presentes, eles se ligam ao domínio do operador a montante do promotor, formando uma sequência pseudo de uma única-strand(a*b*) capaz de deslocar a gaiola através do deslocamento mediado do DNA. (B) Este domínio a*b* pode deslocar a gaiola via TMDSD para iniciar a transcrição. Este número foi modificado de Chou e Shih27. Abreviaturas: TF = fator de transcrição; RNAP = polimerase de RNA; TMDSD = deslocamento de fios de DNA mediados por toehold. Clique aqui para ver uma versão maior desta figura.
{kind=link}
O uso de fatores de transcrição nucleicos baseados em ácido para regulação transcricional in vitro permite a implementação escalável de comportamentos sofisticados de circuito, como lógica digital, feedback e cascata de sinais. Por exemplo, pode-se construir cascatas lógicas do portão projetando sequências de ácido nucleico de tal forma que as transcrições de um gene a montante ativam um gene a jusante. Uma aplicação que explora a cascata e multiplexing capazes por essa tecnologia proposta é o desenvolvimento de circuitos de computação molecular mais sofisticados para diagnósticos portáteis e processamento de dados moleculares. Além disso, integrar os recursos de computação molecular e síntese de novo RNA pode permitir novas aplicações. Por exemplo, um circuito molecular pode ser projetado para detectar uma ou uma combinação de RNAs definidas pelo usuário como insumos e saída de RNAs terapêuticos ou mRNAs codificando peptídeos funcionais ou proteínas para aplicações médicas de ponto de cuidado.
Protocolo
1. Preparação do buffer
NOTA: A preparação do tampão de purificação de proteínas pode ocorrer em qualquer dia; aqui, foi feito antes de começar os experimentos.
- Prepare o tampão de lise/equilíbrio contendo 50 mM tris (hidroximetil)aminometano (Tris), cloreto de sódio de 300 mM (NaCl), 5% de glicerol e 5 mM β-mercaptoethanol (BME), pH 8. Adicione 1,5 mL de 1M Tris, 1,8 mL de 5M NaCl, 1,5 mL de glicerol, 25,2 mL de água desionizada (ddH2O) em um tubo centrífuga de 50 mL e adicione 10,5 μL de 14,2 M BME pouco antes de usar.
NOTA: Tris pode causar toxicidade aguda; portanto, evite respirar seu pó, e evite a pele e o contato visual. A BME é tóxica e só deve ser usada em um capô de fumaça. É importante adicionar BME por último, pouco antes da ressuspensão e lise celular. Consulte a Tabela 1 para obter a fórmula do tampão de lise. - Prepare o tampão de lavagem (pH 8) contendo 50 mM Tris, 800 mM NaCl, 5% glicerol, 5 mM BME e 20 mM imidazol. Adicione 1,5 mL de 1 M Tris, 4,8 mL de 5 M NaCl, 1,5 mL de glicerol e 22,2 mL de ddH2O em um tubo centrífuga de 50 mL. Pouco antes de usar, adicione 7 μL de 14,2 M BME e 200 μL de 2 M imidazol a 20 mL da solução acima.
NOTA: Para prevenir a toxicidade aguda devido ao imidazol, utilize equipamentos de proteção individual. É importante adicionar BME e imidazol por último, pouco antes de lavar a proteína para fora da coluna. Consulte a tabela 2 para obter a fórmula do buffer de lavagem. - Prepare o tampão de elução (pH8) contendo 50 mM Tris, 800 mM NaCl, 5% glicerol, 5 mM BME e 200 mM imidazol. Adicione 0,5 mL de 1 M Tris, 1,6 mL de 5 M NaCl, 0,5 mL de glicerol e 6,4 mL de ddH2O a um tubo centrífuga de 15 mL. Pouco antes de usar, adicione 3,5 μL de 14,2 M BME e 1 mL de imidazol de 2 M a 10 mL da solução acima.
NOTA: É importante adicionar BME e imidazol por último, pouco antes de eluia da proteína para fora da coluna. Consulte a tabela 3 para obter a fórmula do buffer de eluição. - Prepare o tampão de armazenamento 2x (a ser misturado 1:1 com glicerol) contendo 100 mM Tris, 200 mM NaCl, 40 mM BME e 2 mM ácido ethylenodiaminatotraacético (EDTA), 0,2% de um surfactante não iônico (ver a Tabela de Materiais). Prepare 50 mL do tampão de armazenamento adicionando 5 mL de 1 M Tris, 2 mL de 5 M NaCl, 42,56 mL de ddH2O, 200 μL de 0,5 M EDTA, 100 μL do surfactante não iônico para um tubo de centrífusuga de 50 mL. Misture até que a solução seja homogênea, filtre o tampão de armazenamento através de um filtro de seringa de 0,2 μm e adicione 140,8 μL de BME à solução acima antes de ser usada.
NOTA: Para evitar a toxicidade aguda devido ao EDTA, evite respirar o pó e evite o contato com a pele e o contato visual. É importante adicionar BME por último e misturar todo o buffer de armazenamento 1:1 com glicerol, pouco antes de armazenar a proteína purificada. Consulte a tabela 4 para obter a fórmula do buffer de armazenamento.
2. Crescimento da cultura durante a noite: Dia 1
- Prepare 1.000x de estoque de kanamicina dissolvendo 500 mg de kanamicina em 10 mL de ddH2O.
NOTA: Use equipamentos de proteção individual para prevenir a toxicidade aguda devido à kanamicina. - Adicione 20 μL do estoque de kanamycina de 1.000x a 20 mL de caldo de lysogenia. Usando uma ponta de pipeta estéril, cutuque um estoque bl21 e. coli gliceol transformado e, em seguida, inocular a cultura introduzindo a ponta no caldo de mídia de crescimento.

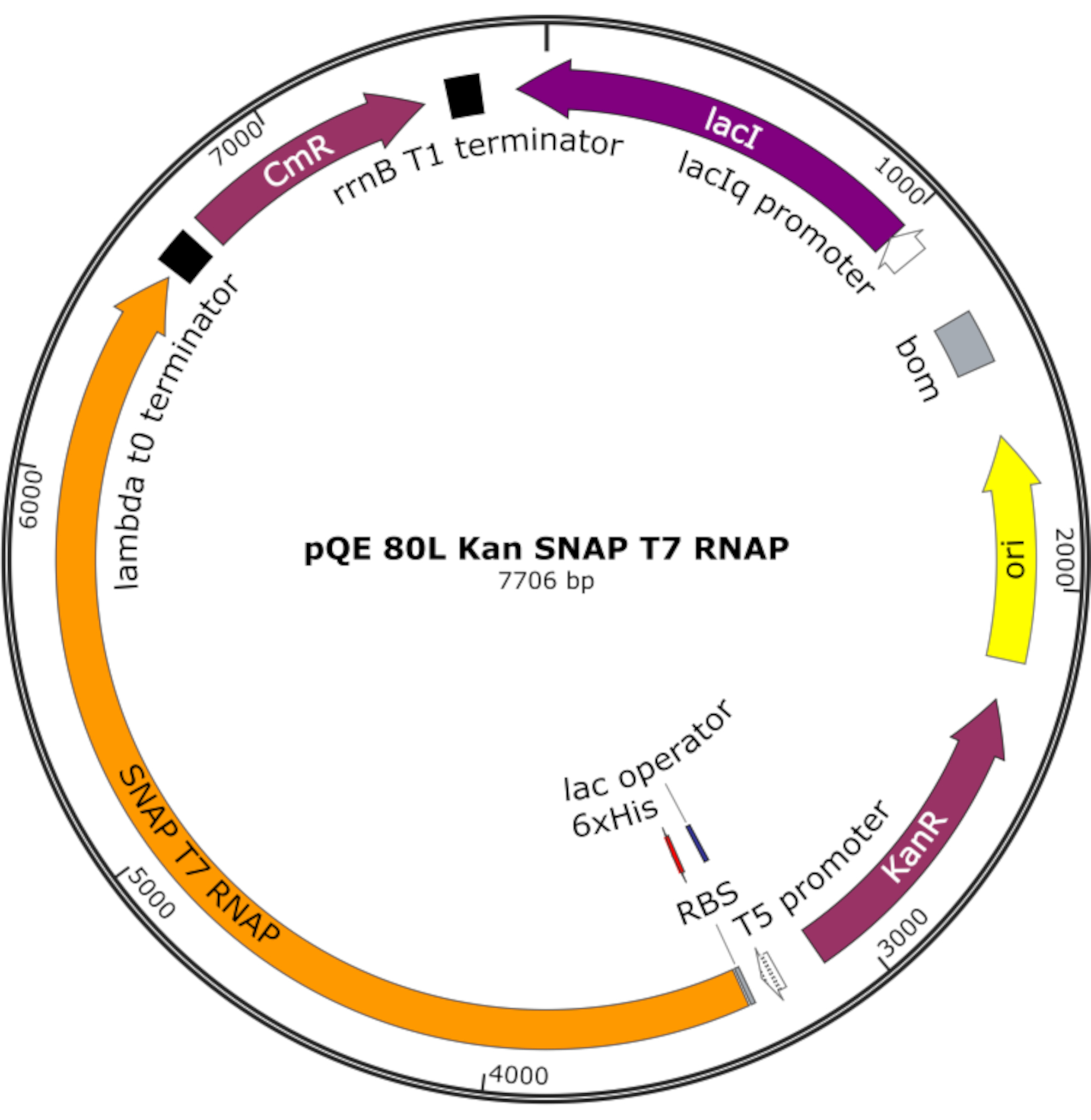
Figura 4: Mapa plasmídeo para SNAP T7 RNAP. O plasmid codifica um T7 RNAP contendo uma tag histidina N-terminal (6x His) e domínio snap-tag (SNAP T7 RNAP) sob um repressor de lac (lacI) em uma espinha dorsal pQE-80L. Outras características incluem a resistência à kanamicina (KanR) e os genes de resistência ao clorofenícol (RMC). Abreviação: RNAP = RNA polymerase. Clique aqui para ver uma versão maior desta figura.
{kind=link}
NOTA: O plasmídeo codifica um T7 RNAP contendo uma tag histidina N-terminal e um domínio SNAP-tag (SNAP T7 RNAP), bem como um gene de resistência à kanamicina sob uma espinha dorsal pQE-80L(Figura 4)25.
- Novamente, adicione 20 μL do estoque de kanamicina de 1.000x a um frasco de cultura separado contendo 20 mL de caldo de lysogenia, e incuba-o como controle.
- Incubar as duas amostras (das etapas 2.2 e 2.3) durante a noite por 12-18 h a 37 °C, enquanto gira a 10 × g.
3. Crescimento celular e indução: Dia 2
- Inocular 400 mL de caldo de liseógeno contendo 400 μL de estoque de kanamicina com 4 mL da cultura de crescimento da noite para o dia a partir do passo 2.4. Incubar os frascos de cultura a 37 °C, enquanto gira a 10 × g.
- Uma vez que a cultura tenha atingido uma densidade óptica (OD) a 600 nm de ~0,5, retire 1 mL de amostra do frasco de crescimento como controle. Armazene a amostra de controle a 4 °C.
- Induzir as células com isopropílico β-D-1-thiogalactopyranoside (IPTG) adicionando 40 μL de 1M IPTG por 100 mL de cultura para alcançar uma concentração final de 0,4 mM IPTG. Incubar a amostra por 3h a 37 °C, girando a 10 × g, e, em seguida, gire a cultura induzida a 8.000 × g por 10 minutos para pelotar as células. Retire o sobrenatante e armazene a pelota a -20 °C até que use mais.
NOTA: Para evitar a toxicidade aguda devido ao IPTG, evite respirar sua poeira e evite o contato com a pele e o contato visual. Se necessário, você pode pausar o experimento aqui e continuar no dia seguinte.
4. Lise celular, purificação de proteínas: Dia 3
- Resuspenque a pelota de célula armazenada com 10 mL de tampão de lise no gelo, e gire suavemente para garantir que toda a pelota seja resuspendida. Em seguida, pipeta 1 mL de amostra em dez tubos de 1,5 mL que são mantidos no gelo.
- Sonicate cada amostra em um ajuste de amplitude de "1", pulsado por 2 s com um ciclo de responsabilidade de 50% durante um período de 30 s. Antes e depois de cada amostra, limpe a ponta de sônica com 70% de etanol e ddH2O. Mantenha todas as amostras no gelo durante e após a sônicação.
NOTA: Mantenha 70% de etanol longe do calor e da chama aberta. - Equilibre uma coluna de rotação de purificação de ácido nitrilotriactico com níquel (Ni-NTA) a uma temperatura de trabalho de 4 °C. Coloque/armazene a coluna a 4 °C e mantenha no gelo durante o uso.
- Centrifugar as dez amostras de 1 mL a 15.000 × g por 20 min a 4 °C. Pipeta cuidadosamente o supernasce contendo o RNAP recombinante sem perturbar a pelota. Se necessário, use um buffer de equilíbrio adicional para ajustar o volume total a ≥ 6 mL.
- Remova suavemente a guia inferior da coluna de giro Ni-NTA para permitir o fluxo através da coluna. Coloque a coluna em um tubo de centrífuga e mantenha-a no gelo.
NOTA: Use um tubo de centrífugas de 50 mL com as colunas de giro Ni-NTA de 3 mL. - Centrifugar a coluna a 700 × g e 4 °C por 2 min para remover o buffer de armazenamento. Equilibre a coluna adicionando 6 mL de tampão de equilíbrio à coluna. Deixe o tampão entrar totalmente na cama de resina.
- Remova o tampão de equilíbrio da coluna por centrifugação a 700 × g e 4 °C por 2 min. Antes de adicionar o extrato celular preparado à coluna, coloque um plugue inferior na coluna para evitar perder qualquer produto. Em seguida, adicione o extrato celular à coluna e misture em uma batedeira orbital por 30 min a 4 °C.
- Remova o plugue inferior da coluna e coloque a coluna em um tubo de centrífuga de 50 mL rotulado de fluxo atravésde . Centrifugar a coluna a 700 × g por 2 min para coletar o fluxo.
- Adicione 6 mL de tampão de lavagem à coluna para lavar a resina. Centrifugar a coluna a 700 × g por 2 min para coletar a fração em um novo tubo de centrífuga rotulado de lavagem 1. Repita esta etapa mais duas vezes para um total de 3 frações separadas e colete as frações em tubos de centrífugas separados(lave 2 e lave 3).
- Adicione 3 mL de tampão de eluição para elutar as proteínas marcadas por Sua da resina. Centrifugar a coluna a 700 × g por 2 min para coletar fração em um novo tubo de centrífuga rotulado eluate 1. Repita esta etapa mais duas vezes para um total de 3 frações separadas, e colete as frações em tubos de centrífuga separados(eluvar 2 e eluia 3).
- Combine os eluatos e realize a desalar para remover sais da solução proteica.
- Pipeta 15 mL de 0,05 % w/v polisorbate 20 sobre uma unidade de filtro centrífugo de 100 kDa. Centrifugar a 4.000 × g por 40 min e descartar o fluxo..
- Use o filtro revestido para concentrar os eluatos 1, 2 e 3 (9 mL do total de proteínas eluadas + 6 mL de tampão de armazenamento) para ~1.500 μL. Centrifugar o filtro a 3.220 × g por 20 min, e lavar suavemente a membrana para evitar a precipitação.
- Diluir a amostra para 15 mL com buffer de armazenamento. Execute uma troca de buffer usando buffer de armazenamento 1:1.000 repetindo o passo 4.11.2 mais duas vezes.
- Quantifique a proteína purificada medindo a absorvência da fração a 280 nm. Em branco o espectrofotômetro com tampão de armazenamento (tampão de armazenamento de 2x a 4 °C). Misture suavemente a amostra dos elunatos combinados e meça sua absorvência.
NOTA: Realize três leituras separadas em diluições de 1x, 10x e 50x da amostra de proteína para mediar e quantificar a proteína. Diluir amostras no buffer de armazenamento. - Ajuste as amostras de proteína para 100 μM usando tampão de armazenamento 2x. Diluir a amostra ajustada 1:1 em volume com 100% de glicerol. Armazene a solução de proteína resultante a -80 °C.
5. Sulfato de sulfato-poliacrilamida de sódio eletroforese gel (SDS-PAGE) análise do produto proteico: Dia 3
- Execute um gel SDS-PAGE para análise de proteínas. Misture 9 μL da amostra com 3 μL de sulfato de dodecyl de lítio 4x (LDS). Aqueça as amostras a 95 °C por 10 minutos.
- Carregue as amostras em uma configuração de gel SDS-PAGE Bis-Tris de 4-12%. Carregue a escada de proteína em poço 1, depois com amostras (da esquerda para a direita): fluxo-through, lavagem 1, lavagem 2, lavagem 3, elução 1, elução 2, elução 3 e eluição total dessalada.
NOTA: A tabela 5 contém uma tabela de carregamento de amostras para o gel SDS-PAGE. - Execute as amostras de gel carregado em 2-(N-morpholino) tampão de ácido esfilfônico (MES) por 35 min a 200 V. Enxágue o gel em uma bandeja limpa três vezes por 10 minutos cada usando 200 mL de ddH2O, com agitação suave para remover qualquer SDS da matriz de gel.
NOTA: Use equipamentos de proteção individual para evitar toxicidade aguda devido ao MES. - Manche o gel com 20 mL de azul Coomassie, e incubar o gel durante a noite à temperatura ambiente com agitação suave. Descolora o gel duas vezes por 1 h cada com 200 mL de ddH2O com agitação suave em um agitador orbital.
NOTA: Lavar o gel por um período mais longo ou substituir com frequência a água aumentará a sensibilidade. Além disso, colocar um tecido de limpeza de tarefas delicadas dobrado no recipiente para absorver o excesso de corante acelerará o processo de des coloração.
6. Verificação funcional do SNAP T7 RNAP via transcrição in vitro
NOTA: Este protocolo usa o modelo de DNA, que codifica para o aptamer RNA de Brócolis fluorescente e permite o uso de fluorescência para monitorar a cinética da transcrição em um leitor de placa de fluorescência.
- Configure três reações de transcrição in vitro (IVT) para comparar a atividade do SNAP T7 RNAP com o T7 RNAP do tipo selvagem (WT) de uma fonte comercial e um controle somente para buffer. Ajuste o volume de cada reação para 20 μL.
- Prepare a reação SNAP T7 RNAP IVT misturando 2 μL de buffer de transcrição de 10x, 0,4 μL de 25 mM ribonucleoside triphosphate (rNTP), 5 μL de modelo de DNA de 500 nM, 2 μL de 500 nM SNAP T7 RNAP e 10,6 μL de ddH2O.
- Prepare a reação WT RNAP IVT misturando 2 μL de tampão de transcrição de 10x, 0,4 μL de 25 mM rNTP mix, 5 μL de 500 nM de modelo de DNA, 2 μL de WT T7 RNAP e 10,6 μL de ddH2O.
- Prepare a reação ivt somente tampão misturando 2 μL de buffer de transcrição de 10x, 0,4 μL de 25 mM rNTP mix, 5 μL de 500 nM de dna modelo e 12,6 μL de ddH2O.
NOTA: Adicione o RNAP por último, mantendo as amostras no gelo até sua introdução. Tabela 6, Tabela 7e Tabela 8 contêm as fórmulas de reação IVT.
- Monitore a cinética de transcrição em um leitor de placa de fluorescência por 2h a 2 minutos a 37 °C usando um comprimento de onda de excitação de 470 nm e um comprimento de onda de emissão de 512 nm.
7. Preparação de oligonucleotídeos modificados pela BG: Dia 1
- Dissolva o oligonucleotídeo com modificação de 3'-amine em ddH2O a uma concentração final de 1 mM. Rotule este S1.
- Misture 25 μL de 1 M de bicarbonato de sódio (NaHCO3),284 μL de sulfoxida de 100% dimetil (DMSO), 125 μL de S1 (oligonucleotídeo estoque), e 66 μL de 50 mM do éster BG-N-hidroxisuccinimide (NHS) diluído com DMSO, ajuste o volume para 500 μL, e incubar durante a noite a temperatura ambiente a 100 × g.
NOTA: Mantenha o DMSO longe do calor e da chama, pois é um líquido combustível. A Tabela 9 contém a fórmula de reação para a conjugação BG ao oligonucleotídeo.
- Misture 25 μL de 1 M de bicarbonato de sódio (NaHCO3),284 μL de sulfoxida de 100% dimetil (DMSO), 125 μL de S1 (oligonucleotídeo estoque), e 66 μL de 50 mM do éster BG-N-hidroxisuccinimide (NHS) diluído com DMSO, ajuste o volume para 500 μL, e incubar durante a noite a temperatura ambiente a 100 × g.
8. Precipitação de etanol/acetona de conjugado BG-oligonucleotídeo: Dia 2
- Centrifugar o produto da etapa 7.1.1. a 13.000 × g por 5 min. Transfira cuidadosamente o supernante para um tubo fresco e descarte qualquer BG precipitado. Divida a reação em duas alíquotas iguais de 250 μL para evitar o estouro e realize as seguintes etapas em ambas as alíquotas.
- Adicione 1/10do volume de acetato de sódio de 3 M (25 μL), seguido por 2,5x o volume em 100% etanol (625 μL). Incubar a -80 °C por 1 h.
NOTA: Use equipamentos de proteção individual ao manusear acetato de sódio (pode causar irritação nos olhos, pele, aparelho digestivo e respiratório) e etanol (extremamente inflamável, causa irritação no contato). Se necessário, pare o experimento aqui e continue no dia seguinte. - Coloque os tubos na centrífuga e marque a borda externa. Centrifugar os tubos a 17.000 × g por 30 min a 4 °C.
NOTA: A pelota de oligonucleotídeo aparecerá na borda marcada do tubo. - Sem perturbar a pelota, descarte o supernatante. Cubra com 750 μL de etanol resfriado de 70%, e gire a 17.000 × g para 10 min a 4 °C.
- Sem perturbar a pelota, descarte o supernatante. Cubra com 750 μL de 100% acetona, e gire a 17.000 × g para 10 min a 4 °C.
NOTA: Use equipamentos de proteção individual ao manusear acetona, pois é extremamente inflamável e causa irritação no contato. - Com a tampa do tubo aberta, o ar seque por 5 minutos para remover qualquer excesso de acetona através da evaporação. Ressume o oligonucleotídeo em 250 μL de 1x Tris-EDTA (TE) para produzir uma solução BG-oligonucleotide de ~850 μM.
- Repita as etapas 8.2 a 8.6 e se dissolva em 70 μL de 1x te buffer. Rotule este S2.
9. Limpeza bg-oligonucleotídeo através de cromatografia de filtragem de gel
- Suspender a matriz invertendo vigorosamente as colunas várias vezes; remover a tampa superior e estalar a ponta inferior da coluna. Coloque a coluna em um tubo de centrífuga de 1,5 mL e centrifugar o tubo a 1.000 × g por 1 min à temperatura ambiente. Descarte o tampão e o tubo de coleta elucidos.
NOTA: É importante evitar a formação de vácuo. Use colunas preparadas imediatamente. - Coloque as colunas embaladas em tubos de centrífugas de 1,5 mL limpos. Adicione 300 μL de tampão 1x TE ao centro da cama da coluna e centrífugas a 1.000 × g por 2 min para trocar a solução tampão. Mais uma vez, descarte o tampão e o tubo de coleta elucidos.
- Coloque as colunas trocadas por buffer em tubos de centrífugas limpos de 1,5 mL. Aplique até 75 μL de amostra no centro da cama. Gire a 1.000 × g por 4 min.
NOTA: Não perturbe a cama ou toque nas laterais da coluna; o ponto mais alto da mídia gel deve apontar para o rotor externo. - Recolhe o eluato do tubo de coleta, pois contém o ácido nucleico purificado. Para quantificar a amostra, meça sua absorção em 260 nm; rotular este S3.
NOTA: Observe o comprimento do caminho utilizado na medição e calcule a concentração usando a lei Beer-Lambert.
10. Análise de desnaturação de PAGE do conjugado BG-oligonucleotídeo
- Lançar um gel Tris-borate-EDTA (TBE)-Urea PAGE de 18%. Dissolver 4,8 g de UREA, 4,5 mL de 40% de acrilamida (19:1), e 1 mL de 10x TBE em 2,8 mL de ddH2O; adicionar 5 μL tetrametilenodiamina (TEMED) e misturar completamente. Repita com 100 μL de 10% de persulfito de amônio (APS). Despeje a solução em um de gel vazio e deixe a polimerização por 40 minutos.
NOTA: Utilize equipamentos de proteção individual adequados ao manusear a ureia (causa irritação nos olhos e na pele), acrilamida (tóxica e cancerígena) e TEMED (tóxica, inflamável, corrosiva). A Tabela 10 contém a fórmula de reação para um gel de poliacrilamida TBE-UREA de 18%. - Micro-ondas 500 mL de tampão TBE (0,5x) por 2 min e 30 s ou até ~70 °C e despeje em um aparelho de gel. Prepare o corante de carregamento formamida (desnaturação) contendo 95% de formamida + 1 mM EDTA e azul bromofenol. Misture o corante de carga com cada amostra e carregue a mistura no gel de poliacrilamida.
NOTA: Use equipamentos de proteção individual adequados ao manusear formamida, pois é cancerígeno. A tabela 11 contém uma mesa de carregamento de gel de amostra. - Execute o gel a 270 V por 35 min, ou até que a frente de corante migre até o final. Coloque o gel em uma caixa de gel e colora com corante de cianinina para ácidos nucleicos por 15 minutos à temperatura ambiente antes da imagem.
NOTA: Use equipamentos de proteção individual adequados ao manusear corante de cianina, pois é combustível.
11. Conjugação de oligonucleotídeo para análise SNAP T7 RNAP e PAGE
- Prepare os reagentes para o acoplamento em escala analítica de BG-oligonucleotídeo ao SNAP T7 RNAP: faça 9 diluições de oligo de DNA mono-encalhado (ssDNA) com ddH2O para criar relações oligo:RNAP que variam de 5:1 a 1:5. Diluir o estoque de proteínas para 50 μM.
NOTA: As razões de exemplo podem ser encontradas na Tabela 12; essas razões são calculadas utilizando-se uma concentração RNAP de 50 μM. - Para cada diluição do oligo ssDNA, faça 10 μL da mistura de reação contendo 2 μL de buffer SNAP, 4 μL de BG-oligonucleotídeo e 4 μL de SNAP T7 RNAP.
NOTA: A tabela 13 contém fórmulas de reação para a reação de rotulagem snap-tag.- Prepare mais duas amostras de controle: 1) um controle RNAP substituindo BG-oligonucleotídeo por ddH2O; 2) um controle de DNA substituindo snap T7 RNAP por ddH2O (para a menor concentração de oligonucleotídeo de SNAP T7 RNAP). Incubar todas as amostras em temperatura ambiente por 1h, e mantenha no gelo até que seja necessário.
- Configure onze reações de 10 μL adicionando 2 μL de cada amostra a 4 μL de tampão SNAP e 2 μL de corante de carga proteica, e aqueça a 70 °C por 10 minutos. Carregue 2 μL de cada amostra no gel de proteína Bis-Tris de 4-12%, e realize eletroforese de gel no gelo a 200 V por 35 min.
NOTA: A tabela 14 contém fórmulas de reação para as amostras de carregamento de gel.- Lave o SDS através de 3x de troca de água em um shaker, cada lavagem com duração de 10 min cada. Mancha com corante de cianinina para ácidos nucleicos por 15 minutos antes da imagem. Manche o gel novamente usando 20 mL de mancha azul Coomassie por 1 h. Destalar com ddH2O por 1h (ou durante a noite) antes da imagem.
NOTA: No gel, uma das reações produzirá a polimerase mais amarrada, juntamente com a menor quantidade de excesso livre bg-oligonucleotídeo; esta é a proporção ideal.
- Lave o SDS através de 3x de troca de água em um shaker, cada lavagem com duração de 10 min cada. Mancha com corante de cianinina para ácidos nucleicos por 15 minutos antes da imagem. Manche o gel novamente usando 20 mL de mancha azul Coomassie por 1 h. Destalar com ddH2O por 1h (ou durante a noite) antes da imagem.
- Prepare os reagentes para a escala preparatória acoplamento BG-oligonucleotídeo ao SNAP T7 RNAP. Realize a reação de acoplamento com a razão ideal encontrada na escala analítica.
NOTA: Minimizar a exposição da proteína à temperatura ambiente colocando a proteína no gelo quando não estiver em uso.
12. Purificação do SNAP-T7 com tethered oligonucleotídeo usando colunas de troca de íons
- Siga as instruções do fabricante para a configuração do tubo se ele se desviar das instruções listadas aqui. Prepare um tampão de purificação com pH maior que o ponto isoelétrico da proteína.
NOTA: Para o exemplo, foi utilizado um tampão de purificação de 10 mM tampão fosfato de sódio (pH 7).- Prepare 1.000 μL de tampão de elução contendo concentrações finais de 50 mM Tris e 0,5 M NaCl. Misture 50 μL de 1 M Tris, 100 μL de 5 M NaCl e 850 μL de ddH2O.
NOTA: A tabela 15 contém a fórmula de reação para o tampão de eluição.
- Prepare 1.000 μL de tampão de elução contendo concentrações finais de 50 mM Tris e 0,5 M NaCl. Misture 50 μL de 1 M Tris, 100 μL de 5 M NaCl e 850 μL de ddH2O.
- Coloque uma coluna em um tubo de centrífugas de 2 mL e lave com tampão de purificação a 2.000 × g por 15 minutos, ou até que todo o buffer tenha sido elucido. Descarte o tampão elucido.
- Diluir cada amostra com tampão de purificação em uma razão tampão de purificação de 3:1:e carregar a amostra na coluna 400 μL de cada vez. Gire a 2.000 × g por 10 min, ou até que todo o buffer tenha sido elucido. Colete o fluxo e rotule-ocomo fluxo.
- Adicione 400 μL de tampão de purificação no centro da coluna. Gire a 2.000 × g por 15 min, ou até que todo o buffer tenha sido elucido. Colete o fluxo e rotule-o como lavagem 1. Repita mais duas vezes para lavar 2 e lave 3.
- Adicione 50 μL de tampão de eluição no centro da coluna. Gire a 2.000 × g por 5 min, ou até que todo o buffer tenha sido elucido. Colete o fluxo e rotule-o como eluado 1. Repita mais duas vezes para eluiar 2 e eluiar 3.
- A piscina eluia 1, 2 e 3 (rotule este elunato total),deixando uma pequena fração de cada eluato para o gel, e mede a absorvância em 260 nm (A260) e 280 nm (A280). Após a medição, adicione glicerol a uma proporção de 1:1 e armazene a -20 °C até que use mais.
- Use uma unidade de filtro centrífugo (0,5 mL; 30 kDa) para trocar o buffer total com buffer de armazenamento 2x (~1:100) (rotule este produto). Meça A260/280 novamente. Adicione glicerol a uma proporção de 1:1 e armazene a -20 °C até usar mais.
- Carregue cada eluato: flow-through, wash 1-3, total eluate e produto em um gel SDS-PAGE 4-12% Bis-Tris, juntamente com uma escada de proteína. Corra a 200 V por 35 min, ou até que a frente de corante migre até o final.
13. Demonstração do controle sob demanda da atividade de polimerase de RNA amarrada
- Prepare o tampão de renas de 5x contendo 25 mM Tris, 5 mM EDTA e 25 mM cloreto de magnésio (MgCl2). Misture 2,4 μL de cada modelo (1 μM) com 5 μL de tampão de renas e 14,2 μL de ddH2O para formar 25 μL de 1 μM dsDNA cage. Incubar esta solução a 75 °C por 2 min. Da mesma forma, anneal o sentido e fios antisentamais do modelo de DNA aptamer verde malachite. Prepare uma solução de 1mM de oxalato verde malachite.
NOTA: A tabela 16 contém a fórmula de reação para tampão de renas de 5x, a Tabela 17 contém a fórmula de reação para a reposição de dois modelos ssDNA. - Incubar o SNAP T7 RNAP amarrado com a gaiola dsDNA em uma relação molar de 1:5 em temperatura ambiente por 15 minutos a uma concentração final de 500 nM RNAP. Mantenha-se no gelo até que seja necessário.
- Pré-aqueça o leitor de placas a 37 °C. Configure três reações de 25 μL IVT no gelo
- Configure uma reação contendo o SNAP T7RNAP enjaulado com fatores de transcrição de ácido nucleico. Misture 2,5 μL de tampão IVT de 10x, 1 μL de mistura rNTP de 25 mM, 1 μL de 1 mM verde malachite, 2,5 μL da mistura RNAP-cage, 2,5 μL cada um dos fios de transcrição de 1 μM A e B oligonucleotídeos, e 3 μL de modelo de aptamer verde malachita de 1 mM em 10 μL de ddH2O.
- Configure uma reação contendo o SNAP T7RNAP enjaulado sem fatores de transcrição de ácido nucleico. Misture 2,5 μL de tampão IVT de 10x, 1 μL de mistura rNTP de 25 mM, 1 μL de 1 mM verde malachite, 2,5 μL da mistura RNAP-cage e 3 μL de 1 mM malachite modelo de aptamer verde malachite em 15 μL de ddH2O.
- Configure apenas uma reação contendo tampão. Misture 2,5 μL de tampão IVT de 10x, 1 μL de mistura rNTP de 25 mM, 1 μL de 1 mM verde malachite e 3 μL de 1 mM malachite modelo de aptamer verde malachita em 17,5 μL de ddH2O.
NOTA: A tabela 18 contém uma referência geral para as reações de transcrição in vitro.
- Transfira cada reação para uma placa de 384 poços. Monitore a transcrição do aptámero verde malachita em um leitor de placa de fluorescência por 2 h a 37 °C e com excitação de 610 nm e emissão de 655 nm. Uma vez terminado, mantenha a placa no gelo até que seja necessário.
- Tampão de micro-ondas 0,5x TBE por 2 min 30 s ou até ~70 °C. Execute os produtos RNA de cada poço em um gel de poliacrilamida TBE-Urea de 12% no tampão TBE aquecido a 280 V por 20 minutos, ou até que a frente de corante chegue ao final. Manche o gel com corante de cianina mancha de ácido nucleico por 10 minutos em um agitador orbital antes da imagem.
NOTA: A tabela 19 contém a fórmula de reação para um gel de 12% de TBE-Urea PAGE de desnaturação.
Resultados

Figura 5: Análise SDS-PAGE da expressão SNAP T7 RNAP e ensaio de transcrição in vitro. (A) ANÁLISE DE PURIFICAÇÃO DE PROTEÍNAS SNAP T7 RNAP, PESO molecular SNAP T7 RNAP: 119.4kDa. FT = fluxo-through da coluna, W1 = frações de eluição de tampão de lavagem contendo impurezas, E1-3 = frações de eluição contendo produto purificad...
Discussão
Este estudo demonstra uma abordagem inspirada em nanotecnologia de DNA para controlar a atividade da polimerase T7 RNA, acoplando um T7 RNAP recombinante com marca DE-T7 com um oligonucleotídeo funcionalizado pela BG, que foi posteriormente usado para programar reações TMDSD. Por design, a tag SNAP foi posicionada no N-terminus da polimerase, já que o C-terminus do tipo selvagem T7 RNAP está enterrado dentro do núcleo da estrutura proteica e faz contatos importantes com o modelo de DNA28. Te...
Divulgações
Não há interesses financeiros concorrentes para declarar por nenhum dos autores.
Agradecimentos
L.Y.T.C reconhece o generoso apoio do New Frontiers in Research Fund-Exploration (NFRF-E), do Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant e da University of Toronto's Medicine by Design Initiative, que recebe financiamento do Canada First Research Excellence Fund (CFREF).
Materiais
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
Referências
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -. J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981 (2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324 (2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -. B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607 (2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C., Doudna, J. A. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -. B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados