
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
In vivo Imagem de Tecido Cerebral Totalmente Ativo em Larvas de Zebrafish Acordados e Juvenis por Remoção de Crânio e Pele
Neste Artigo
Resumo
Aqui apresentamos um método para a imagem do cérebro embrionário de zebrafish in vivo upto larval e juvenil estágios. Este procedimento microinvasivo, adaptado a partir de abordagens eletrofisiológicas, fornece acesso a detalhes celulares e subcelulares de neurônio maduro e pode ser combinado com estudos optogenéticos e neurofarmaológicos para caracterização da função cerebral e intervenção medicamentosa.
Resumo
Compreender as alterações efêmeras que ocorrem durante o desenvolvimento e amadurecimento cerebral requer imagens detalhadas de alta resolução no espaço e no tempo na resolução celular e subcelular. Os avanços nas tecnologias moleculares e de imagem nos permitiram obter numerosas informações detalhadas sobre os mecanismos celulares e moleculares do desenvolvimento cerebral no embrião transparente de zebrafish. Recentemente, os processos de refinamento da conectividade neuronal que ocorrem em estágios larvais posteriores várias semanas após a fertilização, que são, por exemplo, o controle do comportamento social, tomada de decisão ou comportamento motivado por motivação, passaram a se concentrar na pesquisa. Nestas fases, a pigmentação da pele do zebrafish interfere com a penetração da luz no tecido cerebral, e soluções para estágios embrionários, por exemplo, inibição farmacológica da pigmentação, não são mais viáveis.
Portanto, é fornecida uma solução cirúrgica minimamente invasiva para o acesso de microscopia ao cérebro de zebrafish acordado que é derivada de abordagens eletrofisiológicas. Em teleosts, a cartilagem da pele e do crânio macio pode ser cuidadosamente removida micro-descascando essas camadas, expondo neurônios subjacentes e tratos axonais sem danos. Isso permite o registro da morfologia neuronal, incluindo estruturas sinápticas e seus conteúdos moleculares, e a observação de alterações fisiológicas como transitórios Ca2+ ou eventos de transporte intracelular. Além disso, o interrogatório desses processos por meio de inibição farmacológica ou manipulação optogenética é viável. Esta abordagem de exposição cerebral fornece informações sobre mudanças estruturais e fisiológicas nos neurônios, bem como a correlação e interdependência desses eventos no tecido cerebral vivo na faixa de minutos ou horas. A técnica é adequada para imagens cerebrais in vivo de larvas de zebrafish até 30 dias após a fertilização, o último estágio de desenvolvimento testado até agora. Assim, fornece acesso a questões tão importantes como refinamento sináptico e dimensionamento, transporte axonal e dendrático, direcionamento sináptico de carga citoesquelética ou expressão local dependente de atividades. Portanto, um uso amplo para esta abordagem de montagem e imagem pode ser antecipado.
Introdução
Nas últimas décadas, o zebrafish (Danio rerio) evoluiu como um dos organismos modelo vertebrados mais populares para estudos de desenvolvimento embrionário e larval. A grande fecundidade das fêmeas de zebrafish aliada ao rápido desenvolvimento ex utero do embrião e sua transparência durante os estágios iniciais do desenvolvimento embrionário são apenas alguns dos fatores-chave que fazem do zebrafish um poderoso organismo modelo para resolver questões de desenvolvimento1. Avanços nas tecnologias genéticas moleculares combinados com estudos de alta resolução in vivo de imagem permitiram abordar mecanismos biológicos celulares subjacentes aos processos de desenvolvimento2. Em particular, no campo da diferenciação neuronal, fisiologia, conectividade e função, o zebrafish lançou luz sobre a interação da dinâmica molecular, funções cerebrais e comportamento organismo em detalhes sem precedentes.
No entanto, a maioria desses estudos são restritos a estágios embrionários e larvais iniciais durante a primeira semana de desenvolvimento, uma vez que a transparência do tecido do sistema nervoso é progressivamente perdida. Nestas fases, o tecido cerebral é impedido de acessar por abordagens de microscopia de alta resolução que se tornam protegidas pela diferenciação do crânio e pigmentação3.
Portanto, questões-chave de diferenciação neuronal, maturação e plasticidade, como o refinamento da conectividade neuronal ou o dimensionamento sináptico são difíceis de estudar. Esses processos celulares são importantes para definir mecanismos celulares que conduzem, por exemplo, comportamento social, tomada de decisão ou comportamento baseado em motivação, áreas para as quais a pesquisa de zebrafish sobre larvas de várias semanas de idade contribuiu recentemente com os principais achados baseados em estudos comportamentais4.
Abordagens farmacológicas para inibir a pigmentação em larvas de zebrafish por várias semanas são pouco viáveis ou podem até causar efeitos prejudiciais5,6,7,8. Cepas mutantes duplas ou triplas com defeitos de pigmentação específicos, como o casper9 ou o cristal10,tornaram-se ferramentas tremendamente valiosas, mas são trabalhosas na reprodução, fornecem poucos descendentes e representam o perigo de acumular malformações genéticas devido ao excesso de endogamia.
Aqui, é fornecido um procedimento invasivo mínimo como alternativa que seja aplicável a qualquer cepa de zebrafish. Este procedimento foi adaptado de estudos eletrofisiológicos para registrar atividade neuronal em larvas vivas e acordadas de zebrafish. Em teleosts, a cartilagem da pele e do crânio macio pode ser cuidadosamente removida através da micro-descascamento dessas camadas, porque elas não estão firmemente entrelaçadas com a vasculatura cerebral. Isso permite expor o tecido cerebral contendo neurônios e tratos axonais sem danos e para o registro de morfologia neuronal, incluindo estruturas sinápticas e seus conteúdos moleculares, que por sua vez incluem a observação de alterações fisiológicas como transitórios ca2+ ou eventos de transporte intracelular por até várias horas. Além disso, além das caracterizações descritivas, o acesso direto ao tecido cerebral permite o interrogatório de funções neuronais maduras por meio da administração de substâncias neurofarmacológicas e abordagens optogenéticas. Portanto, as relações de função da verdadeira estrutura podem ser reveladas no cérebro juvenil de zebrafish usando essa estratégia de exposição cerebral.
Protocolo
Todo o trabalho animal descrito aqui está de acordo com as regulamentações legais (Diretiva UE 2010/63). A manutenção e manuseio de peixes foram aprovadas pelas autoridades locais e pelo representante do bem-estar animal da Technische Universität Braunschweig.
1. Preparação de fluido espinhal cerebro artificial (ACSF), agarose de baixo derretimento e agulhas de vidro afiadas
- Prepare o ACSF dissolvendo os produtos químicos listados nas seguintes concentrações em água destilada. 134 mM NaCl (58,44 g/mol), 2,9 mM KCl (74,55 g/mol), 2,1 mM CaCl2 (110,99 g/mol), 1.2 mM MgCl2 6x H2O (203,3 g/mol), 10 mM HEPES (238,31 g/mol) e 10 mM d-Glicose (180,16 g/mol).
NOTA: Para MgCl2, CaCl2e KCl, as soluções de estoque de 1 M são preparadas em água estéril salgada e armazenadas a 4 °C para posterior preparação de ACSF fresco. Glicose, HEPES e NaCl são dissolvidos como compostos sólidos na solução ACSF fresca. Para dissolver produtos químicos, siga as instruções do fabricante. - Ajuste o pH do ACSF para 7,8 com 10 M NaOH. A preparação do ACSF requer medição precisa de produtos químicos e ajuste fino do pH, pois substitui o fluido espinhal cerebro e mantém as condições fisiológicas necessárias para que os neurônios sejam totalmente funcionais, pois pode causar erro de funcionamento cerebral e morte neuronal.
- Armazene o ACSF recém-preparado a 4 °C para uma máxima de 4 semanas. Para condições de trabalho, aliquotar o volume necessário de ACSF para o dia/experimento e pré-aquecimento em 25-28 °C (e opcionalmente oxigená-lo, passo 2.5)
NOTA: ASCF recém-preparada está bem por 1 dia. Se planeja usá-lo durante vários dias, o ACSF precisa ser filtrado estéril. - Para anestesia posterior das larvas, prepare uma solução de estoque de 50 mM de d-Tubocurarina em água destilada e armazene a solução a -20 °C como alíquotas de 100 μL no congelador até que seja necessário.
- Para incorporar o peixe, prepare 2,5% de baixo derretimento (LM) agarose dissolvendo 1,25 g LM-agarose(Tabela de Materiais) em 50 mL ACSF e ferva até que a agarose seja completamente dissolvida.
NOTA: Alternativamente, concentrações maiores ou menores de LM-agarose podem ser usadas dependendo da configuração experimental. No entanto, se a agarose for muito macia, não será capaz de manter o peixe em posição ao abrir o crânio. - Armazene a agarose a 37 °C de banho de água, para evitar a solidificação e porque essa temperatura também não prejudicará as larvas ao incorporar. Depois que a agarose cozida for resfriada até 37 °C no banho d,, adicione a quantidade necessária de d-Tubocurarine à agarose aliquoted necessária para o dia para atingir uma concentração de trabalho de 10 μM. Para uso futuro, armazene a agarose de sobra a 4 °C para evitar contaminação.
- Prepare agulhas de vidro afiadas e finas de capilares de vidro(Figura Suplementar 1) usando um puxador de micropipette com as seguintes configurações.
- Puller I, Capilar tipo 1: Calor 1: 65,8; Calor 2: 55.1; Puxar 2 passos
Puller II, Tipo capilar 2: Calor = 700; Fil = 4; Vel = 55; Del = 130; Pul = 55; 1 passo puxando.
NOTA: As unidades são específicas para cada puxador e capilar de vidro utilizado aqui, respectivamente (ver Tabela de Materiais). Outros capilares e puxadores também podem ser usados para preparar as agulhas de vidro. Mas as agulhas de vidro não devem ser muito finas, pois podem quebrar quando entrarem em contato com o crânio. Capilar: comprimento: 100 mm (4 polegadas); OD: 1,5 mm; ID: 0,84 mm; filamento: Sim
- Puller I, Capilar tipo 1: Calor 1: 65,8; Calor 2: 55.1; Puxar 2 passos
2. Anestesia de larvas e preparações para incorporação
- Ao iniciar o experimento do dia, transfira os animais que são necessários com uma pipeta Pasteur de plástico para uma placa de Petri de 90 mm de diâmetro, que é preenchida com Danieau (para larvas que ainda são mantidas em uma placa de Petri com Danieau) ou água da instalação de peixes (para larvas que são maiores que 7 dpf e são mantidas na instalação de peixes).
- Ao enchimento de peixes com mais de 2 semanas, certifique-se de que a abertura da pipeta seja grande o suficiente para evitar ferir os peixes ao transferi-los. Não use uma rede porque ela vai danificar fisicamente especialmente as larvas mais jovens.
- Adicione rotifera ou artemia nauplii adequado para o tamanho das larvas mantidas na placa de Petri, para garantir o acesso gratuito aos alimentos e o estado máximo de saúde das larvas e reduzir o estresse.
- Para incorporar, transfira as larvas selecionadas para uma placa de Petri de 35 mm de diâmetro cheia de ACSF. Adicione o volume necessário de d-Tubocurarine para atingir uma concentração de trabalho/dose eficaz de 10 μM e espere por alguns minutos até que as larvas estejam completamente imobilizadas11.
NOTA: Quando o peixe envelhece ou se for necessária uma anestesia completa mais rápida (menos de 5 min), é possível aumentar a concentração de d-Tubocurarine (LD50 para camundongos é de 0,13 mg/kg por via intravenosa12). Também é possível usar um anestésico diferente, como α-bungarotoxina (concentração de trabalho: 1 mg/mL), que tem o mesmo efeito que curare e também mantém o cérebro totalmente ativo13. Se um cérebro totalmente ativo não é necessário para o assunto de interesse, tricaine em uma dose não letal (0,02%) também é uma opção para anestesiar totalmente as larvas. No entanto, tricaine bloqueia canais de sódio, prejudicando assim a atividade cerebral14. - Prepare a câmara de montagem pegando a tampa da placa petri de 35 mm de diâmetro, vire a tampa de cabeça para baixo e coloque uma mancha de vidro quadrado (24 x 24 mm) na parte inferior da tampa. Consulte a Figura 1 (parte superior) para obter uma descrição esquemática dessas etapas. A superfície mais suave do vidro evita escapar do bloco de agarose, que contém as larvas durante o procedimento de abertura do crânio.
- Aliquotar a quantidade de ACSF necessária para o dia em um frasco apropriado (por exemplo, tubo de 50 mL, béquer, garrafa de Schott, etc.) e oxigená-lo com carbogen (5% CO2, 95% O2). Se a imagem apenas de morfologia (por exemplo, padrões de fluorescência) ACSF ainda é necessária para garantir a integridade do cérebro e que as células não são influenciadas negativamente por efeitos de osmolaridade, mas a oxigenação do ACSF não é necessária. Esta etapa só precisa ser realizada quando a atividade cerebral completa é necessária para a imagem.
NOTA: Para uma saturação de oxigênio ideal do meio, adicione uma pedra de ar na extremidade do tubo de carbogen. Para garantir um nível de oxigênio suficientemente alto, é necessário trocar o ACSF nas câmaras de imagem com ACSF recém-oxigenado a cada 20-60 minutos, dependendo do número e idade das larvas embutidas na mesma câmara de imagem (por exemplo, para uma única troca de larvas incorporadas ACSF a cada hora é suficiente. Para seis larvas com mais de 14 dpf embutidas em paralelo, é necessário trocar ACSF a cada 20 minutos) então planeje a quantidade necessária de ARSF saturada de oxigênio de acordo com o experimento planejado.
3. Incorporação das larvas
- Transfira as larvas totalmente anestesiadas com uma pipeta pasteur de plástico para a câmara de montagem preparada (na etapa 2.4). Em seguida, remova cuidadosamente o excesso médio para evitar a diluição da Agarose LM. Todas as etapas a seguir devem ser executadas sob um microscópio estéreo com ampliação suficiente.
NOTA: Inclinar a câmara de montagem pode ajudar a remover totalmente o meio. - Prossiga imediatamente para o próximo passo, adicionando uma gota de LM-agarose suficientemente grande em cima das larvas (cerca de 1 mL, dependendo do tamanho das larvas) para proteger os animais de secar e reduzir o estresse desnecessário.
- Oriente as larvas em posição antes que a agarose se solidifique. Certifique-se de que a parte dorsal das larvas seja direcionada para cima. Além disso, certifique-se de incorporar as larvas o mais perto possível da superfície da agarose.
NOTA: Dependendo do tamanho e número de larvas planejadas para incorporar ao mesmo tempo, é possível ajustar a concentração de ágarose. Por exemplo, para 1-3 larvas de 30 dpf de idade, recomenda-se uma concentração de 1,8%-2% LM-agarose. Para 1-4 larvas que são de 7 dpf de idade, é mais praticável usar 2,5% de LM-agarose, enquanto, para 5-8 larvas, 2% é mais adequado. Se for necessário um cérebro totalmente ativo, recomenda-se incorporar apenas três peixes ao mesmo tempo para reduzir o tempo necessário para operar as larvas. Em geral, recomenda-se o uso de concentrações menores (1,8%-2%) quanto mais velhas as larvas ficam ou mais larvas são planejadas para serem incorporadas ao mesmo tempo. - Se as imagens forem gravadas usando um microscópio invertido, corte o bloco de agarose contendo as larvas em uma pequena forma cuboide. Isso é importante para transferir as larvas para a câmara de imagem mais tarde. Se usar um microscópio vertical, esse corte não é necessário, pois a câmara de montagem também pode ser usada como a câmara de imagem. Na Figura 1 (parte superior), pode-se encontrar uma descrição esquemática dessas etapas.
4. Expor o cérebro
NOTA: Todas as etapas a seguir devem ser realizadas com o maior cuidado para não ferir desnecessariamente as larvas. Se um cérebro totalmente ativo for necessário para o experimento, tenha em mente que a cada segundo que passa, enquanto o peixe ainda está totalmente montado em agarose e tem um crânio aberto sem ACSF oxigenado, o cérebro sofrerá de falta de oxigênio e também secará. Os efeitos da deficiência de oxigênio se tornarão ainda mais dramáticos, quanto mais velhas as larvas incorporadas. Por isso, é importante realizar a cirurgia não apenas no menor tempo possível, mas também com a máxima precisão para não evocar danos cerebrais mecânicos com a agulha. Quando treinado, as etapas 4.2-4.4 não devem levar mais de 30 s por peixe.
- Comece a cirurgia assim que a agarose se solidificar. Primeiro, aparar todo o excesso de agarose acima da região do cérebro de interesse para obter acesso livre à cabeça e um espaço de trabalho claro. Se a parte dorsal da cabeça já está saindo da agarose, pule esta etapa.
- Dependendo da região de interesse, escolha um lugar para começar com a cirurgia. Pegue a agulha de vidro e faça uma pequena incisão através da pele, mas sem penetrar muito fundo no tecido. Este será o ponto de partida para descascar a pele sobreposta.
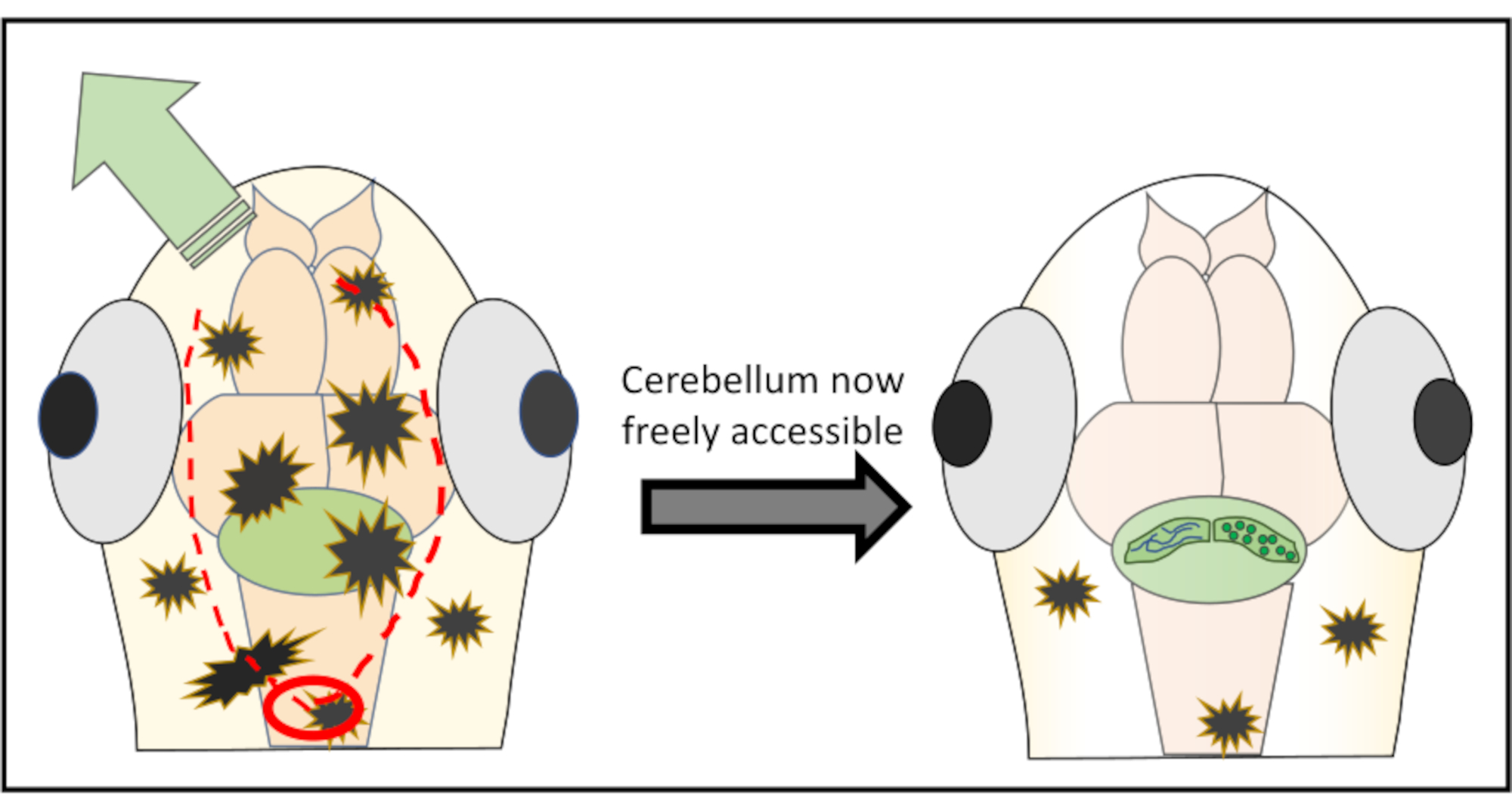
NOTA: Para obter resultados ótimos, nunca comece diretamente acima da região de interesse para reduzir o risco de danificar estruturas importantes. Se necessário, é possível até mesmo iniciar posteriormente para o cérebro traseiro e a partir daí trabalhar para a frente até que a área indesejada da pele seja descascada. - Continue com cortes muito pequenos ao longo da parte da pele com o objetivo de remover, movendo mal a agulha logo abaixo da superfície. Na maioria das vezes não é necessário mover-se completamente ao redor do cérebro e cortar um pedaço de pele e crânio em círculo, mas sim apenas fazer duas incisões ao longo da cabeça e, em seguida, empurrar a pele para um ou outro lado. A Figura 2 mostra uma representação esquemática da estratégia de corte ideal para obter acesso livre ao cerebelo.
NOTA: Esta microcirurgia é um procedimento delicado e provavelmente precisará de algum treinamento para remover perfeitamente a pele sem danificar o cérebro subjacente. Também é recomendado descobrir a estratégia de corte ideal para a região de interesse do cérebro e ficar com ela durante o período do experimento. - Imediatamente após remover a pele de todas as larvas incorporadas, proceda derramando ACSF (oxigenado) sobre a agarose para inundar partículas de pele e sangue indesejados e para manter o cérebro totalmente ativo e protegê-lo de secar.
NOTA: Se um cérebro saudável for necessário para o experimento, recomenda-se ir para um máximo de três peixes por vez. - Se estiver usando um microscópio vertical, comece diretamente com a imagem.
- Ao usar um microscópio invertido, deslize uma pequena espátula sob o bloco de agarose cuboide (passo 3.4).
- Adicione uma pequena gota de LM-agarose ao fundo da câmara de imagem (por exemplo, prato de fundo de vidro) e vire imediatamente o bloco de agarose contendo as larvas com a espátula por 180° e empurre-a suavemente para o fundo da câmara de imagem, enquanto a gota de agarose líquida age como cola.
- Quando a agarose se solidificar, encha a câmara de imagem com ACSF (oxigenado) e comece a fotografar. Consulte a Figura 1 (parte inferior) para obter uma descrição esquemática.
- Quando a atividade cerebral completa for necessária para o experimento, certifique-se sempre de que o ACSF na câmara de imagem tenha um nível de oxigênio suficientemente alto. Para garantir isso, troque o meio cuidadosamente com ACSF recém-oxigenado quando possível a cada 20-60 min (dependendo do número e tamanho do peixe, tamanho e superfície da câmara de imagem e duração da imagem).

Figura 1: Procedimento esquemático para a preparação de zebrafish de crânio aberto para imagens in vivo de forma stepwise. As instruções de trabalho para as diferentes etapas podem ser encontradas no próprio gráfico. Gráfico projetado por Florian Hetsch e adaptado por Paul Schramm. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Representação esquemática detalhada da microcirurgia realizada para remover pedaços de pele e crânio acima da região cerebral de interesse. O círculo vermelho marca o local onde o primeiro corte precisa ser feito. A linha pontilhada vermelha delineia o caminho ideal para cortar junto com a agulha para obter acesso livre ao cerebelo sem danificá-lo. O arqueiro verde marca a direção em que os pedaços excessivos de pele e crânio podem ser facilmente empurrados para longe. Certifique-se de nunca penetrar no tecido cerebral durante todo o procedimento. Depois de descascar com sucesso a pele, a região de interesse cerebral (aqui, cerebelo) estará livremente acessível para qualquer tipo de imagem in vivo de alta resolução. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Resultados
Figura 3AC mostrar uma larva dpf de 14 dpf da linha transgênica Tg [-7.5Ca8:GFP]bz12[15] com o crânio ainda intacto. As células pigmentadas na pele sobreposta estão distribuídas por toda a cabeça e estão interferindo com o sinal de fluorescência na região de interesse (aqui, cerebelo). Com a larva nesta condição, não é possível obter imagens de alta resolução do cérebro.
Discussão
O método apresentado fornece uma abordagem alternativa ao isolamento cerebral ou ao tratamento de larvas de zebrafish com fármacos inibindo pigmentação para registro de imagens de alta resolução de neurônios em seu ambiente in vivo. A qualidade das imagens registradas com este método é comparável às imagens de cérebros explantados, mas em condições naturais.
Além disso, evita-se uma perda de intensidade de fluorescência, pois não há necessidade de tratamento com fixa...
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Agradecemos especialmente a Timo Fritsch pelo excelente cuidado animal e Hermann Döring, Mohamed Elsaey, Sol Pose-Méndez, Jakob von Trotha, Komali Valishetti e Barbara Winter pelo apoio útil. Também somos gratos a todos os outros membros do laboratório Köster por seu feedback. O projeto foi financiado em parte pelo projeto da Fundação Alemã de Pesquisa (DFG, KO1949/7-2) 241961032 (para a RWK) e pelo Bundesministerium für Bildung und Forschung (BMBF; Era-Net NEURON II CIPRESS projeto 01EW1520 para JCM) é reconhecido.
Materiais
| Name | Company | Catalog Number | Comments |
| Calcium chloride | Roth | A119.1 | |
| Confocal Laser scanning microscope | Leica | TCS SP8 | |
| d-Glucose | Sigma | G8270-1KG | |
| d-Tubocurare | Sigma-Aldrich | T2379-100MG | |
| Glass Capillary type 1 | WPI | 1B150F-4 | |
| Glass Capillary type 2 | Harvard Apparatus | GC100F-10 | |
| Glass Coverslip | deltalab | D102424 | |
| HEPES | Roth | 9105.4 | |
| Hoechst 33342 | Invitrogen (Thermo Fischer) | H3570 | |
| Imaging chamber | Ibidi | 81156 | |
| Potassium chloride | Normapur | 26764298 | |
| LM-Agarose | Condalab | 8050.55 | |
| Magnesium chloride (Hexahydrate) | Roth | A537.4 | |
| Microscope Camera | Leica | DFC9000 GTC | |
| Needle-Puller type 1 | NARISHIGE | Model PC-10 | |
| Needle-Puller type 2 | Sutter Instruments | Model P-2000 | |
| Pasteur-Pipettes 3ml | A.Hartenstein | 20170718 | |
| Sodium chloride | Roth | P029.2 | |
| Sodium hydroxide | Normapur | 28244262 | |
| Tricain | Sigma-Aldrich | E10521-50G | |
| Waterbath | Phoenix Instrument | WB-12 | |
| 35 mm petri dish | Sarstedt | 833900 | |
| 90 mm petri dish | Sarstedt | 821473001 |
Referências
- Embryology. Zebrafish Development Available from: https://embryology.med.unsw.edu.au/embryology/index.php/Zebrafish_Development (2020)
- Sassen, W. A., Köster, R. W. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. Dove Medical Press. 2015 (5), 151-163 (2015).
- Singh, A. P., Nüsslein-Volhard, C. Zebrafish stripes as a model for vertebrate colour pattern formation. Current Biology. 25 (2), 81-92 (2015).
- Kalueff, A. V., et al. Time to recognize zebrafish 'affective' behavior. Brill: Behaviour. 149 (10-12), 1019-1036 (2012).
- Karlsson, J., von Hofsten, J., Olsson, P. -. E. Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Marine Biotechnology. 3, 522-527 (2001).
- Bohnsack, B. L., Gallina, D., Kahana, A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle development to changes in retinoic acid and IGF signaling. PloS One. 6, 22991 (2011).
- Elsalini, O. A., Rohr, K. B. Phenylthiourea disrupts thyroid function in developing zebrafish. Development Genes and Evolution. 212, 593-598 (2003).
- Baumann, L., Ros, A., Rehberger, K., Neuhauss, S. C. F., Segner, H. Thyroid disruption in zebrafish (Danio rerio) larvae: Different molecular response patterns lead to impaired eye development and visual functions. Aquatic Toxicology. 172, 44-55 (2016).
- White, R., et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2, 183-189 (2008).
- Antinucci, P., Hindges, R. A crystal-clear zebrafish for in vivo imaging. Scientific Reports. 6, 29490 (2016).
- Burr, S. A., Leung, Y. L. Curare (d-Tubocurarine). Encyclopedia of Toxicology (3rd Edition). , 1088-1089 (2014).
- Gesler, H. M., Hoppe, J. 3,6-bis(3-diethylaminopropoxy) pyridazine bismethiodide, a long-acting neuromuscular blocking agent. The Journal of Pharmacology and Experimental Therapeutics. 118 (4), 395-406 (1956).
- Furman, B. . Alpha Bungarotxin. Reference Module in Biomedical Sciences. , (2018).
- Attili, S., Hughes, S. M. Anaesthetic tricaine acts preferentially on neural voltage-gated sodium channels and fails to block directly evoked muscle contraction. PLoS One. 9 (8), 103751 (2014).
- Namikawa, K., et al. Modeling neurodegenerative spinocerebellar ataxia type 13 in zebrafish using a Purkinje neuron specific tunable coexpression system. Journal of Neuroscience. 39 (20), 3948-3969 (2019).
- Hennig, M. Theoretical models of synaptic short term plasticity. Frontiers in Computational Neuroscience. 7 (45), (2013).
- Wang, Y., et al. Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 137, 3119-3128 (2010).
- Hobro, A., Smith, N. An evaluation of fixation methods: Spatial and compositional cellular changes observed by Raman imaging. Vibrational Spectroscopy. 91, 31-45 (2017).
- Knogler, L. D., Kist, A. M., Portugues, R. Motor context dominates output from purkinje cell functional regions during reflexive visuomotor behaviours. eLife. 8, 42138 (2019).
- Hsieh, J., Ulrich, B., Issa, F. A., Wan, J., Papazian, D. M. Rapid development of Purkinje cell excitability, functional cerebellar circuit, and afferent sensory input to cerebellum in zebrafish. Frontier in Neural Circuits. 8 (147), (2014).
- Scalise, K., Shimizu, T., Hibi, M., Sawtell, N. B. Responses of cerebellar Purkinje cells during fictive optomotor behavior in larval zebrafish. Journal of Neurophysiology. 116 (5), 2067-2080 (2016).
- Harmon, T. C., Magaram, U., McLean, D. L., Raman, I. M. Distinct responses of Purkinje neurons and roles of simple spikes during associative motor learning in larval zebrafish. eLife. 6, 22537 (2017).
- Zehendner, C. M., et al. Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein disruption. PLoS One. 8 (12), 82823 (2013).
- Dhabhar, F. S. The short-term stress response - mother nature's mechanism for enhancing protection and performance under conditions of threat, challenge, and opportunity. Frontiers of Neuroendocrinology. 49, 175-192 (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados