
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Utilizando a eletroporação pós-natal in vivo para estudar a morfologia do neurônio do grânulo cerebelar e o desenvolvimento da sinapse
Neste Artigo
Erratum Notice
Resumo
Aqui descrevemos um método para visualizar a sinaptogênese de neurônios de grânulos no cerebelo do rato ao longo do curso de tempo do desenvolvimento cerebral pós-natal, quando essas células refinam suas estruturas sinápticas e formam sinapses para se integrarem ao circuito cerebral geral.
Resumo
Os neurônios sofrem mudanças dinâmicas em sua estrutura e função durante o desenvolvimento do cérebro para formar conexões apropriadas com outras células. O cerebelo de roedores é um sistema ideal para rastrear o desenvolvimento e a morfogênese de um único tipo de célula, o neurônio do grânulo cerebelar (CGN), ao longo do tempo. Aqui, a eletroporação in vivo de progenitores de neurônios de grânulos no cerebelo de camundongos em desenvolvimento foi empregada para marcar esparsamente as células para análises morfológicas subsequentes. A eficácia desta técnica é demonstrada em sua capacidade de mostrar os principais estágios de desenvolvimento da maturação do CGN, com um foco específico na formação de garras dendríticas, que são estruturas especializadas onde essas células recebem a maioria de suas entradas sinápticas. Além de fornecer instantâneos das estruturas sinápticas da CGN ao longo do desenvolvimento cerebelar, esta técnica pode ser adaptada para manipular geneticamente os neurônios dos grânulos de maneira autônoma celular para estudar o papel de qualquer gene de interesse e seu efeito na morfologia da CGN, no desenvolvimento de garras e na sinaptogênese.
Introdução
O desenvolvimento do cérebro é um processo prolongado que se estende desde a embriogênese até a vida pós-natal. Durante esse tempo, o cérebro integra uma combinação de estímulos intrínsecos e extrínsecos que esculpem a fiação das sinapses entre dendritos e axônios para, finalmente, orientar o comportamento. O cerebelo de roedores é um sistema modelo ideal para estudar como as sinapses se desenvolvem porque o desenvolvimento de um único tipo de neurônio, o neurônio do grânulo cerebelar (CGN), pode ser rastreado à medida que transita de uma célula progenitora para um neurônio maduro. Isso se deve, em parte, ao fato de que a maioria do córtex cerebelar se desenvolve no pós-natal, o que permite fácil manipulação genética e marcação celular após o nascimento1.
Em mamíferos, a diferenciação de CGN começa no final do desenvolvimento embrionário, quando um subconjunto de células proliferativas no rombencéfalo migra sobre o lábio rômbico para formar uma zona germinativa secundária na superfície do cerebelo 2,3,4. Embora estejam totalmente comprometidas com uma identidade de progenitor de neurônios de grânulos (PNB), essas células continuam a proliferar dentro da porção externa da camada externa de grânulos (EGL) até o dia 14 pós-natal (P14). A proliferação dessa camada resulta em uma expansão maciça do cerebelo, pois essas células dão origem exclusivamente a CGNs5. Uma vez que os CGNs recém-nascidos saem do ciclo celular no EGL, eles migram para dentro em direção à camada interna de grânulos (IGL), deixando para trás um axônio que se bifurcará e viajará na camada molecular do cerebelo, formando fibras paralelas que fazem sinapse nas células de Purkinje6. A posição dessas fibras dentro da camada molecular depende do tempo de saída do ciclo celular.
Os CGNs que se diferenciam primeiro deixam suas fibras paralelas em direção ao fundo da camada molecular, enquanto os axônios dos CGNs que se diferenciam posteriormente são agrupados no topo 7,8. Uma vez que os corpos celulares CGN atingem o IGL, eles começam a elaborar dendritos e formar sinapses com neurônios inibitórios e excitatórios próximos. A árvore dendrítica madura de um CGN exibe uma arquitetura estereotipada com quatro processos principais. Ao longo da maturação da CGN, as estruturas no final desses dendritos formam uma garra que se torna enriquecida com proteínas pós-sinápticas 9,10. Essas estruturas especializadas, chamadas garras dendríticas, contêm a maioria das sinapses nos neurônios dos grânulos e são importantes para receber tanto entradas excitatórias de inervações de fibras musgosas originárias da ponte, bem como entradas inibitórias de células locais de Golgi. Uma vez totalmente configuradas, as conexões sinápticas dos CGNs permitem que essas células retransmitam entradas de núcleos pré-cerebelares para células de Purkinje, que se projetam do córtex cerebelar para os núcleos cerebelares profundos.
A eletroporação pós-natal in vivo de PNB é vantajosa em relação a outros métodos baseados em marcação, como infecção viral e geração de linhagens transgênicas de camundongos, porque a expressão das construções desejadas pode ser alcançada em uma linha do tempo rápida, e o método tem como alvo uma pequena população de células, útil no estudo dos efeitos autônomos celulares. Este método já foi utilizado em estudos prévios para estudar o desenvolvimento morfológico de CGNs; no entanto, esses estudos têm se concentrado em um único ponto de tempo ou em uma curta janela de tempo 9,10,11,12,13. Este método de marcação foi emparelhado com a análise de imagens para documentar as mudanças na morfologia do CGN que ocorrem ao longo de todo o curso de tempo da diferenciação do CGN nas primeiras três semanas de vida pós-natal. Esses dados revelam a dinâmica do desenvolvimento de dendritos CGN subjacentes à construção de circuitos cerebelares.
Protocolo
NOTA: Todos os procedimentos foram realizados sob protocolos aprovados pelo Duke University Institutional Animal Care and Use Committee (IACUC).
1. Preparação de DNA para eletroporação in vivo ou IVE (1 dia antes da cirurgia)
- Reunir os seguintes materiais: DNA purificado (0,5-25 μg por animal), acetato de sódio 3 M, etanol, corante Fast Green, água destilada ultrapura, solução tampão fosfato (PBS) (ver Tabela de Materiais).
NOTA: Para o DNA, uma construção expressando proteína fluorescente verde (GFP) sob um promotor de ubiquitina humana foi obtida de Addgene (FUGW, https://www.addgene.org/14883/). Qualquer construto que expresse GFP ou outra proteína fluorescente sob o controle de um promotor onipresente deve funcionar. A marcação específica de CGN com esta técnica não depende do construto, mas sim da eletroporação. - Preparar o DNA para eletroporação misturando a quantidade desejada de DNA, 10% em volume de acetato de sódio 3 M e 250% em volume de etanol 100% gelado. Observe que o DNA se precipitará para fora da solução imediatamente.
- Continuar a precipitar a mistura de ADN durante a noite a -20 °C ou durante uma hora a -80 °C.
- Pellet precipitou DNA em uma centrífuga de mesa a > 16.000 × g e lavou duas vezes com etanol a 70%.
- Deixe o pellet de DNA secar completamente e reconstitua-se em uma solução PBS + 0,02% Fast Green a 1x.

Figura 1: Limitando a profundidade de injeção a 1,5 mm usando um espaçador. (A) Um segmento de 11,2 mm é cortado de uma pipeta de carregamento usando uma lâmina de barbear. (B) O espaçador é montado na ponta da seringa Hamilton (o comprimento total é de 1,27 cm ou 0,5 pol.) e fixado com adesivo ou parafilme. A ponta exposta deve ter 1,5 mm de comprimento. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
2. Eletroporação in vivo de progenitores de neurônios de grânulos em camundongos pós-natais de sete dias de idade
NOTA: Todas as cirurgias de eletroporação foram realizadas em uma sala cirúrgica estéril e altamente ventilada, e todo o pessoal usava equipamentos completos de proteção individual, incluindo luvas, máscara facial, capô de cabelo, avental e capas de sapato. Alternativamente, as cirurgias podem ser realizadas em um exaustor ventilado e estéril.
- Reúna os seguintes materiais: DNA para eletroporação, tesoura cirúrgica pequena, pinça cirúrgica pequena, seringa Hamilton personalizada, aplicador de ponta de algodão, almofada de aquecimento, betadina, etanol a 70%, PBS 1x, parafilme, adesivo tecidual (cianoacrilato de n-éster butílico), isoflurano, eletroporador e eletrodos do tipo trezer (consulte a Tabela de Materiais).
- Corte um espaçador de uma ponta de carga esterilizada para caber sobre a seringa de Hamilton para limitar a profundidade de injeção a 1,5 mm (Figura 1A,B). Prenda o espaçador com adesivo ou parafilme.
- Anestesiar o filhote P7 em uma câmara de isoflurano a uma taxa de entrega de 0,8 L/min. Confirme a anestesia completa monitorando o animal quanto à diminuição da respiração e à falta de uma resposta de beliscão do dedo do pé ou da cauda (Figura 2A).
- Uma vez que o animal esteja totalmente anestesiado, coloque os filhotes em um pedestal equipado com um cone nasal, fornecendo isoflurano constante a 4% a uma taxa de entrega de 0,8 L / min. Limpe o topo da cabeça do filhote 3 vezes com um cotonete estéril de betadina e depois 70% de etanol, alternando entre os dois, para preparar o local. Deixe a solução secar antes de prosseguir.
- Usando uma tesoura esterilizada, faça uma pequena incisão com um corte que se estenda da distância do topo até a base das orelhas para revelar o rombencéfalo (Figura 2B).
- Localize o cerebelo (Figura 2C), insira a ponta exposta da seringa de Hamilton através do crânio, perpendicular ao cérebro, e injete 1,5 μL de mistura de DNA no parênquima cerebelar empurrando lentamente o êmbolo traseiro da seringa. Após a entrega da mistura de DNA, puxe lentamente a agulha de volta para evitar derramamentos nas costas e permita que a solução de DNA se difunda por 30 s.
- Desligue o isoflurano e coloque o filhote em uma almofada de aquecimento a 37 °C. Prepare o eletrodo do tipo tweezer para eletroporação mergulhando ambas as extremidades em PBS estéril 1x.
NOTA: Molhar o eletrodo do tipo tzezer evitará queimaduras de contato na pele do filhote durante a administração dos pulsos elétricos. - Orientar o eléctrodo-ezezer acima do local de injecção com a extremidade positiva virada para baixo e a extremidade negativa acima da cabeça do animal (Figura 2D). Administre cinco pulsos elétricos do eletroporador com as seguintes configurações: intervalo entre pulsos de 50 ms, 130 V e 950 ms.
NOTA: Se necessário, execute uma injeção de teste para garantir que o local de injeção esteja localizado no vermis cerebelar (Figura 2E). - Aperte a incisão fechada e sele a ferida com um adesivo de tecido de cianoacrilato de n-butil-éster não tóxico. Limpe a ferida com etanol a 70%, pois qualquer vestígio de sangue aumenta a probabilidade de infanticídio parental e canibalismo.
- Permitir que o animal recupere numa almofada de aquecimento de 37 °C antes de devolver o filhote à mãe. Monitore o(s) filhote(s) a cada 30 minutos por pelo menos 2 h após a cirurgia para garantir a recuperação completa.
NOTA: O infanticídio por qualquer um dos pais é bastante comum. Para evitar o canibalismo , coloque o touro em uma gaiola diferente antes de iniciar a eletroporação e sempre devolva os filhotes limpos e recuperados (ou seja, sem manchas de sangue, totalmente móveis) para a gaiola original na cama original. Os filhotes também podem ser limpos com excrementos da gaiola original para minimizar o cheiro de sangue. O uso de uma barragem substituta pode ser necessário se a barragem original continuar a canibalizar seus filhotes.

Figura 2: Eletroporação cerebelar in vivo de progenitores de neurônios de grânulos em filhotes de camundongos do tipo selvagem P7. (A) Os filhotes são anestesiados com isoflurano a 4% administrado a uma taxa de 0,8L/min para garantir a anestesia durante toda a injeção da solução de DNA. O isoflurano é administrado a uma taxa de 0,8 L / min. (B) Depois de esterilizar o rato 3 vezes com betadine e etanol a 70%, é feita uma incisão que se estende pela distância das orelhas, revelando o rombencéfalo. (C) Uma imagem ampliada de uma demarcação branca no crânio, um marco para o local da injeção. O construto de ADN deve ser injectado a menos de 1 mm acima da marca; linhas pontilhadas delineiam a demarcação, e seta preta denota o local da injeção. As cristas do vermis cerebelar podem ser visíveis e podem ser úteis para encontrar o local da injeção. (D) Orientação do eletrodo do tipo pinça para eletroporação eficiente. A extremidade Plus (+) deve ser orientada para baixo para puxar o DNA carregado negativamente para o parênquima cerebelar antes da administração de pulsos elétricos. (E) A injeção de teste de 1 μL de um corante Fast Green a 0,02% mostra que a injeção está localizada no meio do vermis cerebelar entre os lóbulos 5-7. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
3. Imuno-histoquímica de CGNs eletroporados
- Recolher os seguintes materiais: isoflurano, 1x PBS, paraformaldeído a 4% (PFA), 30% de sacarose, soro de cabra normal, detergente não iónico, lâminas de vidro, lâminas de vidro, esmalte, meios de montagem, corante nuclear Hoechst e anticorpos primários e secundários adequados (ver Tabela de Materiais).
- Anestesiar o animal experimental com isoflurano e confirmar a anestesia completa com uma pitada no dedo do pé e na cauda.
- Realizar uma perfusão transcárdica injetando lentamente 1x PBS e 4% de PFA no ventrículo esquerdo do coração do animal. Deixe o sangue drenar do animal cortando a veia cava.
- Fixe o cérebro durante a noite, submergindo-o em 4% de PFA a 4 °C. No dia seguinte, lave rapidamente o cérebro com 1x PBS e transfira o cérebro para 30% de sacarose em 1x PBS para crioproteção por pelo menos 24 h.
- Se necessário, corte o cérebro ao meio ao longo do eixo rostral-caudal e confirme a expressão da construção do repórter transfectado usando um microscópio de dissecação fluorescente vertical.
NOTA: Mantenha o cérebro submerso em 1x PBS em um prato pequeno para evitar que ele seque. - Monte o cérebro em um micrótomo congelante, corte seções sagitais de 25 μm e permita que as seções se desdobrem em uma mistura 1:1 de 1x PBS e glicerol.
NOTA: As secções podem ser armazenadas nesta solução crioprotetora a -20 °C para armazenamento a longo prazo. - Lave as seções três vezes em 1x PBS por 10 min cada para remover o crioprotetor e bloqueie o tecido em 1x PBS + 10% de soro de cabra normal + 0,2% de detergente não iônico em um agitador orbital à temperatura ambiente por 1 h.
- Preparar solução primária de anticorpos: 1x PBS, soro de cabra 10% normal, detergente não iônico a 0,2% e anticorpo anti-GFP e centrifugar a solução por 5 min a >16.000 × g. Incubar secções na solução de anticorpos a 4 °C num agitador orbital durante 48 h.
- Lave a solução de anticorpos primários por 15 min cinco vezes com 1x PBS + detergente não iônico a 0,2%.
- Preparar solução de anticorpos secundários: 1x PBS, soro de cabra 10% normal, detergente não iônico a 0,2% e um anticorpo secundário apropriado para detectar GFP; centrifugar a solução a >16.000 × g. Incubar seções na solução de anticorpos em um agitador orbital à temperatura ambiente por 2-3 h. Proteja as seções da exposição à luz para evitar o branqueamento.
- Lave a solução de anticorpos secundários três vezes com 1x PBS + detergente não iônico a 0,2% por 15 minutos de cada vez. Incubar secções em 1x PBS + Hoechst durante 5 min para corar núcleos.
- Lave a solução Hoechst com 1x PBS + detergente não iónico a 0,2% e monte em lâminas de vidro. Cubra as seções com meios de montagem, cubra as lâminas e sele a lâmina com esmalte para evitar a evaporação.
4. Análises morfológicas de CGNs - reconstrução tridimensional (3D) e área de superfície e volume celular
- Imagem de CGNs eletroporados únicos em um microscópio confocal a 63x objetiva com zoom de 2x, tirando imagens z-stack a 0,5 μm por pilha. Imagem de uma célula por janela de imagem para permitir uma fácil análise e reconstrução da imagem.
- Instale o plug-in Simple Neurite Tracer para FIJI usando o seguinte link (https://imagej.net/Simple_Neurite_Tracer:_Basic_Instructions) para rastrear de forma fácil e eficiente a estrutura de CGNs eletroporados, no espaço tridimensional (3D).
Observação : há uma versão atualizada do plug-in (https://imagej.net/SNT). - Analise o comprimento da neurite e a formação de garras dendríticas de forma cega usando o Simple Neurite Tracer. Carregue imagens z-stack de canal único de CGNs eletroporados em FIJI e clique em Plugins | Segmentação | Traçador de neurite simples (Figura 3D).
- Acesse o menu suspenso e selecione Create New 3D Viewer (Figura 3D).
- Role até a base de um dendrito, onde ele se conecta ao soma da célula e inicie um caminho clicando na junção. Trace manualmente o caminho clicando nas seções onde o sinal de preenchimento de célula é mais brilhante, pressionando [y] para manter o rastreamento. Trace até o final do dendrito se ele não contiver uma garra ou até a base da garra e confirme o caminho pressionando [f] (Figura 4D).
- Em seguida, trace a garra iniciando um caminho na base da estrutura e traçando até o final da neurita mais longa. Rastreie ramificações secundárias e terciárias mantendo pressionada a tecla [ctrl] no Windows ou [alt] em um Mac OS e clicando no caminho. Confirme o caminho pressionando [f].
- Observe que as medições para os vestígios são visíveis em uma janela separada; somar todas as medidas dos ramos das garras (primária, secundária, terciária) para obter o comprimento total de cada garra.
- Para analisar a área de superfície e o volume celular de CGNs eletroporosos, baixe o software de análise celular Imaris (https://imaris.oxinst.com/).
NOTA: FIJI também pode ser usado para reconstruir células em 3D a partir de imagens z-stack usando plug-ins prontamente disponíveis e gratuitos. Além disso, há um recurso de renderização volumétrica no Simple Neurite Trator, mas o Imaris foi usado pelas razões descritas abaixo. - Carregue a imagem z-stack de um CGN eletroporado no Imaris. Acesse o kit de ferramentas de reconstrução 3D pressionando Surpass.
- Para reconstruir o CGN, pressione Superfícies e selecione uma região de interesse que englobe a totalidade da célula dentro da janela de imagem. Quando terminar, pressione a seta azul para a frente no canto inferior direito em Criar.
- Se a imagem contiver vários canais para sinais diferentes, selecione o canal que contém o CGN eletroporado e pressione a seta azul para a frente.
- Usando a barra deslizante, defina um limite desejado que se ajuste com mais precisão ao sinal da célula eletroporosa. Amplie mais perto da superfície da célula para determinar com precisão o limite. Uma vez terminado, pressione a seta verde dupla para reconstruir a célula e obter a área de superfície e o tamanho do volume a partir dos metadados.

Figura 3: Análise imuno-histoquímica e reconstrução tridimensional de neurônios de grânulos eletroporosos. Camundongos P7 CD-1 foram eletroporados, com um construto expressando GFP. Os cérebros foram coletados e submetidos à imuno-histoquímica, microscopia confocal e reconstrução 3D para análise morfológica. (A) Linha do tempo desde a eletroporação até o processamento de imagem de um mouse de 10 DPI. (B) Imagem de projeção máxima de uma seção transversal sagital do cerebelo eletroporoso 10-DPI; linhas brancas demarcam camadas cerebelares, e a barra de escala é de 25 μm. (C) Imagem de projeção máxima de um único neurônio de grânulo eletroporoso 10-DPI e o traço 3D correspondente, barra de escala é de 10 μm. (D) As reconstruções 3D foram geradas usando o plugin FIJI Simple Neurite Tracer. Todas as medidas foram rastreadas através da pilha z, seguindo o sinal de enchimento celular. As medidas do eixo e da garra foram traçadas separadamente para cada dendrito; linha pontilhada denota porção de dendrito dentro do plano atual. Abreviaturas: 3D = tridimensional; GFP = proteína verde fluorescente; DPI = dias após a injeção; PSD-95 = proteína de densidade pós-sináptica 95; PNB = progenitores de neurónios de grânulos; PFA = paraformaldeído. Por favor, clique aqui para ver uma versão maior desta figura.
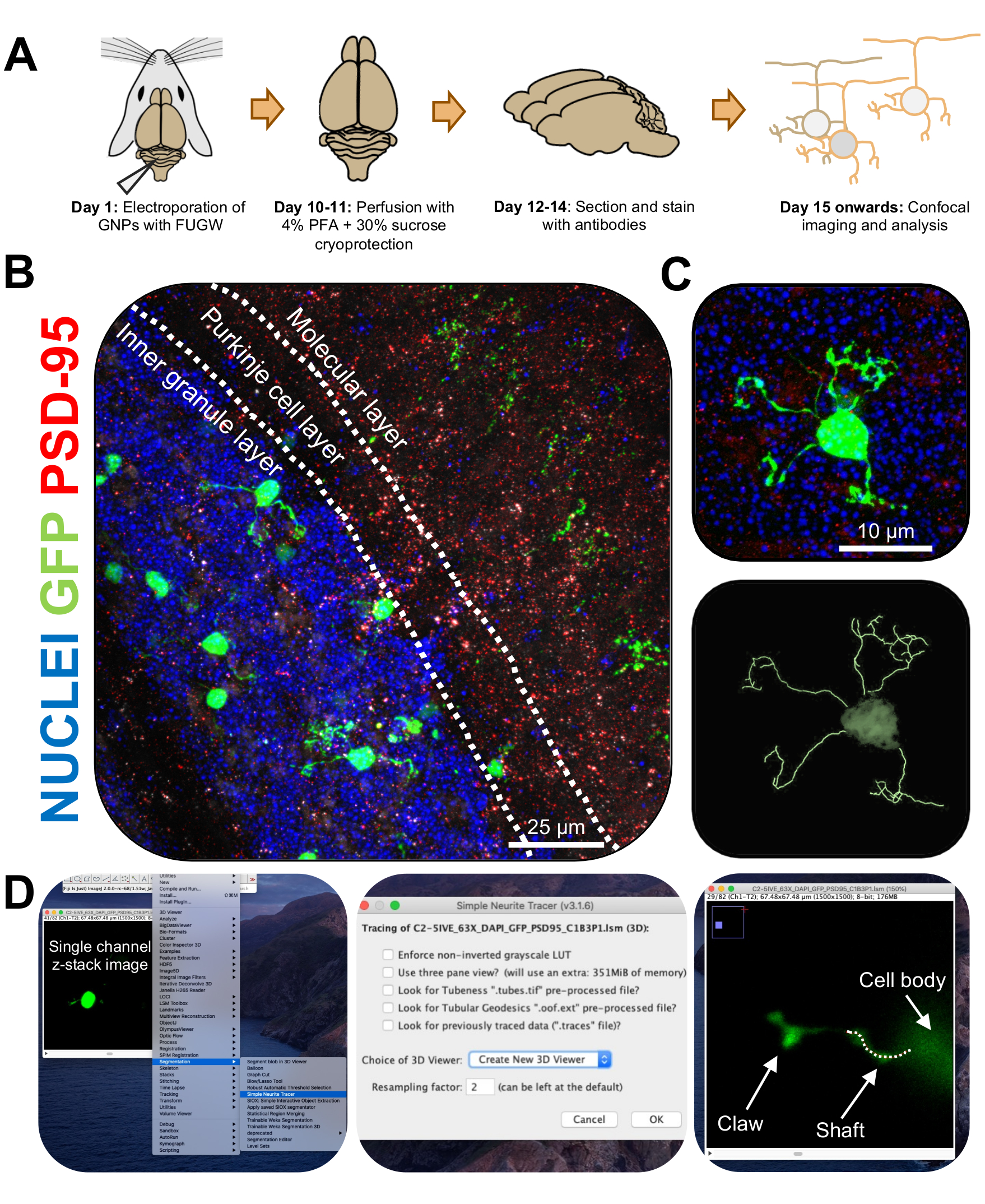{kind=link}
Resultados

Figura 4: Análise da morfologia do neurônio granulado durante o desenvolvimento cerebelar. (A) Imagens de projeção máxima de CGNs eletroporados, de 3-DPI a 14-DPI (idade pós-natal P10 a P21), núcleos (azul) e GFP (verde); as pontas das setas indicam dendrito individual, e a barra de escala é de 10 μm. (B) Número médio de dendritos. (C
Discussão
Os neurônios dos grânulos cerebelares são os neurônios mais abundantes no cérebro dos mamíferos, representando quase 60-70% da população total de neurônios no cérebro de roedores 1,14. O cerebelo tem sido amplamente utilizado para elucidar mecanismos de proliferação celular, migração, formação de dendritos e desenvolvimento de sinapses 6,9,10,11,15,16,17,18,19,20
Divulgações
Os autores declaram não haver conflitos de interesse.
Agradecimentos
O trabalho foi apoiado pelos subsídios do NIH R01NS098804 (A.E.W.), F31NS113394 (U.C.) e Programa de Neurociência de Verão da Duke University (D.G.).
Materiais
| Name | Company | Catalog Number | Comments |
| Betadine | Purdue Production | 67618-150-17 | |
| Cemented 10 µL needle | Hamilton | 1701SN (80008) | 33 gauge, 1.27 cm (0.5 in), 4 point style |
| Chicken anti-GFP | Millipore Sigma | AB16901 | Our lab uses this antibody at a 1:1000 concentration |
| Cotton-tip applicator | |||
| Donkey anti-chicken Cy2 | Jackson ImmunoResearch | 703-225-155 | Our lab uses this antibody at a 1:500 concentration |
| Ethanol (200 proof) | Koptec | V1016 | |
| Electroporator ECM 830 | BTX Harvard Apparatus | 45-0052 | |
| Fast Green FCF | Sigma | F7252-5G | |
| FUGW plasmid | Addgene | 14883 | |
| Glass slides | VWR | 48311-703 | Superfrost plus |
| Glycerol | Sigma-Aldrich | G5516 | |
| Heating pad | Softheat | ||
| Hoescht 33342 fluorescent dye | Invitrogen | 62249 | |
| Imaris | Bitplane | ||
| Isoflurane | Patterson Veterinary | 07-893-1389 | |
| Micro cover glass | VWR | 48382-138 | |
| Nail polish | Sally Hansen | Color 109 | |
| Normal goat serum | Gibco | 16210064 | |
| O.C.T. embedding compound | Tissue-Tek | 4583 | |
| Olympus MVX10 Dissecting Scope | Olympus | MVX10 | |
| P200 pipette reach tip | Fisherbrand | 02-707-138 | Used for needle spacer |
| Parafilm | Bemis | PM-996 | |
| PBS pH 7.4 (10x) | Gibco | 70011-044 | |
| Simple Neurite Tracer | FIJI | https://imagej.net/Simple_Neurite_Tracer:_Basic_ Instructions | |
| Sucrose | Sigma | S0389 | |
| Surgical tools | RWD Life Science | Small scissors and tweezers | |
| Triton X-100 | Roche | 11332481001 | non-ionic detergent |
| Tweezertrodes | BTX Harvard Apparatus | 45-0489 | 5 mm, platinum plated tweezer-type electrodes |
| Ultrapure distilled water | Invitrogen | 10977-015 | |
| Vectashield mounting media | Vectashield | H1000 | |
| Vetbond tissue adhesive | 3M | 1469SB | |
| Zeiss 780 Upright Confocal | Zeiss | 780 |
Referências
- Altman, J., Bayer, S. A. . Development of the cerebellar system : in relation to its evolution, structure, and functions. , (1997).
- Rahimi-Balaei, M., Bergen, H., Kong, J., Marzban, H. Neuronal migration during development of the cerebellum. Frontiers in Cellular Neuroscience. 12, 484 (2018).
- Alder, J., Cho, N. K., Hatten, M. E. Embryonic precursor cells from the rhombic lip are specified to a cerebellar granule neuron identity. Neuron. 17 (3), 389-399 (1996).
- Hatten, M. E., Heintz, N. Mechanisms of neural patterning and specification in the developing cerebellum. Annual Review of Neuroscience. 18, 385-408 (1995).
- Ben-Arie, N., et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 390 (6656), 169-172 (1997).
- Borghesani, P. R., et al. BDNF stimulates migration of cerebellar granule cells. Development. 129 (6), 1435-1442 (2002).
- Espinosa, J. S., Luo, L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. Journal of Neuroscience. 28 (10), 2301-2312 (2008).
- Markwalter, K. H., Yang, Y., Holy, T. E., Bonni, A. Sensorimotor coding of vermal granule neurons in the developing mammalian cerebellum. Journal of Neuroscience. 39 (34), 6626-6643 (2019).
- Shalizi, A., et al. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. Journal of Neuroscience. 27 (37), 10037-10046 (2007).
- Shalizi, A., et al. A Calcium-regulated MEF2 sumoylation switch controls poststynaptic differentiation. Science. 311 (5763), 1012-1017 (2006).
- Konishi, Y., Stegmuller, J., Matsuda, T., Bonni, S., Bonni, A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 303 (5660), 1026-1030 (2004).
- Holubowska, A., Mukherjee, C., Vadhvani, M., Stegmuller, J. Genetic manipulation of cerebellar granule neurons in vitro and in vivo to study neuronal morphology and migration. Journal of Visualized Experiments: JoVE. (85), e51070 (2014).
- Yang, Y., et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 353 (6296), 300-305 (2016).
- Herculano-Houzel, S. Coordinated scaling of cortical and cerebellar numbers of neurons. Frontiers in Neuroanatomy. 4, 12 (2010).
- Wilson, P. M., Fryer, R. H., Fang, Y., Hatten, M. E. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. Journal of Neuroscience. 30 (25), 8529-8540 (2010).
- Kokubo, M., et al. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. Journal of Neuroscience. 29 (28), 8901-8913 (2009).
- Schwartz, P. M., Borghesani, P. R., Levy, R. L., Pomeroy, S. L., Segal, R. A. Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron. 19 (2), 269-281 (1997).
- Segal, R. A., Pomeroy, S. L., Stiles, C. D. Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. Journal of Neuroscience. 15 (7), 4970-4981 (1995).
- Zhou, P., et al. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 55 (1), 53-68 (2007).
- Dhar, M., Hantman, A. W., Nishiyama, H. Developmental pattern and structural factors of dendritic survival in cerebellar granule cells in vivo. Scientific Reports. 8 (1), 17561 (2018).
- Ito, M. Synaptic plasticity in the cerebellar cortex and its role in motor learning. Canadian Journal of Neurological Sciences. 20, 70-74 (1993).
- Jorntell, H., Hansel, C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 52 (2), 227-238 (2006).
- Nakanishi, S. Genetic manipulation study of information processing in the cerebellum. Neuroscience. 162 (3), 723-731 (2009).
- Chang, C. H., et al. Atoh1 controls primary cilia formation to allow for SHH-triggered granule neuron progenitor proliferation. Developmental Cell. 48 (2), 184-199 (2019).
Erratum
Formal Correction: Erratum: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development
Posted by JoVE Editors on 4/06/2023. Citeable Link.
An erratum was issued for: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development. A figure was updated.
Figure 2 was updated from:
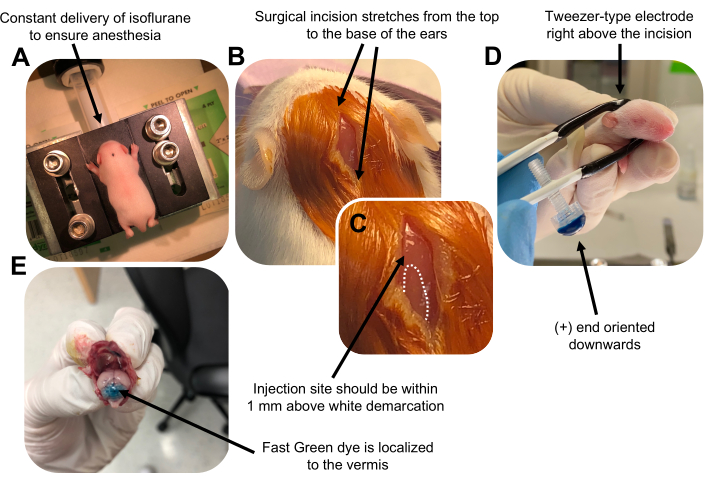
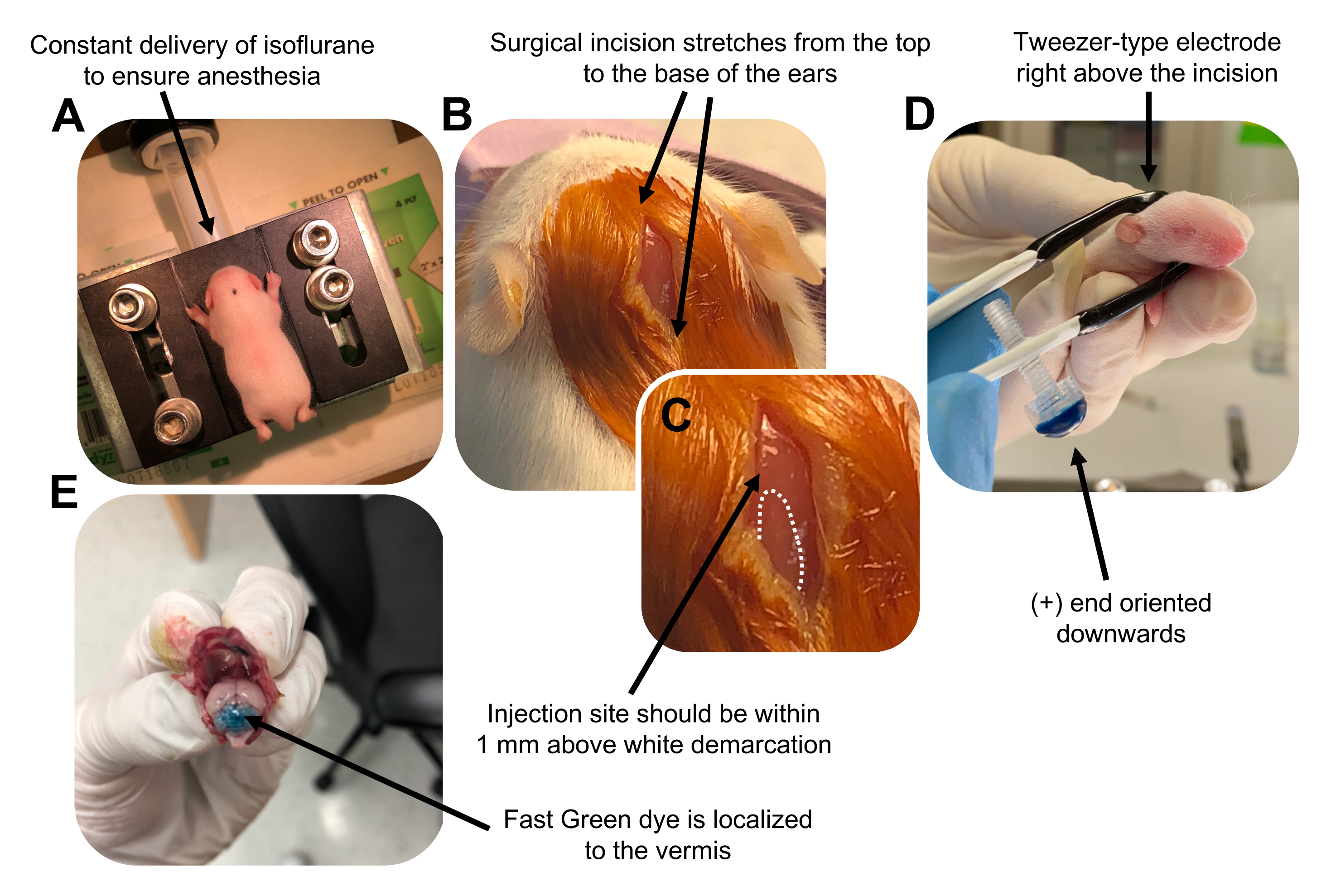
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
{kind=link}
to:
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados