
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Ablações de volume profunda e espacialmente controladas usando um microscópio de dois fótons no Gás de Peixe-Zebra
Neste Artigo
Resumo
O desenvolvimento embrionário requer coordenação em larga escala do movimento celular. A ablação a laser mediada por dois fótons permite a ablação espacialmente controlada em três dimensões de grandes grupos de células profundas. Além disso, essa técnica pode sondar a reação de células migratórias coletivamente in vivo a perturbações em seu ambiente mecânico.
Resumo
A morfogênese envolve muitos movimentos celulares para organizar células em tecidos e órgãos. Para o desenvolvimento adequado, todos esses movimentos precisam ser rigorosamente coordenados, e acumular evidências sugere que isso é alcançado, pelo menos em parte, através de interações mecânicas. Testar isso no embrião requer perturbações físicas diretas. As ablações a laser são uma opção cada vez mais utilizada que permite aliviar restrições mecânicas ou isolar fisicamente duas populações celulares uma da outra. No entanto, muitas ablações são realizadas com um laser ultravioleta (UV), que oferece resolução axial limitada e penetração tecidual. Um método é descrito aqui para ablto de volumes profundos, significativos e espacialmente bem definidos usando um microscópio de dois fótons. As ablações são demonstradas em uma linha transgênica de zebrafish expressando a proteína fluorescente verde no mesendoderme axial e usadas para cortar o mesendoderme axial sem afetar o ectoderme sobreceptório ou a célula de gema subjacente. O comportamento celular é monitorado por imagens ao vivo antes e depois da ablação. O protocolo de ablação pode ser usado em diferentes estágios de desenvolvimento, em qualquer tipo de célula ou tecido, em escalas que variam de alguns mícrons a mais de cem mícrons.
Introdução
As interações célula-célula desempenham papéis vitais no desenvolvimento. As células fornecem sinais que seus vizinhos diretos, ou células mais distantes, podem perceber, influenciando assim seu destino e/ou comportamento. Muitos desses sinais são de natureza química. Por exemplo, nos eventos de indução bem caracterizados, um grupo celular produz moléculas difusíveis que afetam o destino de outra população celular1. Outros sinais, no entanto, são mecânicos; as células exercem forças e restrições sobre seus vizinhos, que os vizinhos percebem e respondem a2.
Uma forma de estudar a importância dessas interações célula-célula in vivo é eliminar algumas células e observar o desenvolvimento subsequente. Infelizmente, as técnicas disponíveis para remover ou destruir células são limitadas. As células podem ser removidas cirurgicamente3,4, usando agulhas ou pequenos fios, mas tais tratamentos são invasivos, não muito precisos, e geralmente realizados sob um estereoscópio, impedindo imagens imediatas sob um microscópio. Além disso, mirar células profundas implica perfurar um buraco em tecidos sobrelvando, criando perturbações indesejadas. Fotosensibilizadores geneticamente codificados, como KillerRed, têm sido usados para induzir a morte celular através da iluminação 5. Fotosensitizers são cromóforos que geram espécies reativas de oxigênio após a irradiação da luz. Sua principal limitação é que eles requerem longas iluminação luminosas (cerca de 15 minutos), o que pode ser difícil de alcançar se as células estão se movendo, e que induzem a morte celular através da apoptose, o que não é imediato.
Finalmente, as ablações a laser foram desenvolvidas e amplamente utilizadas nos últimos 15 anos6,7,8,9,10,11,12. Um raio laser é focado na célula/tecido alvo. Induz sua ablação através do aquecimento, fotoblação ou ablação induzida pelo plasma; o processo envolvido depende da densidade de energia e do tempo de exposição13. A maioria dos protocolos de ablação usam lasers UV para sua alta energia. No entanto, a luz UV é absorvida e espalhada por tecidos biológicos. Assim, mirar células profundas requer uma alta potência laser, que então induz danos em tecidos mais superficiais e fora do avião. Isso limita o uso de lasers UV para estruturas superficiais e explica sua resolução axial relativamente baixa. A óptica não linear (a chamada microscopia de dois fótons) usa propriedades não lineares de luz para excitar um fluoróforo com dois fótons de aproximadamente metade da energia no domínio infravermelho. Quando aplicado a ablações, isso tem três principais vantagens. Primeiro, a luz infravermelha é menos dispersa e menos absorvida do que a luz UV por tecidos biológicos14, permitindo alcançar estruturas mais profundas sem aumentar a potência laser necessária. Em segundo lugar, o uso de um laser pulsado femtosegundo fornece densidades de energia muito altas, criando uma ablação através da indução plasmática, que, ao contrário do aquecimento, não difunde espacialmente15. Em terceiro lugar, a densidade de energia induzindo a formação plasmática é alcançada apenas no ponto focal. Graças a essas propriedades, as ablações a laser de dois fótons podem ser usadas para atingir precisamente células profundas sem afetar o ambiente tecidual circundante.
As migrações coletivas são um excelente exemplo de processos de desenvolvimento nos quais as interações célula-células são fundamentais. As migrações coletivas são definidas como migrações celulares nas quais as células vizinhas influenciam o comportamento de uma célula16. A natureza dessas interações (químicas ou mecânicas) e como elas afetam a migração celular podem variar muito e muitas vezes não são totalmente compreendidas. A capacidade de remover células e observar como isso afeta os outros é fundamental para desvendar ainda mais esses processos coletivos. Há alguns anos, estabelecemos — usando abordagens cirúrgicas — que a migração do polster durante a gastrusão de zebrafish é uma migração coletiva17. O polster é um grupo de células que constitui as primeiras células internalizadoras no lado dorsal do embrião18. Essas células, rotuladas em verde na linha transgênica Tg(gsc:GFP), estão localizadas profundamente no embrião, abaixo de várias camadas de células epiblastos. Durante a gastruação, esse grupo lidera a extensão do mesoderme axial, migrando do organizador embrionário para o polo animal19,20,21,22,23 (Figura 1A). Estabelecemos que as células precisam de contato com seus vizinhos para orientar sua migração na direção do polo animal. No entanto, entender melhor as bases celulares e moleculares dessa migração coletiva envolve a remoção de algumas células para ver como isso influencia as demais. Nós, portanto, desenvolvemos ablações de grandes e profundos volumes usando uma configuração de microscopia de dois fótons. Aqui, demonstramos o uso deste protocolo para cortar o polster em seu meio e observar as consequências na migração celular rastreando núcleos rotulados com Histone2B-mCherry.
Protocolo
Todo o trabalho animal foi aprovado pelo Comitê de Ética n 59 e pela Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche sob o número de arquivo APAFIS#15859-2018051710341011v3. Algumas das etapas descritas abaixo são específicas para nossos equipamentos e software, mas podem ser facilmente adaptadas a diferentes equipamentos.
1. Preparação para injeção
- Prepare 75 mL de solução de 1% de agarose em Embrião Médio (EM).
- Coloque o molde de injeção em uma placa de Petri de 90 mm e despeje aproximadamente 50 mL de agarose, o suficiente para o molde flutuar. Deixe a agarose solidificar e remover o molde de injeção.
- Prepare uma placa revestida de agarose derramando 1 mL de agarose em uma placa de Petri de 30 mm.
- Prepare 4 μL de 30 ng/μL Solução de mRNA Histone2B-mCherry diluindo a solução de estoque em água sem RNase e mantenha no gelo.
NOTA: Tome cuidado para usar luvas enquanto manipula mRNA para evitar a degradação mediada pelo RNase. - Puxe uma agulha de injeção de um capilar usando o puxador de micropipette.
2. Preparação do embrião
- Uma vez que os peixes tenham colocado ovos, colete, enxágue e colhe em uma placa de Petri de 90 mm em EM. Coloque os embriões em uma incubadora de 28,5 °C.
- Espere 20 min para que a primeira célula se torne visível.
- Transfira 30 embriões para a placa de injeção preenchida com EM. Esprema os embriões nas ranhuras usando fórceps ligeiramente contundentes e oriente-os com o polo animal para cima.
- Usando uma ponta de microcarregador, encha uma agulha de injeção com 2 μL de solução mRNA. Insira a agulha no suporte capilar colocado em um micromedetor conectado com tubos de politetrafluoroetileno (PTFE) a um injetor de ar.
- Sob o estereótipo, quebre cuidadosamente a ponta da agulha.
- Injete a solução mRNA nos embriões do estágio de 1 célula inserindo a agulha na célula.
NOTA: O volume injetado é de aproximadamente um terço do volume da célula. - Coloque de volta embriões injetados na incubadora de 28,5 °C.
3. Preparação do microscópio de dois fótons
NOTA: Dois lasers são usados neste protocolo. Um deles é usado para imagem GFP (a 920 nm) e realizar ablations (a 820 nm). Será referido como o laser verde/ablação. O outro é usado a 1160 nm para imagem mCherry. Será referido como o laser vermelho.
- Defina o laser verde/ablação para 820 nm (comprimento de onda de ablação) e o laser vermelho para 1160 nm (excitação mCherry).
- Usando espelhos móveis no caminho óptico, alinhe raios verdes/ablação e laser vermelhos tanto na entrada quanto na saída da cabeça de varredura.
NOTA: Isso aumenta o foco do feixe de laser e minimiza o volume focal para excitação e ablação. - Meça a potência máxima do laser verde/ablação a 820 nm sob o objetivo. Para isso, coloque o medidor de energia sob o objetivo, feche a câmara preta, coloque a potência laser verde/ablação em 100%, e abra as persianas. Calcule a porcentagem de energia laser necessária para atingir 300 mW.
- Recue o laser verde/ablação para 920 nm (excitação GFP) e defina a potência laser para 7%. Defina a potência do laser vermelho para 15%.
- Ativar detectores de tubos epi-fotomultiplier (PMT) para linhas verdes e vermelhas; definir sensibilidade PMT linha verde e vermelha para 65.
- Defina o campo de visão para 400 x 400 μm, resolução de imagem para 512 x 512 pixels e frequência de digitalização para 800 Hz.
- Selecione o modo de imagem timelapse 3D . Em seguida, crie uma pasta e ative o Autosave para obter dados após cada aquisição.
- Monte a câmara de aquecimento e coloque-a a 28 °C. Espere pelo menos 10 min para a câmara e o objetivo de aquecer.
4. Montagem do embrião
- Sob um estereómico fluorescência, identifique embriões a 70% epiboly que expressam GFP.
NOTA: Selecione embriões com um sinal brilhante no mesoderme axial e sem fluorescência de fundo para melhor qualidade de imagem. - Transfira três para quatro embriões selecionados no prato revestido de agarose (etapa 1.3) usando uma pipeta pasteur de plástico e cuidadosamente descorra-los usando fórceps finos.
NOTA: Embriões desobriados são muito delicados e vão estourar após o contato com ar ou plástico. - Despeje 1 mL de 0,2% de agarose em 1x penicilina-estreptomicina EM em um pequeno frasco de vidro. Coloque o frasco em um aquecedor de bloco seco pré-aquecido de 42 °C.
NOTA: As seguintes etapas devem ser executadas rapidamente para permitir a orientação do embrião antes dos conjuntos de agarose. - Transfira um embrião descorioado no frasco de vidro 0,2% agarose usando uma pipeta de vidro polida com fogo. Tome cuidado para não adicionar muito EM na agarose para evitar diluí-lo. Descarte o EM restante da pipeta e aspire o embrião de volta junto com agarose suficiente para cobrir o slide do prato de fundo de vidro antes que o embrião caia da pipeta.
- Soprar a agarose eo embrião no escorregador de vidro do prato. Tome cuidado para não deixar o embrião tocar o ar ou o lado plástico do prato. Em seguida, encha a câmara ao redor do deslizamento de vidro com agarose.
- Use um cílio para orientar o embrião para que a região alvo esteja no topo (Figura 1B).
NOTA: Ao orientar embriões, tome cuidado para tocar apenas o blastoderm, não a gema muito frágil. Agarose vai definir em torno de 1 min, dependendo da temperatura ambiente. - Aguarde ~5 min para que a agarose se estabilize completamente e, em seguida, adicione algumas gotas de penicilina-estreptomicina EM.
5. Localização do embrião e imagem de pré-ablação
- Coloque o prato de fundo de vidro sob o objetivo na câmara aquecida. Mergulhe o objetivo em penicilina-estreptomicina EM e feche a câmara aquecida.
- Mova o controle deslizante para definir o caminho da luz para oculares. Em seguida, usando oculares, lâmpadas fluorescentes e controle de palco, encontre um embrião e coloque o foco na superfície do embrião.
- Desligue a lâmpada de fluorescência, afina o caminho da luz para os PMTs e feche a câmara preta.
NOTA: Tenha cuidado para desligar todas as fontes de luz na câmara preta, pois pode danificar os PMTs. - Inicie imagens ao vivo e localize o mesoderme axial. Ajuste os poderes verde/ablação e laser vermelho para ter um bom sinal (ou seja, entre 1.000 e 20.000 fótons por pixel para áreas de expressão de GFP). Use o canal vermelho para mover o palco para a parte superior do embrião e definir esta posição como Z = 0.
- Escolha um passo de 1 minuto e um passo Z de 2 μm. Um Z-course de 110 μm é suficiente para abranger todo o polster e é adquirido em menos de 1 min com essas configurações. Coloque a primeira fatia 15 μm acima do mesoderme axial (no ectoderme mais superficial).
NOTA: O polster se move ao longo de uma linha curva de modo que a fatia inferior da pilha Z deve ser definida 30 μm mais profunda do que a posição mais profunda do polster para acomodar seu movimento durante a imagem de lapso de tempo (Figura 1E). - Grave 10-15 minutos de filme de pré-ablação.

Figura 1: Resultado bem sucedido de ablações a laser. (A) Esquema de um embrião gastrulating a 70% epiboly na visão dorsal; pAM: mesoderme axial posterior; a seta preta marca a direção da migração de polster; quadrado preto indica um campo de visão típico para ablações no polster. (B) Esquema de montagem de embrião para corte de polster. Vista lateral. O embrião é montado de tal forma que o plano do polster é perpendicular ao eixo óptico. (C) sobrevivência e (D) morfologia de controle e embriões ablados a 24 h pós-fertilização. A barra de escala é de 300 μm. (E) Sequência de tempo da ablação a laser no polster de um embrião Tg(gsc:GFP) expressando Histone2B-mCherry. As visualizações com o canal verde são apenas projeções máximas. O close-up exibe a área ablada contendo detritos celulares. As vistas com canais verde e vermelho (exibidos como magenta) são fatias XY e XZ antes e depois da ablação (o raio verde representa ablação). As fatias XZ mostram que os tecidos sobreladas (núcleos magenta sem expressão GFP) não foram afetados pela ablação das estruturas subjacentes. A caixa amarela dashed corresponde ao ROI selecionado para tratamento de ablação a laser. A barra de escala é de 50 μm em vistas grandes e 25 μm no close-up. Clique aqui para ver uma versão maior desta figura.
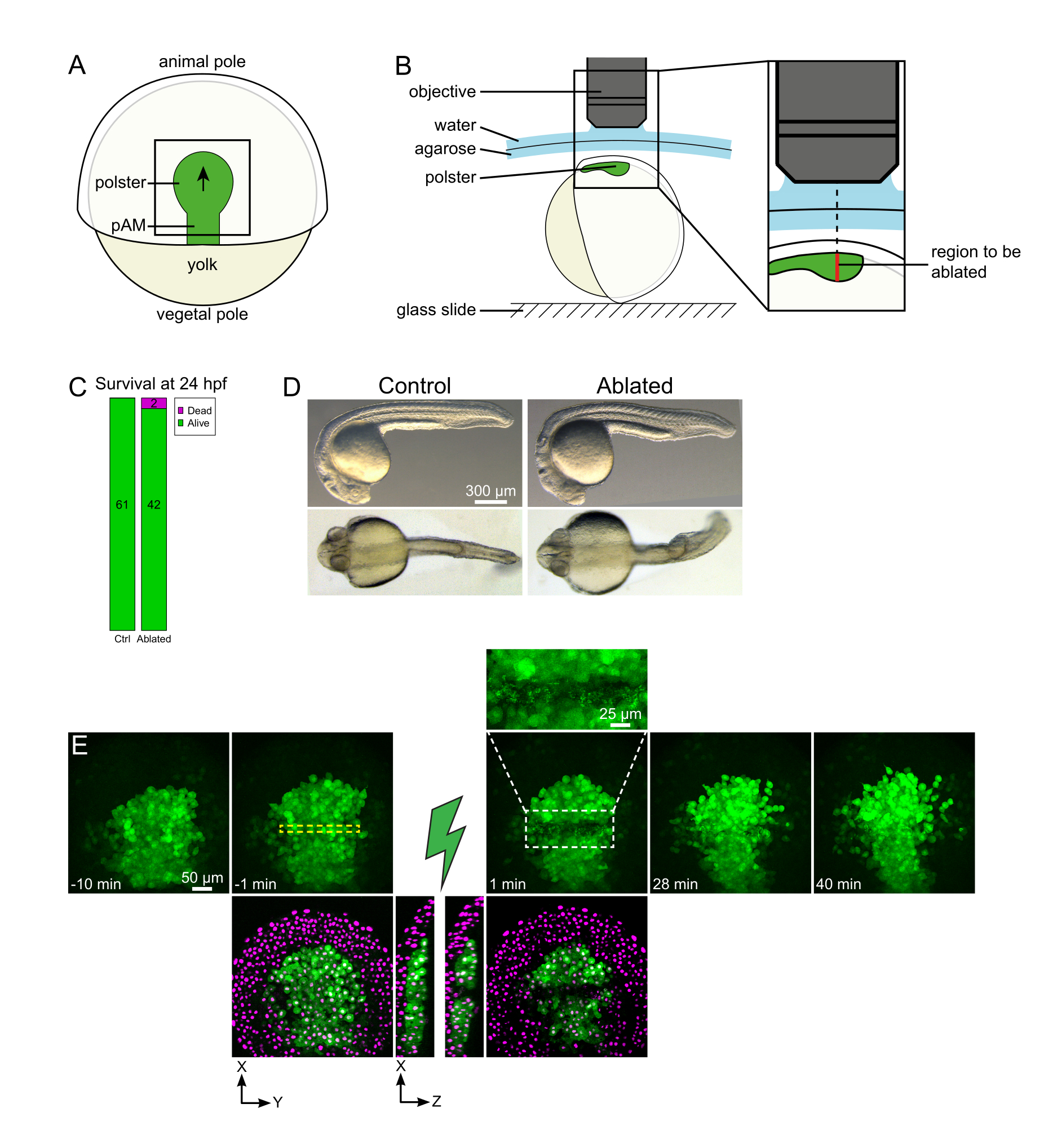{kind=link}
6. Localização do alvo e ablação a laser
- Localize o contorno polster em imagens ao vivo e, usando a ferramenta EOM Modulador Região de Interesse (EOM ROI) eletro-óptica, desenhe um retângulo de 20 pixels (15 μm) de grande porte que abrange a largura do polster. Coloque este retângulo no meio do polster (Figura 1E).
- Observe a posição axial dos planos mais altos e mais baixos contendo células polster. As jagas serão realizadas a cada 10 μm entre esses dois aviões. Tome cuidado para que o ROI não sobreponha a célula da gema em nenhum desses planos.
- Coloque o estágio na posição Z mais baixa do intervalo. As jagas devem ser realizadas de baixo para cima, pois os detritos absorvem a luz.
- Defina o comprimento de onda laser verde/ablação para 820 nm e defina a Porcentagem de Potência para obter uma potência de saída de 300 mW (passo 3.3).
- Defina a Frequência de Imagem para 200 Hz.
- Defina eOM de imagem a laser verde/ablação para 0 e selecione o modo ROI-Treat .
- Ligue o EOM e defina o tratamento para iniciar imediatamente (após 0 quadro).
- Defina o modo de imagem como timelapse e desativar autosave.
- Defina o passo de tempo para o modo rápido.
- Defina o número de quadros de tratamento e o número de quadros ao valor correspondente à profundidade alvo (Tabela 1).
| Profundidade (μm) | Quadros de tratamento |
| -30 | 1 |
| -35 | 1-2 |
| -40 | 1-2 |
| -45 | 2 |
| -50 | 2-3 |
| -55 | 3 |
| -60 | 3-4 |
| -65 | 4 |
| -70 | 4 |
| -75 | 4-5 |
| -80 | 4-5 |
| -85 | 5 |
| -90 | 5 |
| -95 | 5-6 |
| -100 | 6 |
| -105 | 6 |
Tabela 1: Número sugerido de quadros de tratamento a laser em função da profundidade celular direcionada no embrião (0 sendo a superfície do embrião).
- Comece a imagem. A aquisição é preta à medida que o obturador de PMT fecha durante o tratamento EOM.
- Subir o palco para a próxima posição Z da lista (etapa 6.2).
- Repetir as etapas 6.10 a 6.12 até que o topo do polster seja atingido.
7. Verificação pós-ablação e imagem
- Defina o laser verde/ablação para 920 nm e 5% de potência. Defina o EOM de imagem a laser verde/ablação para 100 e selecione o modo Fullfield .
- Defina a frequência de imagem para 800 Hz. Desligue o EOM.
- Passe por toda a pilha no modo ao vivo para verificar se cada avião foi incendiado. Se não for esse o caso, volte ao passo 6.2.
NOTA: A ablação às vezes induz uma mudança vertical de tecidos vizinhos para que a pilha Z possa ter que ser redefinida. - Defina o modo de imagem como timelapse 3D e reativar o Autosave. Recorde de 40-60 min de filme pós-ablação.
- Verifique, no filme pós-ablação, se as células alvo foram efetivamente abladas. A recuperação da fluorescência, ou células-alvo ocupando espaço e impedindo que as células seguidoras se movam, indicam que as células-alvo foram apenas fotobleachadas e não abladas (Figura 1E e Figura 2A).

Figura 2: Resultados negativos de ablações a laser. (A) Exemplos típicos de possíveis falhas na ablação a laser. Grandes vistas XY são projeções máximas, a vista XZ é uma seção reconstruída. A área tratada a laser está localizada entre as duas pontas de flechabra. Três planos focais são destacados na seção reconstruída e exibidos à direita. Eles correspondem a três tipos diferentes de falhas. O plano 1 mostra que as células acima do polster foram abladas. Isso pode ser identificado pela presença de detritos autofluorescentes neste plano focal (ver de perto) acima do polster (ver posição do plano 1 na seção reconstruída). Isso provavelmente resulta de uma definição incorreta da região a ser ablada. O plano 2 mostra células que foram branqueadas, mas não abladas. Eles podem ser identificados como o sinal de baixa fluorescência ainda revela contornos celulares intactos (ver close-up). O plano 3 exibe células intactas, que dificilmente foram branqueadas pelo tratamento a laser. Isso pode resultar de uma definição incorreta da região a ser ablada ou de mau tratamento. Nas situações retratadas nos planos 2 e 3, é possível reaplicar o tratamento de ablação às células-alvo não abladas. A barra de escala é de 50 μm em grandes visualizações e 20 μm em close-ups. (B) Um exemplo típico de bolhas (marcadas por asteriscos brancos) formados por cavitação por causa de um tratamento a laser muito intenso. Tais bolhas não se limitam a um plano Z, às vezes até mesmo abrangendo toda a altura do polster, deformando tecidos vizinhos. A barra de escala é de 50 μm. Por favor, clique aqui para ver uma versão maior desta figura.
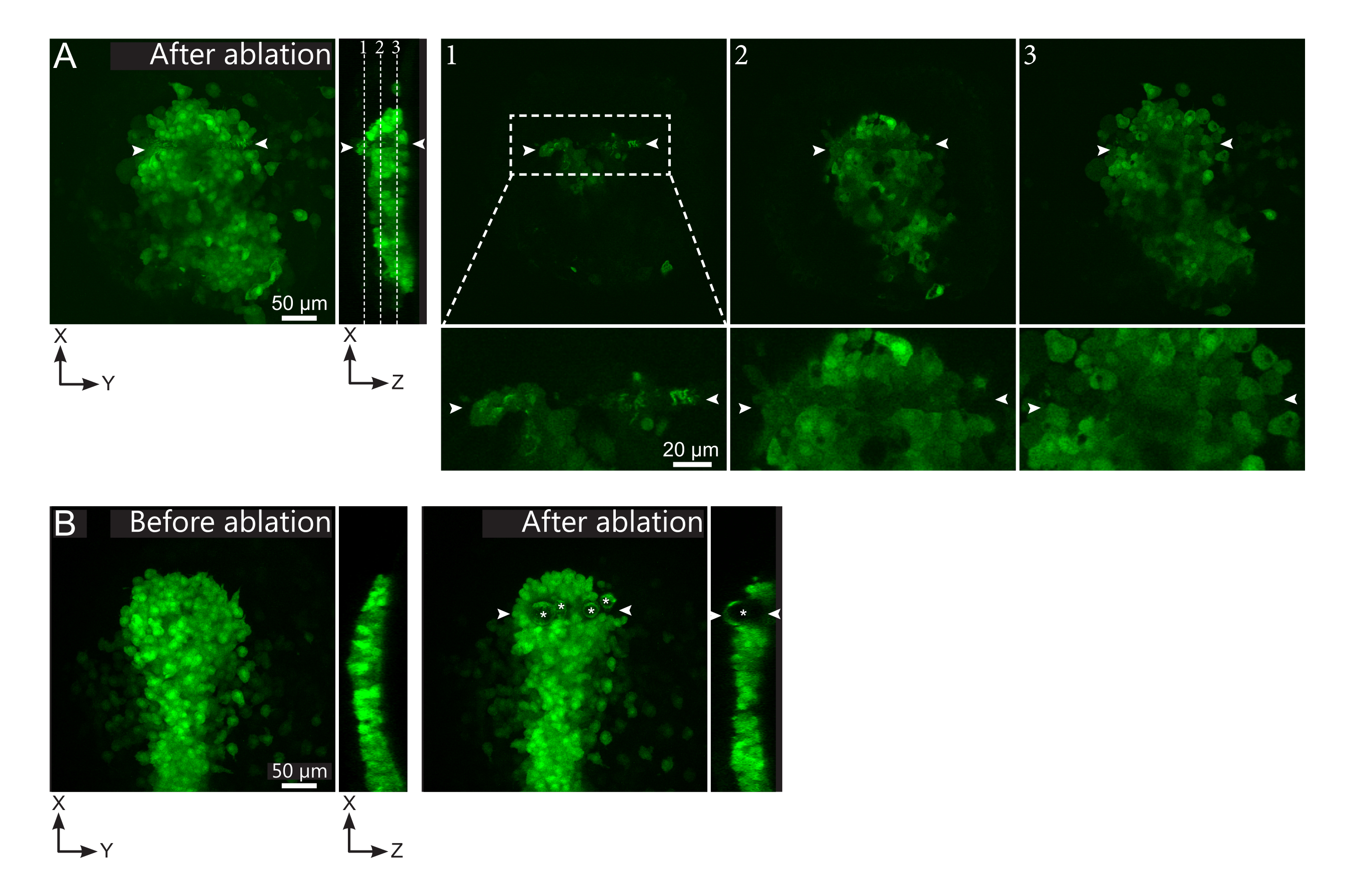{kind=link}
8. Análise de dados
- Abra série de lapso de tempo com o software de análise de imagem e defina o tamanho correto do pixel.
- Na função Spot , defina o Tamanho do Objeto para 10 μm, pois este é o tamanho médio do núcleo durante a gastrulação. Em seguida, execute a função Spot para detectar e rastrear os núcleos.
NOTA: A detecção pode ser ligeiramente melhorada considerando a resolução axial inferior, encaixando uma forma elipsoidal de 12 μm de comprimento ao longo do eixo Z. - Use filtros para remover falsos positivos. Na linha Tg(Gsc:GFP), as células do eixo embrionário e algumas células endodérmicas são rotuladas em verde. Assim, filtrar a intensidade verde permite uma rápida seleção dessas células (Figura 3A).
- Defina a distância máxima entre pontos consecutivos para um valor compatível com a velocidade das células.
NOTA: Tenha cuidado ao considerar o intervalo de tempo entre dois quadros. As células polster migram a 2,8 ± 0,8 μm/min. Assim, permitir 4 μm de deslocamento máximo para uma etapa de tempo de 1 min remove a maioria das faixas artefatosas. - Permitir lacunas em um ou dois pontos de tempo fornece faixas contínuas mais longas, mas pode introduzir erros de rastreamento. Se um núcleo não for detectado corretamente em um ponto único, considere re-executar a detecção de manchas com diferentes parâmetros/filtros.
- Verifique visualmente as faixas e, se necessário, corrija-as.
- Exporte os resultados como um arquivo .xlsx. Processe o arquivo usando rotinas de planilhas publicadas24 (Figura 3B) e rotinas personalizadas em softwares de análise de dados (disponíveis sob consulta).

Figura 3: O isolamento da metade anterior do polster afeta a direcionalidade celular. (A) Reconstruções 3D um embrião Tg(gsc:GFP) expressando Histone2B-mCherry (exibido em magenta), antes e depois de uma ablação a laser cortando o polster em seu meio. Núcleos pertencentes à metade anterior do polster são marcados com um ponto magenta e rastreados ao longo do tempo antes e depois da ablação (ver Filme S1). A barra de escala é de 50 μm. (B) Como medida de persistência migratória, direção auto-correlação de células pertencentes à parte anterior do polster antes e depois da ablação. As células exibem um movimento contínuo antes da ablação, que diminui drasticamente após a ablação, indicando perda de migração orientada coletivamente. A auto-correlação de direção também foi medida em células que formam a metade anterior do polster de um embrião não ablado, como controle. Os envelopes gráficos indicam erro padrão. Clique aqui para ver uma versão maior desta figura.
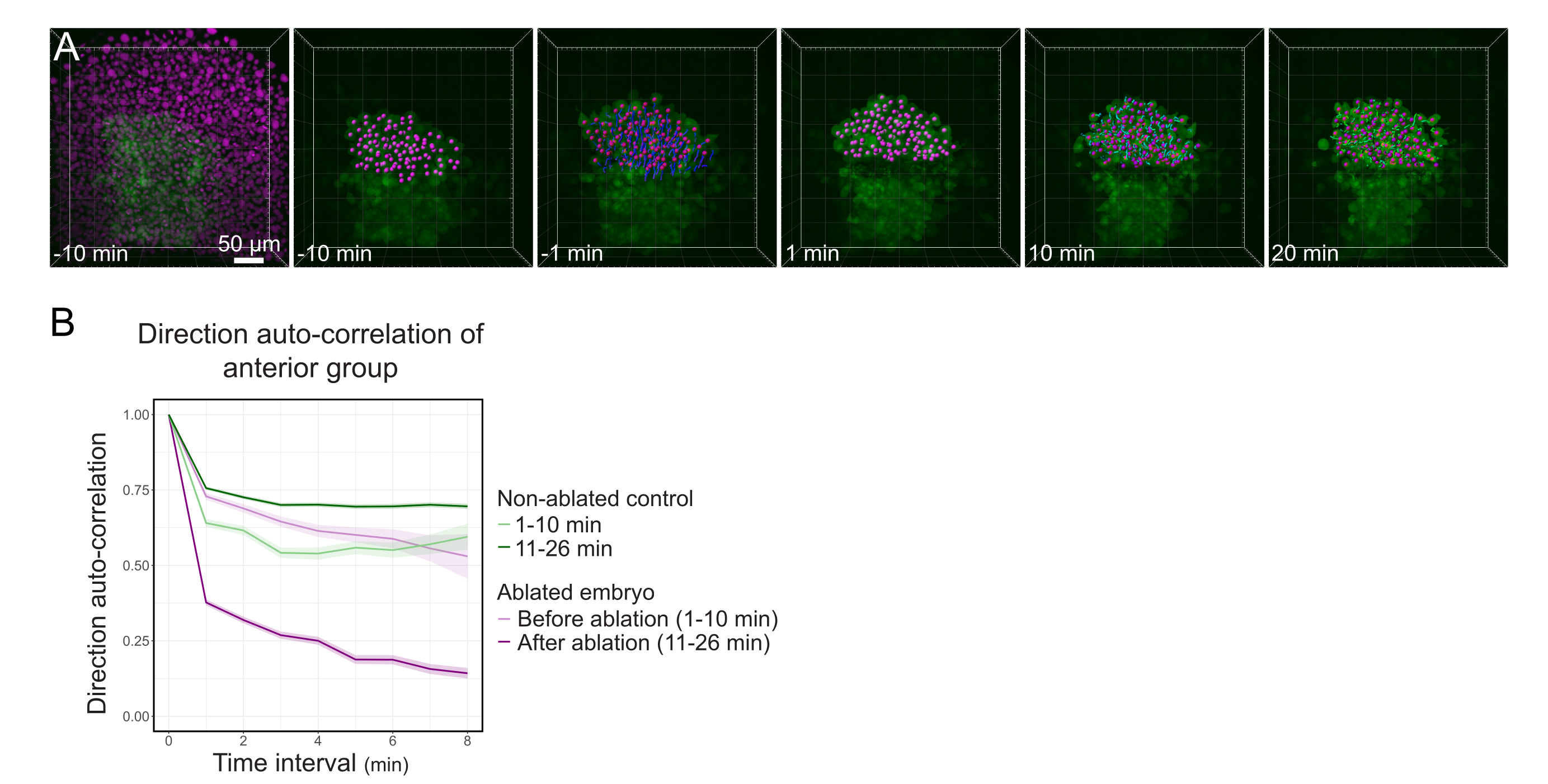{kind=link}
Resultados
Para cortar o polster em seu meio, um embrião Tg(gsc:GFP), injetado com Histone2B-mCherry mRNAs foi montado no estágio de 70% epiboly, como descrito na etapa 4. O polster foi identificado pela expressão GFP, e o embrião foi montado para que o plano do polster seja perpendicular ao eixo óptico (Figura 1B). Inclinar o embrião para longe desta posição complicará o procedimento. A luz terá que passar por mais tecidos para alcançar os planos de ablação, e os aviões de abla?...
Discussão
Aqui, descrevemos um protocolo que usa óptica não linear para realizar ablações de volume profundas e espacialmente bem definidas. O passo mais crítico do protocolo é encontrar condições de tratamento que forneçam energia suficiente para permitir ablações, mas não muita energia, para evitar detritos excessivos ou cavitação. A quantidade de energia entregue no local alvo depende principalmente de: (1) a potência de saída a laser, (2) a qualidade do alinhamento a laser, (3) a natureza do tecido através do ...
Divulgações
Os autores não declaram interesses concorrentes.
Agradecimentos
Agradecemos a Emilie Menant pelo cuidado com os peixes, o Polytechnique Bioimaging Facility, em particular Pierre Mahou, pela assistência com imagens ao vivo em seus equipamentos, em parte apoiados por Région Ile-de-France (interDIM) e Agence Nationale de la Recherche (ANR-11-EQPX-0029 Morphoscope2, ANR-10-INBS-04 France BioImaging). Este trabalho foi apoiado pelas bolsas ANR 15-CE13-0016-1, 18-CE13-0024, 20-CE13-0016, e o programa de pesquisa e inovação Horizon 2020 da União Europeia sob o acordo de subvenção Marie Skłodowska-Curie No 840201, a Ministère de l'Enseignement Supérieur et de la Recherche e o Centre National de la Recherche Scientifique.
Materiais
| Name | Company | Catalog Number | Comments |
| 25x water immersion objective | Olympus | XLPLN25XWMP2 | |
| Agarose | PanReac AppliChem | A8963,0500 | |
| Data analysis software : Matlab | Math Works | ||
| Electro-optic modulator (EOM) | ConOptics | 350-80LA | |
| Embryo Medium (EM) solution | Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 5th Edition. University of Oregon Press, Eugene (Book). (2000). | ||
| Environmental chamber chamber | Okolab | H201-T-UNIT-BL | |
| EOM driver | ConOptics | 302RM | |
| Fluorescence source | Lumencor | SOLA | |
| Glass bottom dishes | MatTek | P35G-0-10-C | |
| Glass capillaries | Harvard Apparatus | 300085 | Outside diameter 1.0 mm, inside diameter 0.58 mm |
| Glass pipettes | Volac | D810 | Tip should be fire polished |
| Green/ablation laser | Spectra Physics | Mai Tai HP DeepSee | |
| Histone2B-mCherry mRNA | Synthesized from pCS2-H2B-mCherry plasmid (Dumortier& al. 2012) | ||
| Image analysis software: IMARIS | Bitplane | ||
| ImSpector software | Abberior Instruments Development Team | ||
| Injection mold | Adapative Science Tools | I-34 | |
| Microloader tips | Eppendorf | 5242956003 | |
| Micromanipulator | Narishige | MN-151 | |
| Micropipette puller | Sutter | P-1000 | |
| mMESSAGE mMACHINE SP6 Transcription Kit | Invitrogen | AM1340 | |
| Penicillin-Streptomycin | Thermofisher | 15140-122 | 10 000 units penicillin and 10 mgstreptomycin per ml |
| Photomultiplier tube (PMT) | Hammamatsu | H7422-40 | |
| PicoPump (Air injector) | World Precision Instrument | PV820 | |
| Red laser | Spectra Physics | OPO/Insight DeepSee | |
| RNAse free water for injection | Sigma | W3500 | |
| Spreadsheet software: Excel | Microsoft | ||
| Stereomicroscope | Nikon | SMZ18 | |
| Tg(gsc:GFP) zebrafish line | Doitsidou, M. et al. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 111 (5), 647–59, doi: doi.org/10.1016/S0092-8674(02)01135-2 (2002). | ||
| TriM Scope II microscope | La Vision Biotech |
Referências
- Slack, J. M. W. Embryonic induction. Mechanisms of Development. 41 (2-3), 91-107 (1993).
- Fernandez-Sanchez, M. -. E., Brunet, T., Röper, J. -. C., Farge, E. Mechanotransduction's impact on animal development, evolution, and tumorigenesis. Annual Review of Cell and Developmental Biology. 31, 373-397 (2015).
- Shih, J., Fraser, S. E. Characterizing the zebrafish organizer: microsurgical analysis at the early-shield stage. Development. 122 (4), 1313-1322 (1996).
- Selleck, M. A. J. Culture and microsurgical manipulation of the early avian embryo. Methods in Cell Biology. 51 (51), 1-21 (1996).
- Bulina, M. E., et al. A genetically encoded photosensitizer. Nature Biotechnology. 24 (1), 95-99 (2006).
- Fang-Yen, C., Gabel, C. V., Samuel, A. D. T., Bargmann, C. I., Avery, L. Laser microsurgery in Caenorhabditis elegans. Methods in Cell Biology. 107, 177-206 (2012).
- Colombelli, J., Grill, S. W., Stelzer, E. H. K. Ultraviolet diffraction limited nanosurgery of live biological tissues. Review of Scientific Instruments. 75 (2), 472-478 (2004).
- Smutny, M., Behrndt, M., Campinho, P., Ruprecht, V., Heisenberg, C. -. P. UV laser ablation to measure cell and tissue-generated forces in the zebrafish embryo in vivo and ex vivo. Methods in Molecular Biology. 1189, 219-235 (2015).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Volpe, B. A., Fotino, T. H., Steiner, A. B. Confocal microscope-based laser ablation and regeneration assay in zebrafish interneuromast cells. Journal of Visualized Experiments: JoVE. (159), (2020).
- Bonnet, I., et al. Mechanical state, material properties and continuous description of an epithelial tissue. Journal of the Royal Society, Interface. 9 (75), 2614-2623 (2012).
- Rauzi, M., Lenne, P. F., Lecuit, T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 468 (7327), 1110-1115 (2010).
- Niemz, M. H. . Laser-Tissue Interactions. Encyclopedia of Biomaterials and Biomedical Engineering, Second Edition - Four Volume Set. , (2019).
- Smith, A. M., Mancini, M. C., Nie, S. Bioimaging: second window for in vivo imaging. Nature Nanotechnology. 4 (11), 710-711 (2009).
- Rauzi, M., Lenne, P. -. F. Cortical forces in cell shape changes and tissue morphogenesis. Current Topics in Developmental Biology. 95, 93-144 (2011).
- Theveneau, E., David, N. B. Migrations cellulaires collectives. Medecine/Sciences. 30 (8-9), 751-757 (2014).
- Dumortier, J. G., Martin, S., Meyer, D., Rosa, F. M., David, N. B. Collective mesendoderm migration relies on an intrinsic directionality signal transmitted through cell contacts. Proceedings of the National Academy of Sciences of the United States of America. 109 (42), 16945-16950 (2012).
- Solnica-Krezel, L., Stemple, D. L., Driever, W. Transparent things: cell fates and cell movements during early embryogenesis of zebrafish. BioEssays. 17 (11), 931-939 (1995).
- Montero, J. -. A., Kilian, B., Chan, J., Bayliss, P. E., Heisenberg, C. -. P. Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Current Biology. 13 (15), 1279-1289 (2003).
- Ulrich, F., et al. Slb/Wnt11 controls hypoblast cell migration and morphogenesis at the onset of zebrafish gastrulation. Development. 130 (22), 5375-5384 (2003).
- Kai, M., Heisenberg, C. -. P., Tada, M. Sphingosine-1-phosphate receptors regulate individual cell behaviours underlying the directed migration of prechordal plate progenitor cells during zebrafish gastrulation. Development. 135 (18), 3043-3051 (2008).
- Smutny, M., et al. Friction forces position the neural anlage. Nature Cell Biology. 19 (4), 306-317 (2017).
- Johansson, M., Giger, F. A., Fielding, T., Houart, C. Dkk1 controls cell-cell interaction through regulation of non-nuclear β-Catenin pools. Developmental Cell. 51 (6), 775-786 (2019).
- Gorelik, R., Gautreau, A. Quantitative and unbiased analysis of directional persistence in cell migration. Nature Protocols. 9 (8), 1931-1943 (2014).
- Grill, S. W., Howard, J., Schäffer, E., Stelzer, E. H. K., Hyman, A. A. The distribution of active force generators controls mitotic spindle position. Science. 301 (5632), 518-521 (2003).
- Desprat, N., Supatto, W., Pouille, P. -. A. A., Beaurepaire, E., Farge, E. Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Developmental Cell. 15 (3), 470-477 (2008).
- Farhadifar, R., Röper, J. -. C., Aigouy, B., Eaton, S., Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Current Biology. 17 (24), 2095-2104 (2007).
- Willier, B. H., Oppenheimer, J. M. . Foundations of Experimental Embryology. , (1964).
- Ashby, W. J., Zijlstra, A. Established and novel methods of interrogating two-dimensional cell migration. Integrative Biology: Quantitative Biosciences from Nano to Macro. 4 (11), 1338-1350 (2012).
- Bosze, B., et al. Pcdh18a regulates endocytosis of E-cadherin during axial mesoderm development in zebrafish. Histochemistry and Cell Biology. 154 (5), 463-480 (2020).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados