
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
DetectSyn: um método fluorescente rápido e imparcial para detectar alterações na densidade da sinapse
Neste Artigo
Resumo
DetectSyn é um ensaio fluorescente rápido e imparcial que mede mudanças no número de sinapse relativa (engajamento pré e pós-sináptico) entre os estados de tratamento ou doença. Esta técnica utiliza uma técnica de ligadura de proximidade que pode ser usada tanto em neurônios cultivados quanto em tecido fixo.
Resumo
Sinapses são o local de comunicação entre neurônios. A força do circuito neuronal está relacionada à densidade sináptica, e a quebra das sinapses é característica de estados da doença como transtorno depressivo grave (MDD) e doença de Alzheimer. Técnicas tradicionais para investigar números de sinapse incluem expressão genética de marcadores fluorescentes (por exemplo, proteína fluorescente verde (GFP)), corantes que preenchem um neurônio (por exemplo, corante de carbocianina, DI) e detecção imunofluorescente de marcadores de coluna (por exemplo, densidade pós-sináptica 95 (PSD95)). Uma grande ressalva a essas técnicas de proxy é que elas só identificam alterações postsináspticas. No entanto, uma sinapse é uma conexão entre um terminal pré-sináptico e uma coluna postsintáptica. O padrão-ouro para medir a formação/eliminação da sinapse requer técnicas demoradas de microscopia eletrônica ou tomografia de matriz. Essas técnicas exigem treinamento especializado e equipamentos caros. Além disso, apenas um número limitado de neurônios pode ser avaliado e são usados para representar alterações em toda uma região cerebral. DetectSyn é uma técnica fluorescente rápida que identifica alterações na formação ou eliminação da sinapse devido a um estado da doença ou atividade medicamentosa. DetectSyn utiliza um ensaio de ligadura de proximidade rápida para detectar proteínas pré e pós-sináptica justtaposed e microscopia fluorescente padrão, uma técnica prontamente disponível para a maioria dos laboratórios. A detecção fluorescente do puncta resultante permite uma análise rápida e imparcial dos experimentos. DetectSyn fornece resultados mais representativos do que microscopia eletrônica porque áreas maiores podem ser analisadas do que um número limitado de neurônios fluorescentes. Além disso, a DetectSyn trabalha para neurônios in vitro cultivados e fatias de tecido fixo. Finalmente, é fornecido um método para analisar os dados coletados dessa técnica. No geral, o DetectSyn oferece um procedimento para detectar alterações relativas na densidade de sinapse em tratamentos ou estados de doença e é mais acessível do que as técnicas tradicionais.
Introdução
As sinapses são a unidade fundamental de comunicação entre os neurônios1. Muitas sinapses entre neurônios dentro das mesmas regiões dão origem a circuitos que mediam o comportamento2. As sinapses consistem em um terminal pré-sináptico de um neurônio que libera neurotransmissores ou neuropeptídeos que retransmitem informações para receptores postsináspicos de outro neurônio. A soma dos sinais pré-sinápticos determina se o neurônio postsinásptico disparará um potencial de ação e propagará a mensagem para outros neurônios.
A sinaptopatologia, a quebra das sinapses, surge em doenças e distúrbios marcados pela diminuição do volume neural, como a doença de Alzheimer e o transtorno depressivo grave, resultando em circuitos que não funcionam maisde forma ideal 3,4,5. Restaurar a densidade da sinapse provavelmente está por trás da eficácia de tratamentos potenciais para esses distúrbios. Por exemplo, foi recentemente demonstrado que o aumento das sinapses está por trás da eficácia comportamental dos antidepressivos rápidos6. Para testar rapidamente possíveis tratamentos de sinaptopatologia, os pesquisadores exigem técnicas que identifiquem rapidamente mudanças nos números da sinapse.
As metodologias atuais são demoradas e caras (microscopia eletrônica, tomografia de matriz), ou examinam apenas alterações postsináspticas sem incorporar engajamento pré-sináptico (análises da coluna vertebral, imunofluorescência/colocalização). Corantes como dii ou proteínas fluorescentes como gfp ajudam a visualizar neurônios e caracterizam espinhas pós-sinápticas. No entanto, a análise da coluna vertebral utiliza razões definidas pelo pesquisador para determinar a morfologia, o que pode diminuir a reprodutibilidade7. Além disso, como as diferentes classes de coluna vertebral se relacionam com sinapses funcionais ainda está sendo descoberto8. A formação da coluna vertebral pode ser transitória e pode refletir plasticidade post-sináptica, mas essas espinhas podem ser eliminadas antes de estabilizar em uma sinapse com um neurônio pré-sináptico9.
A colocalização fornece um proxy melhor para sinapses do que a análise da coluna vertebral, porque pode-se imunossuer proteínas pré-sinápticas e pós-sinápticas. No entanto, as proteínas sinápticas podem produzir baixos valores de colocalização porque as proteínas são justapostas e podem não se sobrepor consistentemente. Assim, como as proteínas não são totalmente sobrepostas, as técnicas de colocação podem não medir com precisão as alterações na formação de sinapse devido a essa informação perdida. Finalmente, embora tanto a microscopia eletrônica (EM) quanto a tomografia de matriz forneçam imagens de alta resolução de sinapses, elas são demoradas. A EM requer ainda equipamentos especializados, e os pesquisadores estão limitados a pequenos volumes de tecido para qualquer experimento. Embora a tomografia de array forneça elegantemente a capacidade de tela para muitas proteínas em seções ultrathin e possa ser combinada com o EM10, essa técnica pode ser muito trabalhosa e além do escopo de experimentos que precisam digitalizar rapidamente para mudanças na formação de sinapsia.
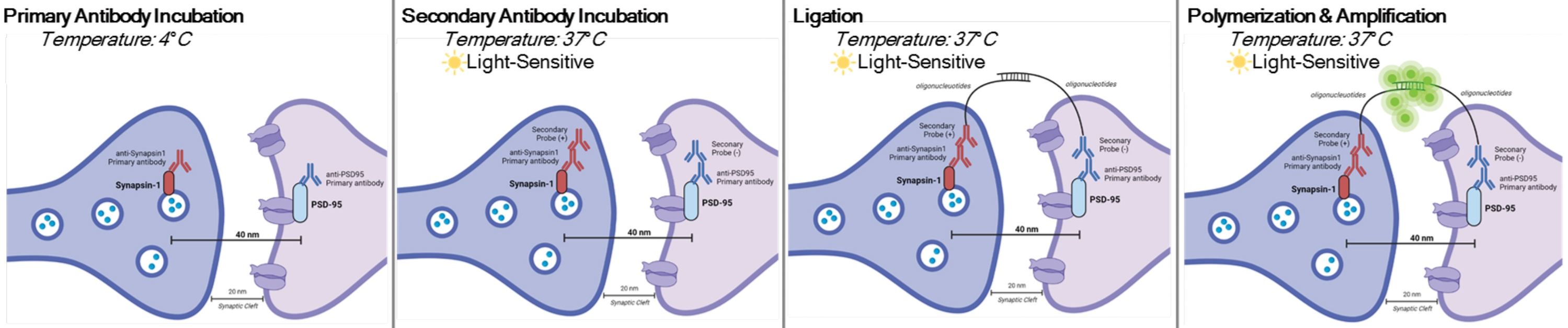
DetectSyn é uma aplicação específica do Ensaio de Ligadura de Proximidade duolink. O ensaio PLA permite a detecção geral de interações proteína-proteína. Detecta as pontes proxy medidas pós-sinápticas amplificando um sinal fluorescente emitido por proteínas pré e pós-sináptica marcadas dentro de 40 nm uma da outra. Se as proteínas sinápticas estiverem dentro de 40 nm, como dentro de uma fenda sináptica, então os anticorpos secundários, que contêm sondas de DNA, hibridizarão em DNA circular. Este DNA circular hibridizado expressa uma sonda fluorescente, que é então amplificada e detectada com técnicas padrão de microscopia fluorescente (ver Figura 1). Crucialmente, ao contrário da em e tomografia de matriz, esta técnica não requer equipamento especializado e leva cerca da mesma quantidade de tempo que a imunohistoquímica padrão. A acessibilidade dessa técnica, assim, permite que pesquisadores fora de instituições intensivas em pesquisa participem de pesquisas sinaptopológicas. Além disso, essa técnica pode examinar alterações na densidade sináptica em múltiplas regiões cerebrais dentro de um único experimento, oferecendo uma representação mais holística de mudanças sinápticas devido a doenças ou tratamento.
Protocolo
O isolamento de células e tecidos de animais foi de acordo com o Guia nacional de Cuidados e Uso de Animais de Laboratório e aprovado pelo Comitê institucional de Cuidados e Uso de Animais da Floresta de Despertar
NOTA: Este protocolo é utilizado em amostras já tratadas e fixas por paradigmas e requisitos experimentais específicos. Para fins de demonstração, a formação de sinapse devido ao tratamento antidepressivo rápido é usada para destacar esta técnica de detecção de sinapse6. Neurônios previamente cultivados em tampas, tratados, fixados em 4% de paraformaldeído (PFA), e armazenados em 1x salina tamponada com fosfato (PBS) serão usados para destacar os procedimentos in vitro. Tecido hipocampal anteriormente fatiado (25 μm de espessura) de camundongos tratados, transcardialmente perfumado com PBS gelado e 4% PFA, e depois armazenado em crioprotetor será usado para destacar os procedimentos de fatia. Consulte11,12 para obter mais informações sobre como cultivar neurônios ou roedores transcárdio perfumados. Consulte a Figura 1 para uma representação gráfica deste procedimento.

Figura 1: Representação gráfica do ensaio DetectSyn. Após a permeabilização das membranas celulares, os anticorpos primários para Synapsin1 e PSD95 se ligam a essas proteínas sinápticas. Secundários com etiquetas de oligonucleotídeos, em seguida, ligam-se aos anticorpos primários. Se Synapsin1 e PSD95 estiverem dentro de 40 nm, como em uma sinapse, então os oligonucleotídeos interagem, e uma tag fluorescente é amplificada. Este sinal fluorescente pode então ser imageado através de microscopia padrão e analisado. Clique aqui para ver uma versão maior desta figura.
{kind=link}
1. Enxágüe amostras
- Enxágüe as amostras com 500 μL de 1x PBS + 0,75% de glicina por 5 min 3 vezes com agitação suave em um agitador orbital para remover PFA residual ou crioprotetor.
2. Bloquear e permeabilize amostras
- Prepare a solução de bloqueio e permeabilização (soro de burro 10% normal, 0,25% Tween 20) em 1x PBS. Prepare-se o suficiente para usar para incubações de bloqueio, primárias e secundárias.
- Às amostras (por exemplo, deslizamentos ou fatias flutuantes livres) em 24 placas de poço, adicione 500 μL de solução de bloqueio e permeabilização. Certifique-se de que cada poço contém uma amostra diferente e é apropriadamente rotulado para evitar que as amostras sejam trocadas.
- Incubar as amostras à temperatura ambiente (RT) por 60 min para células cultivadas ou 2 h para tecido fatiado. Use um agitador orbital para agitação suave.
3. Incubar amostras em anticorpos primários
- Prepare os anticorpos primários no buffer de bloqueio:
- Preparar densidade postina 95 (PSD95; 1:500, polclonal de coelho), Sinapsin1 (1:500, monoclonal do rato), MAP2 (1:400, policlonal de frango)
- Prepare uma alíquota de controle negativo que omita um dos pares sinápticos (por exemplo, sem PSD95)
- Remova cuidadosamente a solução de bloqueio com uma pipeta pasteur de plástico. Tente remover o máximo possível sem perturbar as células ou rasgar tecido.
- Para células cultivadas:
- Forme uma grande placa de petri de plástico com parafilm. Transfira cuidadosamente as tampas para o parafilm usando fórceps.
- Adicione cuidadosamente 60 μL da solução de anticorpos primários ao topo das tampas. Certifique-se de não derramar a solução de anticorpos primária sobre o lado da tampa.
- Para fornecer umidade e evitar que as amostras sequem durante o período de incubação, adicione água ultrauso a uma placa de Petri menor e organize cuidadosamente a pequena placa de Petri ao redor das tampas.
- Cubra a grande placa de Petri e incuba as células cultivadas por 1 h na RT.
- Para tecido fatiado:
- Adicione cuidadosamente 250 μL da solução de anticorpos primários a fatias flutuantes em uma placa de 24 poços.
- Cubra a placa e incuba o tecido durante a noite a 4 °C com agitação suave em um agitador orbital.
4. Lave amostras e incuba em anticorpos secundários
- Prepare anticorpos secundários no buffer de bloqueio:
- Prepare o anti-rato burro (1:5), anti-coelho de burro (1:5), anti-frango de burro (1:400).
- Nesta etapa, controles técnicos adicionais podem ser obtidos preparando uma alíquota secundária que omite o anti-rato ou anti-coelho secundário.
- Para células cultivadas:
- Usando fórceps, toque cuidadosamente a solução primária de tampas em uma toalha de papel
- Usando fórceps, transfira cuidadosamente as tampas de volta para a placa original de 24 poços preenchidas com 500 μL de 1x PBS.
- Para tecido fatiado:
- Remova cuidadosamente a solução de anticorpos primários com uma pipeta pasteur de plástico. Tente remover o máximo possível sem rasgar tecido.
- Adicionar 500 μL de 1x PBS
- Lave as amostras por 10 min 3 vezes em 1x PBS com agitação suave em um agitador orbital. Durante este tempo, leve todos os buffers de lavagem para RT.
- Durante este tempo, mude o parafilm na grande placa de Petri
- Para células cultivadas:
- Usando fórceps, transfira cuidadosamente as tampas de volta para a placa de Petri grande parafilmada
- Adicione cuidadosamente 40 μL da solução de anticorpos secundários à parte superior das tampas. Certifique-se de não derramar a solução secundária de anticorpos sobre o lado da tampa.
- Se necessário, adicione mais água ultrauso a uma placa de petri menor e organize cuidadosamente a pequena placa de petri em torno de tampas.
- Cubra a grande placa de Petri
- Para tecido fatiado:
- Adicione cuidadosamente 250 μL da solução de anticorpos secundários a fatias flutuantes em uma placa de 24 poços.
- Cubra a placa
NOTA: A partir de agora, proteja as amostras da luz, envolvendo os topos das placas com papel alumínio.
- Incubar as amostras a 37 °C por 1 h
5. Ligadura
- Misture o estoque de ligadura 1:5 em água de grau molecular.
- Como na seção 4, transfira cuidadosamente as tampas e remova a mistura secundária do tecido fatiado.
- Lave as amostras em 500 μL de tampão de lavagem A
- Para células cultivadas, lave 2 vezes por 5 minutos. Para tecido fatiado, lave 2 vezes por 10 minutos. Use agitação suave em um agitador orbital para ambos.
- Durante este tempo, mude o parafilm na grande placa de Petri
- Mantendo a ligase em um bloco frio, diluir o ligase 1:40 no estoque de ligaduras a partir do passo 5.1. Realize esta diluição imediatamente antes de adicionar o ligase às amostras.
- Como na seção 4, remova o máximo possível do tampão de lavagem A das amostras antes de adicionar o ligase.
- Para células cultivadas: A transferência cobre clipes de volta para a placa de Petri parafilmada. Adicione 40 μL da mistura de ligadura aos deslizamentos, organize pequenas placas de Petri cheias de água ao redor das tampas e cubra a grande placa de Petri.
- Para tecido fatiado: Adicione 250 μL da mistura de ligadura da etapa 5.4 para cada poço e cubra a placa.
- Incubar as amostras por 30 min a 37 °C.
6. Amplificação
- Misture o estoque de amplificação 1:5 em água de grau molecular.
- Como na seção 4, transfira cuidadosamente as tampas e remova a mistura de ligadura do tecido fatiado.
- Lave amostras em 500 μL de tampão de lavagem A
- Para células cultivadas, lave 2 vezes por 2 minutos. Para tecido fatiado, lave 2 vezes por 10 minutos. Use agitação suave em um agitador orbital para ambos.
- Durante este tempo, mude o parafilm na grande placa de Petri
- Realize esta diluição imediatamente antes de adicionar a polimerase às amostras. Enquanto mantém a polimerase em um bloco frio, dilui a polimerase
- Para células cultivadas, diluir a polimerase 1:80 no estoque de amplificação a partir da etapa 6.1.
- Para tecido fatiado, diluir a polimerase 1:40 no estoque de amplificação a partir da etapa 6.1.
- Como na etapa 4, remova o máximo possível do tampão de lavagem A das amostras antes de adicionar a polimerase.
- Para células cultivadas: Transfira as tampas de volta para a placa de Petri parafilmada. Adicione 40 μL da mistura de amplificação da etapa 6.4.1 às tampas, organize pequenas placas de Petri cheias de água ao redor das tampas e cubra a grande placa de Petri. Incubar as amostras por 100 min a 37 °C.
- Para tecido fatiado: Adicione 250 μL da mistura de amplificação da etapa 6.4.2 para cada poço e cubra a placa. Incubar as amostras por 2h a 37 °C.
NOTA: Durante este tempo, prepare e rotule slides.
7. Montagem
- Como na etapa 4, transfira cuidadosamente as tampas e remova a mistura de amplificação do tecido fatiado.
- Lave as amostras em 500 μL de tampão de lavagem B 2 vezes por 10 minutos com agitação suave em um agitador orbital.
- Lave as amostras em 500 μL de 1% de tampão de lavagem B por 1 min com agitação suave em um agitador orbital.
- Para células cultivadas:
- Solte 3 μL de mídia de montagem em um slide
- Toque no excesso de tampão de lavagem do deslizamento de tampas e, em seguida, coloque o deslizamento (com células voltadas para baixo) na mídia de montagem. Sele as laterais com uma pequena quantidade de esmalte transparente para selar a mancha no lugar.
- Para tecido fatiado:
- Transfira cuidadosamente uma fatia de tecido para o slide preparado e organize-a, de modo que a fatia esteja plana. Solte entre 5-10 μL de mídia de montagem (a quantidade dependerá do tamanho da fatia) na fatia de tecido
- Coloque cuidadosamente uma mancha de vidro sobre a fatia de tecido e selado com uma pequena quantidade de esmalte transparente ao longo da borda para selar a mancha no lugar.
- Aguarde pelo menos 15 minutos antes de analisar sob o microscópio ou armazene a -20 °C.
8. Obtenha imagens digitais com um microscópio confocal
- Otimize as configurações de aquisição (por exemplo, potência laser, ganho, deslocamento) em amostras de todos os tratamentos. Certifique-se de que a otimização inclui a diminuição do ruído de fundo e o aumento do sinal sem saturar demais a intensidade dos sinais fluorescentes. Uma vez determinadas as configurações, aplique as mesmas configurações de aquisição em todas as imagens obtidas.
NOTA: Os seguintes detalhes de aquisição podem ser usados com um microscópio confocal Nikon A1 e o software Nikon NIS AR Elements. - Coloque o slide com a amostra no palco e encontre o plano focal para a amostra usando DAPI através da ocular.
- Desligue a porta dos olhos clicando na porta Eye e escolha um botão de configuração óptica para ajustar as configurações.
- Ajuste o ganho, deslocamento e potência laser para cada canal fluorescente para diminuir o ruído de fundo e melhorar o sinal fluorescente. Certifique-se de que o sinal fluorescente não fique supersaturado como mencionado nas etapas 8.5-8.6.
- Monitore a supersaturação usando um pseudocolor para o sinal fluorescente. Na parte inferior da imagem ao vivo, clique com o botão direito do mouse na guia rotulada com o canal fluorescente atualmente sendo usado.
- Em seguida, escolha o Canal Coloração e escolha um pseudocolor como Rainbow Dark para visualizar a intensidade da fluorescência em um pseudocolor de mapa de calor. Em Rainbow Dark, cores mais frias indicam menos intensidade fluorescente, e cores mais quentes indicam mais intensidade fluorescente.
- Uma vez que todos os canais fluorescentes sejam otimizados, clique com o botão com o botão direito de configuração escolhido anteriormente e escolha Atribuir configuração de câmera atual para este botão.
- Verifique se as configurações escolhidas são suficientes para uma amostra aleatória de cada grupo de tratamento. Se as configurações escolhidas sobressaturarem qualquer uma dessas amostras, repita o passo 8.4 para eliminar a sobresaturação.
- Para neurônios cultivados, siga os passos 8.10-8.16.
- Usando a porta ocular, procure por um neurônio com dendritos que tenham sobreposição mínima com outros dendritos.
- Desligue a porta dos olhos e use o canal DAPI para visualizar o corpo celular do neurônio escolhido. Clique duas vezes no centro da soma para centralizar o neurônio no meio do campo de visão.
- Usando o canal MAP2, encontre o melhor plano de foco para o sinal MAP2 com varredura ao vivo.
- Na guia ND Acquisition , clique em Salvar para Arquivar e escolha um arquivo para salvar a imagem em "Procurar". Em seguida, insira o nome do arquivo.
- Na guia Z , selecione a opção Modo Simétrico Definido por Intervalo . Defina o foco para o melhor plano MAP2 e clique no botão Relativo para definir este plano focal como o meio da pilha z.
- Defina o intervalo para 5 μm com passos de 1 μm e certifique-se de verificar o obturador ativo durante o movimento Z. Na guia Comprimento de onda , selecione o nome do botão óptico com as configurações de aquisição previamente configuradas em Optical Conf. Em seguida, clique em Executar agora.
- Repetir as etapas 8.1.10-8.1.15 para cerca de 10 neurônios por deslizamento/tratamento.
- Para tecido fatiado, siga as etapas 8.18-8.22.
- Usando o olho-porta, procure a região de interesse. Por exemplo, localize CA1 do hipocampo.
- Desligue a porta dos olhos e use o canal MAP2 para encontrar o melhor plano de foco para o sinal MAP2 com varredura ao vivo.
- No menu Adquirir , escolha Digitalizar grande imagem. Em seguida, selecione o nome do botão óptico com as configurações de aquisição previamente configuradas sob o painel Capturando do painel que abre. Além disso, certifique-se de selecionar o objetivo correto neste painel.
- Sob o painel Área e a ocular, use as teclas de seta para definir os limites da região de interesse. Em seguida, clique em Salvar imagem grande para arquivar e criar um nome de arquivo de caminho de salvar para a imagem.
- No painel Configuração , certifique-se de que o Multichannel Capture seja verificado e, em seguida, escolha o nome do botão óptico com as configurações de aquisição previamente configuradas em Optical Conf.
NOTA: Uma pilha de z para uma imagem grande é possível, mas aumentará o tempo de varredura.
9. Análise
- Semelhante às configurações de aquisição, use amostras de todos os tratamentos para otimizar as configurações de limiar. Certifique-se de que a otimização do limiar se concentre em diminuir o ruído de fundo e melhorar o sinal sem sobressaturar a intensidade dos sinais fluorescentes. Uma vez determinadas essas configurações, aplique as mesmas configurações de limiar em todas as imagens usadas para análise, conforme descrito nas etapas 9.2-9.3.
- No ImageJ, a opção limiar está localizada no menu Imagem > Ajustar > Limiar. Escolha a opção Fundo Escuro se a imagem tiver um fundo escuro.
- Em seguida, ajuste os limites superior e inferior do limiar por configurações de limiar otimizados previamente determinadas e clique em Aplicar.
- Para células cultivadas, use o canal MAP2 e uma ferramenta de região de interesse (ROI) para desenhar um ROI para cada neurônio, incluindo dendritos e soma. Para tecido fatiado, desenhe um ROI à mão livre dentro da imagem de fatia que encapsula a área de interesse (por exemplo, radiador de estrato do CA1 dentro do hipocampo).
- Obtenha a área do ROI. No ImageJ, meça a área sob o menu Analisar > Medida.
- Detecte o número de puncta dentro de cada ROI usando uma ferramenta de detecção automática como análise de partículas no ImageJ seguindo as etapas 9.7-9.9.
- Encontre a opção Análise de partículas no menu Analisar > analisar partículas. Primeiro, defina o diâmetro do tamanho do puncta, tipicamente de 0,1-3 μm2.
- Em seguida, escolha a opção Máscaras de Sobreposição no menu suspenso do Show e verifique a opção Resultados de exibição. Em seguida, clique em OK.
- Se puncta não for detectado com a faixa de diâmetro escolhida, ajuste a faixa até que todos os puncta sejam detectados com esta análise. Certifique-se de usar as mesmas configurações de análise de partículas para todas as imagens.
- Divida o número de puncta pela área de uma região de interesse individual seguindo as etapas 9.11-9.13.
- Copie e cole os resultados de cada imagem do pop-up resultados do ImageJ em uma planilha.
- Primeiro, identifique de qual arquivo e amostra os dados foram obtidos. Em seguida, divida a área do ROI pelo número de puncta.
- Em seguida, limpe os dados do pop-up resultados e repita as etapas 9.2-9.12.
- Normalizar resultados para controlar amostras: Média dos resultados (número de puncta/área de ROI) para as amostras de controle. Em seguida, divida os resultados obtidos de todas as amostras pela média do controle para obter os resultados normalizados. A nova média das amostras de controle deve ser igual a 1.
Resultados
Os dados modificados de Heaney et al.6 são apresentados para demonstrar um experimento onde é esperada maior formação de sinapse (consulte6 para obter mais informações e uma discussão mais aprofundada do mecanismo). Anteriormente, foi demonstrado que antidepressivos rápidos requerem ativação do receptor metabotrópico inibidor, GABAB (subtipo B de ácido gama-aminobutírico), para ser eficaz13. Além disso, dados anteriores indicaram que an...
Discussão
DetectSyn é um ensaio rápido que usa um ensaio de ligadura de proximidade para detectar proteínas dentro de 40 nm uma da outra, o que permite a detecção da formação de sinapse. Esta técnica melhora os ensaios fluorescentes atuais, que servem apenas como medidas proxy para formação de sinapse. DetectSyn detecta alterações quantificáveis em proteínas sinápticas localizadas dentro de 40 nm, ou seja, dentro da fissura sináptica, umas das outras. Além disso, o DetectSyn é mais econômico e leva menos tempo d...
Divulgações
Os autores não relatam conflito de interesses.
Agradecimentos
Este trabalho foi apoiado pelos Institutos Nacionais de Saúde NINDS R01 NS105005 (KRG) e NS105005-03S1 (KRG), Departamento de Defesa USAMRMC W81XWH-14-1-0061 (KRG), NIAAA R01AA016852, NIAAA T32AA007565 (CFH), e uma bolsa da FRAXA Research (CFH) e da Associação de Alzheimer, AARG-NTF-21-852843 (KRG), AARF-19-614794-RAPID (KRG).
Materiais
| Name | Company | Catalog Number | Comments |
| 10x PBS | Fisher Scientific | BP39920 | PBS made in house works, as well. |
| 24 well plates | Fisher Scientific | FB012929 | For tissue slices, pre-sterilized plates may be unnecessary. |
| 50 mL conical tubes | Fisher Scientific | 14-432-22 | |
| Aluminium foil | Fisher Scientific | 15-078-290 | |
| Chicken anti-MAP2 antibody | Abcam | ab5392 | |
| Clear nail polish | Fisher Scientific | NC1849418 | Other clear nail polish works, as well. |
| Cold block | Fisher Scientific | 13131012 | |
| Computer workstation | HP | ||
| Confocal or fluorescent microscope | Nikon | A1R HD25 | |
| Donkey anti-chicken FITC | Fisher Scientific | SA1-72000 | |
| Duolink donkey anti-Mouse PLUS | Sigma | DUO92001 | |
| Duolink donkey anti-Rabbit MINUS | Sigma | DUO92005 | |
| Duolink In Situ Detection Reagents Far Red | Sigma | DUO92013 | Contains ligation stock, amplification stock, ligase, and polymerase. |
| Duolink In Situ Mounting Medium with DAPI | Sigma | DUO82040 | |
| Duolink In Situ Wash Buffers, Fluorescence | Sigma | DUO82049 | Contains Wash Buffer A and Wash Buffer B; dilute Wash Buffer B to 1% in diH20 for 1% Wash Buffer B. |
| Fine-tipped paintbrush | Fisher Scientific | NC9691026 | Sable hair, size 00 or 000, can also find at craft stores |
| Fisherbrand Cover Glasses: Rectangles | Fisher Scientific | 12545MP | Cover glass is unnecessary for cultured neurons already on glass coverslips. |
| Fisherbrand Superfrost Plus Microscope Slides | Fisher Scientific | 1255015 | For cultured neurons already on glass coverslips, Superfrost slides may be unnecessary. |
| Freezer, -20°C | VWR | 76449-108 | |
| Glass coverslips | Fisher Scientific | 125480 | |
| Glycine | Fisher Scientific | BP381-1 | |
| Image processing software | e.g. NIS Elements, ImageJ | ||
| Incubator | Fisher Scientific | 15-015-2633 | |
| Large petri dish, 100mm | Fisher Scientific | FB0875712 | |
| Molecular grade water | Fisher Scientific | BP24701 | |
| Mouse anti-Synapsin1 antibody | Synaptic Systems | 106-011 | |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 | |
| Orbital shaker | Fisher Scientific | 02-106-1013 | |
| Parafilm | Fisher Scientific | 13-374-10 | |
| Pipette tips | Fisher Scientific | 02-707-025 | |
| Pipettes | Fisher Scientific | 14-388-100 | Working volumes range from 3 µL to 500 µL |
| Plastic pasteur pipette | Fisher Scientific | 02-708-006 | |
| Precision tweezers/foreceps | Fisher Scientific | 12-000-122 | |
| Rabbit anti-PSD95 antibody | Abcam | ab18258 | Other antibody pairs may work, as well, with optimization. |
| Refrigerator | VWR | 76470-402 | |
| Small petri dish, 60 mm | Fisher Scientific | FB0875713A | |
| Timer | Fisher Scientific | 14-649-17 | |
| Tween 20 | Fisher Scientific | BP337-100 |
Referências
- Südhof, T. C. Towards an understanding of synapse formation. Neuron. 100 (2), 276-293 (2018).
- Bliss, T. V., Collingridge, G. L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 361 (6407), 31-39 (1993).
- Heaney, C. F., Raab-Graham, K. F. Dysregulated protein synthesis in major depressive disorder. The Oxford Handbook of Neuronal Protein Synthesis. , 510-532 (2018).
- Masliah, E., Crews, L., Hansen, L. Synaptic remodeling during aging and in Alzheimer's disease. Journal of Alzheimer's Disease. 9, 91-99 (2006).
- van Spronsen, M., Hoogenraad, C. C. Synapse pathology in psychiatric and neurologic disease. Current Neurology and Neuroscience Reports. 10 (3), 207-214 (2010).
- Heaney, C. F., Namjoshi, S. V., Uneri, A., Bach, E. C., Weiner, J. L., Raab-Graham, K. F. Role of FMRP in rapid antidepressant effects and synapse regulation. Molecular Psychiatry. 26 (6), 2350-2362 (2021).
- Pchitskaya, E., Bezprozvanny, I. Dendritic spines shape analysis-Classification or clusterization? Perspective. Frontiers in Synaptic Neuroscience. 12, 31 (2020).
- Alvarez, V. A., Sabatini, B. L. Anatomical and physiological plasticity of dendritic spines. Annual Review of Neuroscience. 30 (1), 79-97 (2007).
- Berry, K. P., Nedivi, E. Spine Dynamics: Are they all the same. Neuron. 96 (1), 43-55 (2017).
- Micheva, K. D., Smith, S. J. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments: JoVE. (65), e3564 (2012).
- Workman, E. R., Niere, F., Raab-Graham, K. F. mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology. 73, 192-203 (2013).
- Li, N., et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 329 (5994), 959-964 (2010).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados