
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Proteômica por Espectrometria de Massas Guiada por Linhagem Celular no Embrião em Desenvolvimento (Rã)
Neste Artigo
Resumo
Descrevemos aqui uma caracterização proteômica baseada em espectrometria de massa de linhagens celulares com destino tecidual conhecido no embrião do vertebrado Xenopus laevis .
Resumo
A caracterização de eventos moleculares à medida que as células dão origem a tecidos e órgãos aumenta o potencial para entender melhor o desenvolvimento normal e projetar remédios eficientes para doenças. Tecnologias que permitam a identificação e quantificação precisas de diversos tipos e grande número de proteínas forneceriam informações ainda escassas sobre os mecanismos moleculares que orquestram o desenvolvimento de tecidos e organismos no espaço e no tempo. Aqui, apresentamos um protocolo baseado em espectrometria de massas que permite a medição de milhares de proteínas em linhagens celulares identificadas em embriões de Xenopus laevis (rã). A abordagem baseia-se em mapas reprodutíveis de destino celular e métodos estabelecidos para identificar, marcar fluorescentemente, rastrear e amostrar células e suas progênies (clones) a partir deste modelo de desenvolvimento de vertebrados. Após a coleta do conteúdo celular por microamostragem ou isolamento de células por dissecção ou classificação celular ativada por fluorescência, as proteínas são extraídas e processadas para análise proteômica ascendente. Cromatografia líquida e eletroforese capilar são usadas para fornecer separação escalável para detecção e quantificação de proteínas com espectrometria de massas de alta resolução (EMAR). Exemplos representativos são fornecidos para a caracterização proteômica de células adiposas do tecido neural. A proteômica da HRMS guiada por linhagem celular é adaptável a diferentes tecidos e organismos. É suficientemente sensível, específico e quantitativo para perscrutar a dinâmica espaço-temporal do proteoma durante o desenvolvimento dos vertebrados.
Introdução
Nossa compreensão da diferenciação celular e da gênese de tecidos e órgãos é o resultado de décadas de elaboradas telas direcionadas de genes e seus produtos. Aumentar nosso conhecimento de todas as biomoléculas e suas quantidades durante eventos celulares importantes ajudaria a desvendar mecanismos moleculares que controlam o padrão espacial e temporal do plano corporal dos vertebrados. Tecnologias que permitem amplificação molecular e sequenciamento são agora capazes de relatar rotineiramente um grande número de genes e transcritos, apoiando estudos orientados por hipóteses em pesquisa biológica básica e translacional. Para entender os sistemas em desenvolvimento, uma complexa relação entre transcrição e tradução defende a análise direta de múltiplas proteínas e suas modificações pós-traducionais. A proteômica global utilizando sistemas biológicos in vitro, como as células-tronco pluripotentes induzidas, começou a delinear mecanismos de indução tecidual 1,2. Em organismos complexos, como o embrião de vertebrados, o desenvolvimento depende de gradientes morfogênicos no contexto do espaço e do tempo3. Conclui-se que obter conhecimento das mudanças proteômicas à medida que as células se diferenciam para formar tecidos especializados, como tecidos neurais, oferece uma chave para desbloquear programas moleculares que controlam o desenvolvimento normal e defeituoso e orientar a terapêutica de próxima geração.
A rã-de-garra vertebrada sul-africana (Xenopus laevis) é um modelo bem estabelecido em biologia celular e de desenvolvimento, neuro e regenerativa. O Prêmio Nobel de Fisiologia ou Medicina de Sir John Gurdon em 2012 4,5 pela descoberta da pluripotência do núcleo somático destacou a importância desse modelo para descobertasem estudos básicos e translacionais. Os embriões de Xenopus desenvolvem-se externamente à mãe, facilitando assim a manipulação direta de células, clones celulares e expressão gênica ao longo de vários estágios de desenvolvimento. A pigmentação assimétrica e as divisões celulares estereotipadas permitiram o mapeamento de mapas reprodutíveis do destino do embrião de 16-6 e 32 célulasem 7,8 estádios. Para proteômica baseada em espectrometria de massa de alta resolução (EMAR), vantagens adicionais do modelo incluem tamanho relativamente grande (~1 mm de diâmetro), que produz conteúdo proteico abundante para análise (~130 μg em embriões em estágio inicial de clivagem, ~10 μg de conteúdo proteico em células isoladas do embrião de 16 células)9,10.
Atualmente, o HRMS é a tecnologia líder de escolha para detectar proteínas. Essa tecnologia permite a detecção e quantificação direta, sensível e específica de múltiplas, geralmente centenas a milhares de proteínas diferentes11. A proteômica ascendente por HRMS envolve uma série de etapas interconectadas. Após a extração da amostra de célula/tecido, as proteínas são digeridas com uma enzima proteolítica, como a tripsina (proteômica de baixo para cima). Os peptídeos resultantes são separados com base em suas diferentes propriedades físico-químicas, incluindo hidrofobicidade (cromatografia líquida de fase reversa, LC), carga líquida (cromatografia de troca iônica), tamanho (cromatografia de exclusão de tamanho) ou mobilidade eletroforética (eletroforese capilar, CE). Os peptídeos são então carregados (ionizados), tipicamente usando ionização por eletrospray (ESI), e íons peptídeos são detectados e sequenciados via fragmentação em fase gasosa por EMHR tandem. Os dados peptídicos resultantes são mapeados para o proteoma do organismo em estudo. Com a intensidade de sinal do peptídeo específico para proteínas (proteotípicas) correlacionando-se com a concentração, a quantificação da proteína pode ser realizada sem rótulos ou baseada em marcadores (quantificação multiplexação). A proteômica da HRMS fornece um rico recurso de informações sobre o estado molecular do sistema em estudo, permitindo a geração de hipóteses e estudos funcionais de acompanhamento.

Figura 1: Proteômica espaço-temporalmente escalável permitindo a proteômica de HRMS guiada por linhagem celular no embrião em desenvolvimento (rã). (A) Visualização do espécime (1) utilizando um estereomicroscópio (2) para injeção de uma célula identificada (inset), utilizando uma micropipeta fabricada (3) sob controle por um estágio de translação (4). (B) Amostragem subcelular da célula D 11 esquerda identificada em um embrião de16 células. (C) Dissecção de uma célula D11 inteira de um embrião de 16 células. (D) Traçado fluorescente (verde) das progênies D111 esquerda e direita de um embrião de 32 células para guiar a dissecção do ectoderma neural (NE) na gástrula (estágio 10) e isolamento do tecido descendente do girino usando FACS. Barras de escala: 200 μm para embriões, 1,25 mm para o frasco para injetáveis. As figuras foram adaptadas com permissão das referências 15,19,21,59. Clique aqui para ver uma versão maior desta figura.
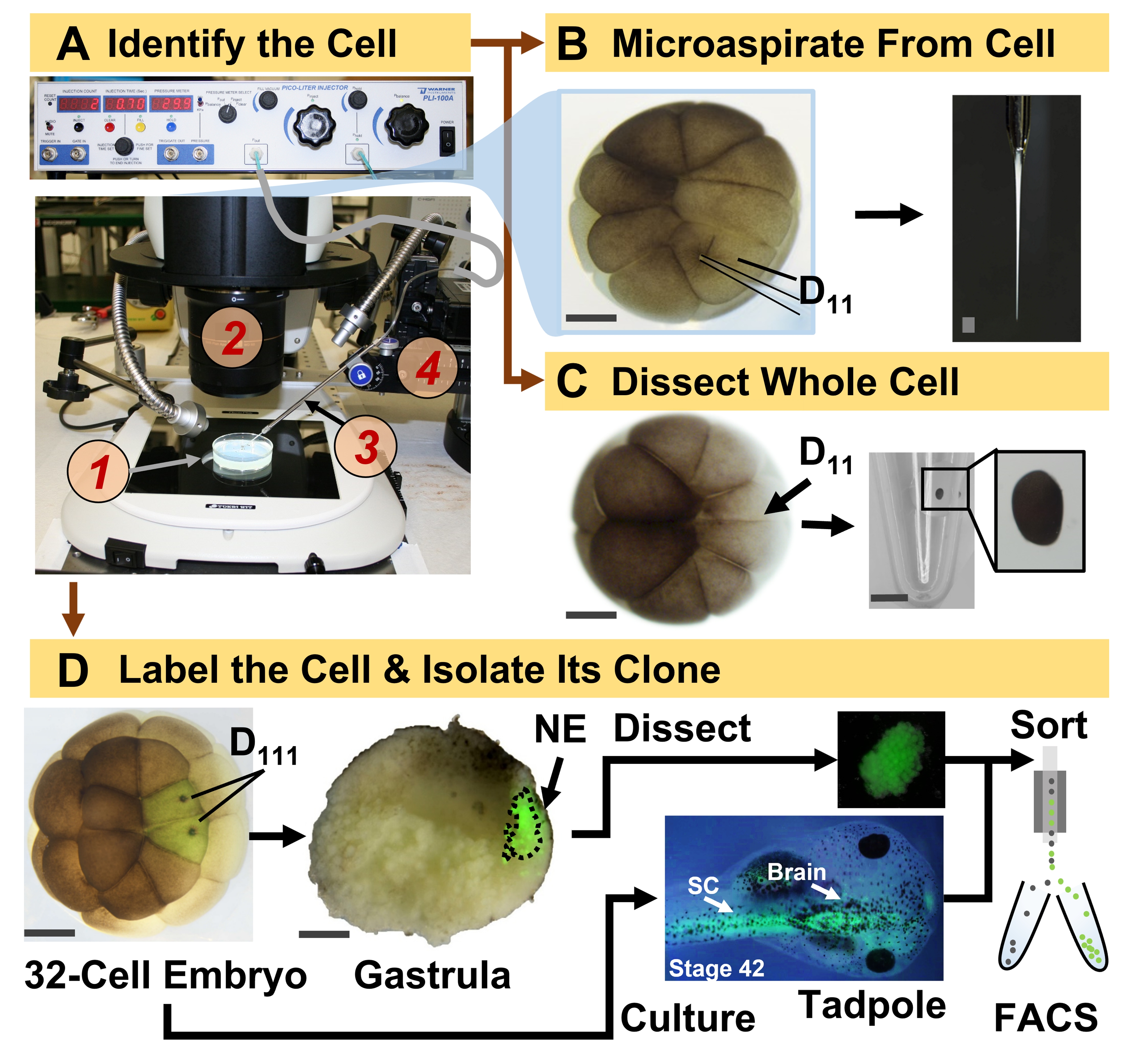{kind=link}
O protocolo aqui apresentado permite a quantificação baseada em HRMS de um grande número de proteínas em células/tecidos identificados em embriões de X. laevis em desenvolvimento. A abordagem baseia-se na identificação celular precisa, mapas de destino celular reprodutíveis e metodologias estabelecidas para rastrear linhagens celulares neste modelo biológico 6,7,8. Como mostrado na Figura 1, estudamos proteomas de células isoladas empregando dissecção de células inteiras ou microamostragem capilar para aspirar o conteúdo celular. O monitoramento da linhagem de uma célula nos permite estudar a evolução espaço-temporal do proteoma à medida que as células formam tecidos durante a gastrulação. A progênie celular é marcada fluorescentemente pela injeção de um fluoróforo conjugado a dextran inerte ou mRNA para proteína fluorescente (por exemplo, proteína fluorescente verde ou GFP). A progênie marcada é isolada nos momentos de desenvolvimento desejados. Durante a gastrulação, clones celulares que estão firmemente agrupados podem ser isolados por dissecção. Após a gastrulação, os clones celulares podem ser distribuídos dentro do embrião devido a movimentos migratórios e podem ser isolados de tecidos dissociados por triagem celular ativada por fluorescência (FACS). As proteínas nessas células e tecidos são medidas via proteômica bottom-up empregando HPLC ou CE para separação e ESI tandem HRMS para identificação. A proteômica da HRMS guiada por linhagens celulares é escalável para diferentes tamanhos e linhagens celulares dentro do embrião e é específica, sensível e quantitativa. Através de exemplos selecionados mostrados aqui, também demonstramos que este protocolo é escalável e amplamente adaptável a diferentes tipos de células e linhagens celulares.

Figura 2: O fluxo de trabalho bioanalítico. A microdissecção e a aspiração capilar, ou FACS, facilitaram a amostragem do conteúdo de proteínas celulares e clonais. A depleção de proteínas vitelínicas abundantes e a separação por eletroforese capilar (CE) ou cromatografia líquida de nanofluxo (LC) aumentaram a sensibilidade de identificação (ID) usando espectrometria de massa de alta resolução (EMRH) com ionização por eletrospray (ESI). A quantificação revelou desregulação, fornecendo novas informações para estudos baseados em hipóteses em conjunto com informações disponíveis na ontologia gênica (GO). As figuras foram adaptadas com permissão da referência15. Clique aqui para ver uma versão maior desta figura.
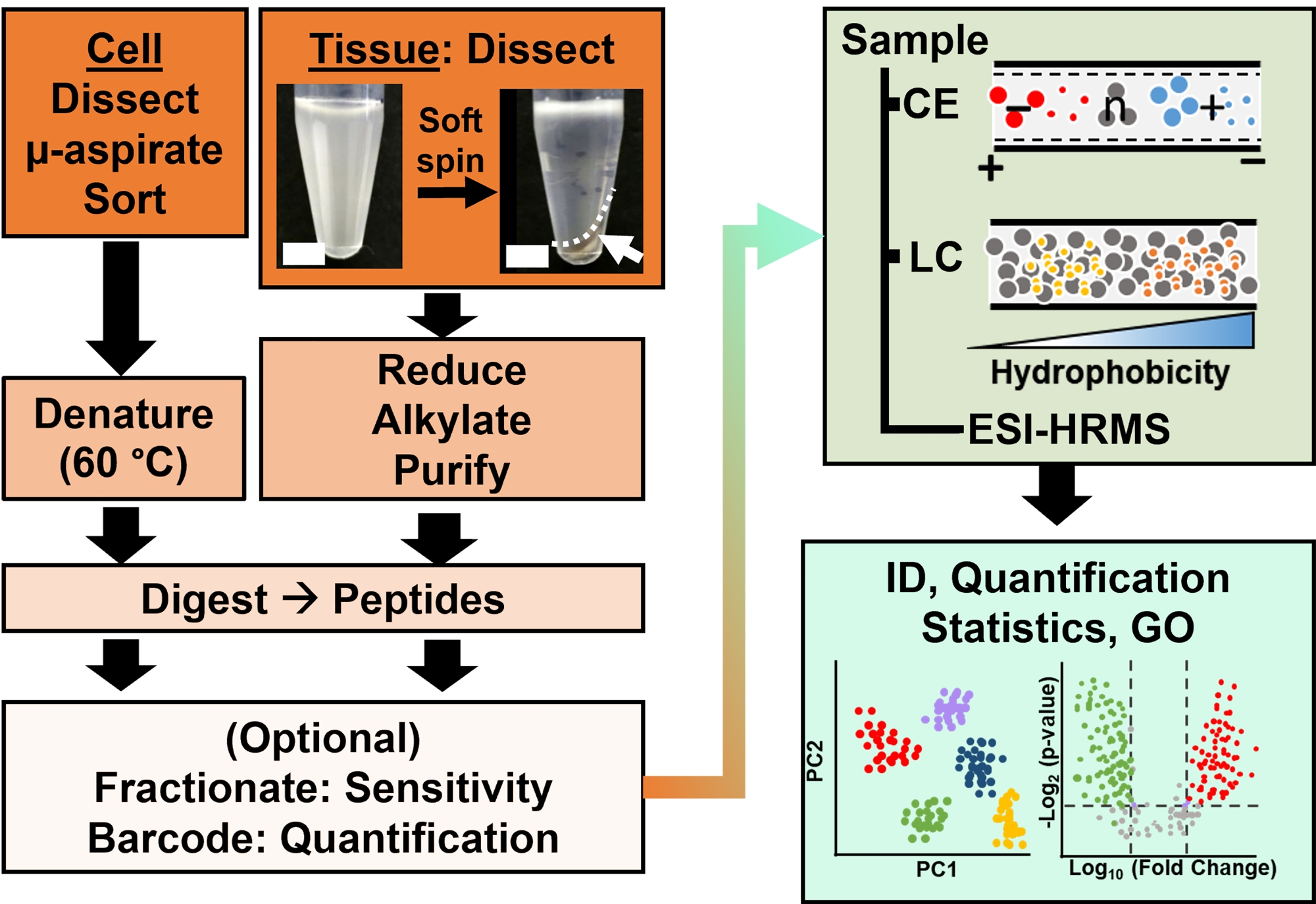{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocolo
Todos os protocolos que garantem a manutenção e o manejo humanizado de rãs adultas de Xenopus laevis foram aprovados pelo Comitê Institucional de Cuidados e Uso de Animais da Universidade de Maryland, College Park (números de aprovação R-DEC-17-57 e R-FEB-21-07).
1. Prepare as soluções
- Para embriologia
- Preparar 1x, 0,5x e 0,2x solução de Steinberg (SS) e, em seguida, autoclavá-los (120 °C por 20 min) para esterilidade seguindo protocolos padrão12.
- Preparar Ficoll a 3% (p/v) em SF 1x esterilizado seguindo protocolos padrão12.
- Para desembaraçar, prepare na hora a solução de cisteína a 2% (p/v) e ajuste o seu pH para 8 adicionando solução de hidróxido de sódio a 10 M gota a gota.
CUIDADO: A exposição à cisteína pode causar danos cutâneos e respiratórios. O hidróxido de sódio é um corrosivo que pode causar sérios danos à pele e aos olhos após exposição direta. Use equipamentos de proteção individual (EPIs) apropriados ao manusear esses produtos químicos, como luvas e jaleco de laboratório. - Para o traçador de linhagem, preparar 0,5% (v/v) de um dextran fluorescente em água deionizada estéril. Alternativamente, prepare uma solução de 0,2 μg/μL de mRNA para proteínas fluorescentes em água deionizada estéril (por exemplo, GFP).
- Para dissociar as células, preparar o tampão Newport 2.0 contendo isotionato de sódio 0,1 M, pirofosfato de sódio 20 mM e CAPS 10 mM e, em seguida, elevar seu pH para 10,513.
CUIDADO: A exposição ao pirofosfato de sódio pode causar irritação na pele e nos olhos. Use EPIs apropriados ao manusear esses produtos químicos.
- Para proteômica de baixo para cima
- Preparar o tampão de lise celular para incluir: 250 mM de sacarose, 1% de substituto de P-40 nonidet (p/v), 20 mM de Tris-HCl, 5 mM de EDTA, 10 μM de citocalasina D e 10 μM de combretastatina 4A. Preparar um estoque de 10% (p/v) de dodecil sulfato de sódio14,15.
NOTA: Tris-HCl foi escolhido para minimizar a contaminação por HEPES durante o nano-fluxo LC (nanoLC)-HRMS.
CUIDADO: A exposição ao substituto nonidet P-40 pode causar irritação da pele. A citocalasina D é teratogênica se consumida e a combretastatina é agudamente tóxica após exposição direta. Use EPIs apropriados ao manusear esses produtos químicos. - Para separar peptídeos por CE, prepare os seguintes solventes (v/v): Solvente de amostra, acetonitrila 75% (ACN) contendo 0,05% de ácido acético (AcOH) em água; solução de bainha, ACN a 10% contendo AcOH a 0,05% em água; eletrólito de fundo (BGE), 25% ACN contendo 1 M de ácido fórmico (FA) em água.
CUIDADO: AcOH e FA são tóxicos quando inalados ou consumidos e podem causar sérios danos à pele e aos olhos após exposição direta. Use EPIs apropriados ao manusear esses produtos químicos. - Para separar peptídeos por nanoLC de fase reversa, prepare (v/v): Fase móvel A (aquosa), água contendo 0,1% de AG; fase móvel B (orgânica), 0,1% de AG na ACN.
NOTA: Todas as misturas devem ser preparadas usando solventes de grau LC-MS para minimizar as interferências químicas durante a detecção de HRMS.
- Preparar o tampão de lise celular para incluir: 250 mM de sacarose, 1% de substituto de P-40 nonidet (p/v), 20 mM de Tris-HCl, 5 mM de EDTA, 10 μM de citocalasina D e 10 μM de combretastatina 4A. Preparar um estoque de 10% (p/v) de dodecil sulfato de sódio14,15.
2. Preparar as ferramentas para microinjeção e dissecção
- Para mover e orientar suavemente os embriões, faça laços de cabelo fixando os cabelos limpos em uma pipeta de Pasteur, como descrito em outroartigo 16.
- Para microinjeção, fabricar agulhas puxando capilares de borossilicato (1 mm/500 μm de diâmetro externo/interno) usando um extrator de pipeta, conforme descrito anteriormente16.
OBS: Aqui, foi utilizado um extrator de pipeta P-1000 com as seguintes regulagens para a confecção das agulhas: calor, 495; puxar, 30, velocidade, 60; tempo, 150; pressão, 200. - Com observação em estereomicroscópio, corte a ponta do capilar usando um par de pinças afiadas para essencialmente fabricar o capilar em uma (micro)agulha (por exemplo, Dumont #5)16.
CUIDADO: Os capilares puxados são muito afiados e devem ser manuseados com cuidado.
NOTA: A ponta da agulha deve ser afiada o suficiente (diâmetro externo 10-15 μm) para ser capaz de perfurar a célula com danos mínimos à membrana celular para que o conteúdo intracelular não vaze e a célula possa curar e continuar a ser viável. - Para segurar os embriões durante a microinjeção, prepare poços em um prato cheio de argila. Em uma placa de Petri de 15 mm, imprima poços de ~1 mm de diâmetro x ~0,5 mm de profundidade em argila plasticina atóxica, conforme descrito anteriormente16.
- Para a microdissecção, prepare pratos revestidos com agarose. Completar agarose a 2% em 1x SS e autoclavá-la para esterilizar a solução (120 °C por 20 min). Encha placas de Petri de 60 mm no meio do caminho e deixe as placas se solidificarem. Faça poços de ~1 mm de diâmetro x ~0,5 mm de profundidade usando uma ferramenta de pipeta Pasteur esférica, conforme descrito anteriormente16.
3. Isole a linhagem celular
Observação : as etapas a seguir são executadas para isolar células únicas identificadas e/ou suas linhagens celulares descendentes. Usualmente, o embrião é cultivado até o estágio de 16 ou 32 células, onde os destinos teciduais de cada célula são mapeados de forma reprodutível6,7,17. As células embrionárias são identificadas com base na morfologia, localização e em referência aos seus mapas de destino. Para a análise unicelular, as células identificadas são isoladas por dissecção manual, ou seu conteúdo intracelular é coletado em uma pipeta capilar e depositado em 5 μL de bicarbonato de amônio 0,5 mM. A amostra resultante é armazenada a -80 °C até a análise (Figura 1)18,19,20,21. Para a análise da linhagem celular, as células identificadas são injetadas com um traçador de linhagem, e seus clones subsequentes são isolados em estágios-chave do desenvolvimento (por exemplo, durante a gastrulação para estudar a indução do tecido, após a neurulação para estudar o comprometimento tecidual). A seguir, são delineados passos para marcar fluorescentemente a linhagem de células identificadas para isolamento por dissecção ou FACS.
- Cultura dos embriões
- Obter embriões via acasalamento natural ou fertilização in vitro (FIV) seguindo protocolos estabelecidos12.
NOTA: O acasalamento natural é logisticamente mais simples, poupa as rãs machos adultas e produz embriões em diferentes estágios de desenvolvimento, enquanto a FIV fornece embriões sincronizados com o desenvolvimento para experimentos que exigem estadiamento preciso. - Dejelly os embriões. Remover a camada de gelatina que envolve os embriões através do tratamento com a solução dejellying conforme descrito anteriormente12,16.
NOTA: Microinjeções e dissecção requerem acesso às células e tecidos, necessitando de dejellying em embriões de X. laevis. - Selecionar embriões de 2 células com pigmentação estereotipada16,22.
NOTA: Esta etapa é importante para garantir precisão e reprodutibilidade na identificação da célula e sua linhagem. - Cultura de embriões para o estágio de desenvolvimento desejado. Transferir os embriões degelatinados para uma placa de Petri contendo 1x SS e incubá-los entre 14-25 °C para controlar a velocidade de desenvolvimento.
NOTA: A dependência da temperatura do desenvolvimento é reprodutível e mapeada para X. laevis, disponível no Xenbase23 (www.xenbase.org). A cultura de lotes de embriões em diferentes temperaturas permite estágios de desenvolvimento escalonados. Isso ajuda a distribuir o número de embriões disponíveis em um determinado momento para experimentação. - Monitorar o padrão de clivagem de embriões e selecionar embriões com padrões estereotipados de pigmentação e clivagem para microinjeção16.
NOTA: Ao selecionar embriões de 16 e 32 células, certifique-se de que as clivagens celulares sejam simétricas para o rastreamento de linhagem reprodutível.
- Obter embriões via acasalamento natural ou fertilização in vitro (FIV) seguindo protocolos estabelecidos12.
- Rotular a(s) célula(s) de interesse
- Configure a agulha de injeção que contém a solução traçadora de linhagem. Monte a agulha de microinjeção em um suporte de micropipeta controlado por um micromanipulador multieixo.
- Conecte o suporte da micropipeta a um microinjetor. Preencher a agulha com o traçador de linhagem, aplicando pressão negativa conforme descrito anteriormente16. A Figura 1A exemplifica a configuração.
- Calibre a agulha. Ajustar o tamanho da ponta da agulha e o tempo de injeção para fornecer ~1 nL da solução traçadora de linhagem, medida em óleo (mineral) seguindo um protocolo disponível em outro lugar16.
NOTA: Capilares com uma ponta mais larga tendem a danificar a membrana celular, fazendo com que o conteúdo subcelular e o traçador de linhagem injetado vazem, enquanto capilares com pontas menores são propensos a entupimentos. Capilares com ~10 μm de diâmetro externo da ponta são ideais, exigindo um pulso de pressão de 40 psi sobre ~300 ms para fornecer ~1 nL. - Inundar a placa de argila de microinjeção com a solução de Ficoll a 3% e transferir ~10 embriões para a placa de argila usando uma pipeta de transferência. Use um laço de cabelo para guiar cada embrião em um poço e posicione-os suavemente de modo que a célula-alvo de interesse esteja em um ângulo reto com a microagulha.
- Identificar a célula precursora da linhagem de interesse seguindo os mapas de destino do tecido de X. laevis. Por exemplo, a Figura 1 demonstra a marcação de clones ectodérmicos neurais com base na injeção de suas células precursoras em embriões de 32 células (D111 à esquerda e à direita).
NOTA: Mapas detalhados do destino para os embriões 7,8 de 16-6 e 32 células estão disponíveis em uma plataforma interativa via Xenbase23. É importante garantir pigmentação estereotipada e clivagens em embriões ao usá-los para experimentos de rastreamento de linhagem. - Injetar a(s) célula(s) de interesse com ~1 nL do dextran fluorescente ou ~200 pg de mRNA, conforme descrito anteriormente16.
NOTA: Use conjugados de dextran que são 10.000-40.000 MW. Conjugados de dextran menores podem passar através de junções de gap, enquanto conjugados de dextran maiores podem não se difundir uniformemente na célula injetada. Planeje injetar células em ~10 embriões para ter tecidos suficientes para análises proteômicas. - Confirme o sucesso da marcação celular sob um estereomicroscópio. Certifique-se de que apenas a célula pretendida é injetada. Descartar embriões contendo células lesionadas ou rotuladas incorretamente seguindo políticas institucionais.
NOTA: Como X. laevis é invasora em muitos ambientes não naturais, os embriões podem ser congelados para garantir a letalidade antes de descartá-los.
- Isole a progênie da célula marcada
- Transferir os embriões injetados para SS 0,5x em uma placa de Petri e cultivá-los entre 14-25 °C até atingir o estágio de desenvolvimento desejado.
NOTA: Consulte os protocolos estabelecidos para estadiar embriões reportados no Xenbase. - Transferir 3-5 embriões para uma placa de ágar com solução de SS 0,2x para microdissecções.
NOTA: Reduzir a concentração de sal da solução de SS de 0,5x para 0,2x ajuda a separar as células durante a dissecção. - Use duas pinças afiadas para remover suavemente a membrana vitelínica ao redor do embrião.
NOTA: Para poupar o clone de interesse de danos, descasque a membrana do lado oposto do clone marcado fluorescentemente. - Isole o clone rotulado por dissecação manual (etapas 3.3.5-3.3.6) ou FACS (etapas 3.3.7-3.3.8) da seguinte maneira.
- Use fórceps para dissecar o clone marcado do embrião.
NOTA: Outras ferramentas, como tesouras microcirúrgicas, agulhas de tungstênio ou facas de cabelo de sobrancelha, podem ser usadas para a dissecção do clone marcado, conforme detalhado em outro artigo16. - Recolher o tecido dissecado com uma pipeta de 0,5-10 μL e depositá-lo num frasco para injetáveis de microcentrífuga. Usando uma pipeta de transferência, aspirar o meio ao redor do tecido coletado para limitar os sais na amostra, que interferem na análise da EMRH em etapas posteriores.
NOTA: Use frascos para injetáveis que minimizem a adsorção de proteínas em superfícies plásticas para minimizar as perdas de proteína nas superfícies dos frascos durante as etapas posteriores do fluxo de trabalho. - Para isolar por FACS, transferir ~5-8 embriões desviados para cada poço de uma placa de 12 poços contendo ~5 mL de tampão Newport 2.0. Dissociar os embriões nutando a placa a 80 rpm por 20-30 min à temperatura ambiente13.
NOTA: Embriões/larvas com idade superior ao estágio 22 possuem abundantes proteínas da matriz extracelular, dificultando a dissociação em células separadas. Abordagens enzimáticas adicionais podem ser adaptadas para dissociar tecidos de embriões mais velhos, como descrito anteriormente24. - Purificar as células fluorescentemente marcadas da suspensão usando FACS como descrito anteriormente24.
- Células de pellet por centrifugação e descarte o sobrenadante.
NOTA: Use baixa velocidade de centrifugação (400 × g) e temperatura (4 °C) para evitar a lise celular. Se estiver usando albumina de soro bovino (BSA) para FACS, lave o pellet de células para reduzir a interferência de BSA durante a detecção de HRMS. Ressuspenda suavemente as células em solução salina tamponada com fosfato (PBS) e centrifugue novamente para as células enxaguadas em pellets. Retire o sobrenadante PBS líquido. - Congelar rapidamente as células isoladas colocando o frasco para injetáveis da amostra em gelo seco ou azoto líquido.
NOTA: Mantenha as amostras (tecidos ou células) resfriadas (por exemplo, em gelo) durante as etapas de processamento. Congele as células com o mínimo possível de meios ao redor da amostra para facilitar o processamento a jusante. - Conservar as amostras a -80 °C até à análise do HRMS.
- Transferir os embriões injetados para SS 0,5x em uma placa de Petri e cultivá-los entre 14-25 °C até atingir o estágio de desenvolvimento desejado.
4. Analisar as proteínas por espectrometria de massas
A caracterização proteômica dos tecidos ou células isoladas é baseada em uma série de etapas estabelecidas na EMAR. A Figura 2 ilustra as etapas do fluxo de trabalho bioanalítico. O protocolo de coleta de amostras usado aqui é compatível com fluxos de trabalho de proteômica bottom-up11, middle-down25 ou top-down26 . A seguir, descreve-se a estratégia bottom-up utilizada neste estudo, que tem se mostrado sensível, quantitativa e adaptável a diversos tipos de espectrômetros de massa. Após a extração e digestão enzimática das proteínas, os peptídeos resultantes são separados, seguindo-se a análise da EMAR.
- Processar os tecidos/células individuais
- Para análise de célula única por CE, aquecer a amostra a 60 °C por ~15 min para desnaturar proteínas e, em seguida, equilibrar a amostra à temperatura ambiente (TR, ~5 min)18,21.
NOTA: Ao contrário do trabalho com tecidos, as etapas de redução e alquilação são ignoradas para limitar as perdas de proteína durante a preparação da amostra a partir de células únicas. O preparo de amostra auxiliado por filtro (FASP)27,28, outras estratégias de pote único 29 e abordagens microfluídicas30 podem ser adotadas para minimizar as perdas proteicas durante o preparo da amostra. - Para análise por nanoLC, lisar até 5 tecidos dissecados em 50 μL de tampão de lise (~100 μg de proteína total). Facilite o processo pipetando a amostra para cima e para baixo algumas vezes.
- Incubar o lisado a 4 °C durante 10 min e, em seguida, pellet os restos celulares e plaquetas da gema por centrifugação a 4.500 × g a 4 °C. Transfira o sobrenadante para um frasco de microcentrífuga limpo e adicione 10% de SDS para obter uma concentração final de 1% de SDS no lisado (v/v).
- Para tecidos, siga os passos 4.1.5-4.1.7.
- Adicionar 0,5 M de ditiotreitol ao lisado para obter uma concentração final de ~25 mM (por exemplo, 2,5 μL de ditiotretol 0,5 M a 50 μL de lisado) e incubar o lisado por 30 min a 60 °C para reduzir quimicamente as ligações dissulfeto nas proteínas.
- Adicionar iodoacetamida 0,5 M para obter uma concentração final de ~75 mM no lisado e incubar a mistura por 15 min no TR no escuro (Figura 2).
- Adicionar 0,5 M de ditiotreitol igual ao volume inicial (por exemplo, 2,5 μL de ditiotretol 0,5 M a 50 μL de lisado) para extinguir os reagentes remanescentes da reação de alquilação.
CUIDADO: Iodoacetamida e ditiotreitol podem causar danos graves à pele e aos olhos após exposição direta. Use EPIs apropriados ao manusear esses produtos químicos. - Purificar proteínas por precipitação. A precipitação à base de clorofórmio-metanol apresenta bom desempenho31. Esse protocolo também é adaptável a outros tipos de abordagens de precipitação32.
NOTA: Para análise de célula única, onde as perdas de proteína são preocupantes, pule a etapa de precipitação para CE-HRMS. - Secar o precipitado proteico num concentrador a vácuo (4-37 °C) e, em seguida, ressuspender o proteoma extraído em 50 μL de bicarbonato de amónio 50 mM. Estimar a concentração de proteína usando um ensaio colorimétrico de proteína total para determinar a quantidade de enzima necessária para a digestão (por exemplo, ensaio de proteína do ácido bicinconínico).
- Digerir as proteínas para peptídeos. Adicionar tripsina (estoque de 1 μg/μL) para obter uma relação protease:proteína de 1:50 e incubar a mistura a 37 °C por até 5 h para amostras unicelulares e até 14 h para amostras de tecido. Consulte as recomendações específicas do fornecedor para a reação.
NOTA: A digestão com tripsina por concentrações superiores a 14 h ou superiores pode introduzir clivagens inespecíficas à sequência da proteína, desafiando a identificação proteica33. - Quantificar a concentração total de peptídeos usando um ensaio colorimétrico.
- OPCIONAL: Para quantificação de multiplexação, marque os peptídeos de cada amostra com uma etiqueta de massa isobárica diferente seguindo instruções específicas do fornecedor. Misture os peptídeos com código de barras em proporções iguais por amostra de peptídeo.
NOTA: Garanta rotulagem e mistura precisas para evitar vieses quantitativos. Para amostras limitadas em quantidade ou amostras de célula única, um canal carreador baseado em TMT composto por tecidos/células agrupados pode ser incluído para minimizar as perdas de amostra durante as etapas subsequentes de separação e aumentar a sensibilidade de proteínas de menor abundância34. - OPCIONAL: Peptídeos de dessal para remover sais e contaminantes (por exemplo, reagentes de etiqueta de massa isobárica não reagidos) em uma coluna/ponta de spin de fase reversa C18 para proteger o sistema LC-MS.
- OPCIONAL: Fracionar (por exemplo, fracionamento de fase reversa de pH médio ou alto) a mistura peptídica para detecção mais profunda do proteoma através de plataformas manuais ou automáticas. Use fase estacionária C18 contendo pontas para fracionar baixas quantidades (1-10 μg) de digestores peptídicos.
- Secar a mistura de peptídeos a 60 °C em um concentrador a vácuo.
- Conservar a mistura de péptidos a -80 °C até à medição.
- Para análise de célula única por CE, aquecer a amostra a 60 °C por ~15 min para desnaturar proteínas e, em seguida, equilibrar a amostra à temperatura ambiente (TR, ~5 min)18,21.
- Separe os peptídeos
NOTA: Após a extração e digestão enzimática das proteínas, os peptídeos resultantes são separados por nanoLC ou CE e ionizados por ESI para sequenciamento por EMAR em tandem. A separação nanoLC de fase reversa é ideal para peptídeos que acumulam ~150 ng a ~1 μg por análise. CE fornece sensibilidade complementar para peptídeos que variam de femtogramas a <100 ng. Various custom-built and commercial CE-ESI interfaces allow for ready coupling of CE to HRMS with robust performance35 e são cada vez mais usados para análise de célula única18,36,37.- Para separar usando CE, siga as etapas 4.2.2-4.2.7.
NOTA: No que se segue, o uso da plataforma CE personalizada para medir os peptídeos é descrito. Protocolos para construir e usar este instrumento CE foram fornecidos anteriormente38, juntamente com um experimento visualizado sobre o uso de pequenas moléculas20. Alternativamente, essas medições podem ser realizadas em um sistema CE comercial, como o AB SCIEX CESI, Agilent 7100 ou equivalente. - Reconstituir o digestor proteico em 1-2 μL do solvente da amostra, vórtice para misturar a amostra e centrifugar-se a 10.000 x g por 2 min para detritos de células pellets.
NOTA: A remoção dos detritos celulares minimiza a probabilidade de entupimento do capilar CE, prolongando assim a vida útil do sistema de separação e aumentando o rendimento da medição. - Inicialize o instrumento CE-ESI lavando o capilar CE com o BGE.
- Validar o desempenho instrumental usando um padrão conhecido (por exemplo, citocromo C ou BSA digest, peptídeos de angiotensina).
NOTA: Recomenda-se avaliar o instrumento em termos de precisão de massa, sensibilidade de detecção, reprodutibilidade e faixa dinâmica linear de quantificação antes de medir amostras preciosas. Notas adicionais sobre validação e solução de problemas de desempenho do CE-ESI-MS estão listadas em outra publicação18,20,38. - Injete ~1-10 nL da amostra no capilar de separação CE.
NOTA: Este estudo usa capilar de sílica fundida de ~1 m de comprimento (diâmetro interno/externo de 40/110 μm) com a configuração de fluxo de bainha eletrocineticamente bombeada. Instrumentos CE comerciais geralmente requerem a apresentação de 5-10 μL de amostra em um microfrasco para injetável. A plataforma CE 18,38 personalizada é compatível com ~250 nL a 1 μL de amostra depositada em um microfrasco de carregamento de amostra. - Transfira a extremidade de entrada do capilar de separação CE para o BGE.
- Inicie a separação eletroforética aumentando gradualmente a tensão de separação CE do solo terrestre (por exemplo, passo a passo ao longo de 1 minuto). Potenciais de 20-28 kV com corrente abaixo de ~10 μA garantem desempenho instrumental estável e reprodutível para análise.
- Para separar usando nanoLC, siga as etapas 4.2.9-4.2.12.
- Ressuspender a amostra de peptídeo na Fase A Móvel. A concentração da amostra e o seu volume injetável dependem do sistema e da coluna LC disponíveis. Neste estudo, ~250 ng-1 μg de proteína digestiva é injetada em 1-20 μL de volume de amostra em uma coluna de leito empacotado C18 (75 μm de diâmetro interno, tamanho de partícula de 2 μm com poros de 100 Å, coluna de separação de 25 cm de comprimento).
- Transfira a amostra para um frasco para injetáveis LC.
NOTA: Certifique-se de que não existem bolhas de ar no frasco para injetáveis, que podem danificar a coluna analítica. Frascos com inserções podem ser usados para amostras de tecido de baixo volume ou de célula única. - Carregue ~200 ng a 2 μg de amostra de peptídeo na coluna analítica C18.
NOTA: Opcionalmente, os peptídeos podem ser carregados em uma coluna de armadilha para dessalinização antes da separação analítica. Por exemplo, uma coluna de armadilha C18 com 0,1 mm de diâmetro interno, tamanho de partícula de 5 μm, tamanho de poro de 100 Å, comprimento de 20 mm. Peptídeos de dessal com tampão A a 100% a uma taxa de fluxo de 5 μL/min por 5 min antes do início do gradiente de separação. - Separe os peptídeos usando eluição de gradiente. A uma taxa de fluxo de 300 nL/min, o gradiente de 120 min utilizado neste estudo é o seguinte: 0-5 min 2% B, 5-85 min 2-35% B, 86-90 min 70% B, 91-120 min 2% B.
- Para separar usando CE, siga as etapas 4.2.2-4.2.7.
- Ionizar os peptídeos por ESI
NOTA: O capilar CE ou nanoLC é mais tipicamente acoplado em uma fonte ESI para ionização. As interfaces CE-ESI de microfluxo (ponta romba) e nanofluxo (ponta cônica39 e eletrocinética de fluxo de bainha bombeada36 ) foram desenvolvidas anteriormente.- Forneça os peptídeos de separação em uma fonte de íons de eletrospray para ionização usando uma interface ESI comercial ou personalizada. Para análise de célula única CE-ESI-MS em embriões de Xenopus , use uma interface de baixo fluxo eletrocineticamente bombeada em que a saída capilar CE é fechada em um emissor de borossilicato puxado.
- Verifique o fluxo de líquido através do emissor de eletrospray usando uma câmera e inspecione visualmente a configuração em busca de possíveis vazamentos.
- Ajuste a tensão de eletrospray para ~2,5 kV para iniciar a fonte ESI (vs. Terra terra).
- Garanta um nanospray estável para análise de HRMS monitorando a corrente total de íons. Ajuste a tensão de eletrospray e a distância do emissor à entrada HRMS para obter uma pulverização estável (<15% de desvio padrão relativo na intensidade total).
- Detectar os peptídeos
NOTA: A detecção de peptídeos segue diferentes considerações instrumentais para peptídeos de massa isobárica marcados e não marcados e depende do tipo de espectrômetro de massa disponível. Este estudo usa um espectrômetro de massa tribrida orbitrap de acordo com as etapas a seguir.- Adquira eventos MS1 com as configurações: Analisador, orbitrap; Resolução espectral, 120.000 de largura total na metade máxima (FWHM); tempo máximo de injeção (TI), 50 ms; controle automático de ganho (AGC), 4 x 105 contagens; microsvarreduras, 1.
- Para sequenciar peptídeos, fragmentos de íons precursores para detecção no analisador de armadilhas de íons utilizando as configurações: modo de fragmentação, dissociação de colisão de maior energia (HCD); gás de colisão de nitrogênio; energia de colisão, 32% energia de colisão normalizada (NCE); TI máxima, 70 ms; AGC, 1 x 104 contagens; microsvarreduras, 1.
- OPCIONAL: Quantificar peptídeos marcados com TMT usando EMRH tandem/multiestágio (MS2/MS3). Para MS3 empregando seleção de precursor síncrono, as configurações instrumentais típicas são as seguintes. Varreduras de estágio único (MS1) que levantam os íons mais abundantes são dissociadas via aquisição dependente de dados usando os parâmetros: modo de fragmentação MS2 , dissociação induzida por colisão (CID); gás de colisão, hélio; energia de colisão, 35% NCE; analisador para íons de fragmento, armadilha de íons seguindo as configurações: TI máximo, 50 ms; AGC, 5 x 104 contagens; microsvarreduras, 1. Selecionar 10 íons MS2 e fragmentá-los com HCD em nitrogênio (65% NCE). Detecte íons de fragmento MS3 usando as seguintes configurações: resolução Orbitrap 15.000 FWHM, TI máxima, 120 ms; AGC, 1 × 105 contagens; microsvarreduras, 1.
NOTA: Diferentes métodos e parâmetros de aquisição de MS podem ser usados para amostras marcadas seguindo as recomendações do fornecedor, conforme descrito em outro lugar11,40.
- Analise os dados
NOTA: As proteínas são identificadas e quantificadas usando pacotes avançados de bioinformática. A fidelidade das identificações é calculada usando um banco de dados de chamariz, expresso como a taxa de descoberta falsa (FDR) no nível de peptídeos e proteínas.- Processar os dados usando pacotes de software comerciais ou de código aberto (revisados na referência41). Combine os dados brutos com um banco de dados que foi preparado concatenando o proteoma Xenopus 9.2 com o banco de dados PHROG derivado de mRNA42.
NOTA: Os parâmetros de busca são: enzima de digestão, tripsina; clivagens perdidas, até 2; modificação variável, oxidação de metionina; modificação estática, carbamidometilação da cisteína; tolerância à massa do precursor, 10 ppm; tolerância à massa dos fragmentos, 0,6 Da; comprimento mínimo do peptídeo, 5; fidelidade de identificação, <1% FDR para peptídeos e proteínas. Sem alquilação a peptídeos, a carbamidometilação como uma modificação estática é excluída durante a busca no banco de dados (por exemplo, para análise de célula única). - Quantificar a abundância de proteínas por meio de estratégias label-free43 ou label-based 44,45.
- OPCIONAL: Anotar proteínas para ontologia gênica. PantherDB46, Reactome47 ou Xenbase23 podem ser usados.
- OPCIONAL: Quantificar a abundância de proteínas e as diferenças nas abundâncias de proteínas entre tipos de células/tecidos usando pacotes de software/ferramentas web, como o Trans-Proteomic Pipeline48, Perseus49 e Orange50.
NOTA: Considerações adicionais sobre o planejamento experimental e as opções de software foram revisadas em outra publicação41,51. - OPCIONAL: Avalie os resultados usando bases de conhecimento, como STRING52 e BioPlex Display 53 para interações proteína-proteína conhecidas e PhosphoSiteplus54 para fosforilações. Para analisar motivos e domínios que são representados no proteoma, use ferramentas web, como Simple Modular Architecture Research (SMART)55.
- Processar os dados usando pacotes de software comerciais ou de código aberto (revisados na referência41). Combine os dados brutos com um banco de dados que foi preparado concatenando o proteoma Xenopus 9.2 com o banco de dados PHROG derivado de mRNA42.
Access restricted. Please log in or start a trial to view this content.
Resultados
Este protocolo possibilitou o estudo de proteínas em células isoladas e suas linhagens à medida que estabelecem tecidos em embriões de X. laevis. A Figura 1 ilustra uma dessas aplicações da abordagem para estudar proteínas em células de tecido neural e o ectoderma neural recém-induzido no embrião. Como mostrado na Figura 1A, o fluxo de trabalho bioanalítico integrou ferramentas tradicionais da biologia celular e do desenvolvimento para identi...
Access restricted. Please log in or start a trial to view this content.
Discussão
Este protocolo possibilita a caracterização da expressão de proteínas em linhagens celulares identificadas em embriões da espécie Xenopus. Derivada do HRMS, a metodologia combina especificidade requintada na identificação molecular, capacidade de detecção de multiproteínas sem sondas moleculares (geralmente centenas a milhares de proteínas diferentes) e capacidade de quantificação. A adaptabilidade a ferramentas clássicas e fluxos de trabalho em (neuro)biologia celular e de desenvolvimento expande...
Access restricted. Please log in or start a trial to view this content.
Divulgações
Os autores declaram não haver interesses concorrentes.
Agradecimentos
Somos gratos a Jie Li (University of Maryland, College Park) pelas valiosas discussões sobre dissociação embrionária e FACS. Agradecemos a Vi M. Quach e Camille Lombard-Banek pelo auxílio na preparação de amostras e coleta de dados em estudos anteriores exemplificando as aplicações proteômicas destacadas neste protocolo. Partes deste trabalho foram apoiadas pela National Science Foundation sob o número de prêmio IOS-1832968 CAREER (para P.N.), os Institutos Nacionais de Saúde sob o número de prêmio R35GM124755 (para P.N.), o Programa de Parceria do Instituto Nacional do Câncer da Universidade de Maryland (para P.N.) e os prêmios de pesquisa da Fundação COSMOS Club (para A.B.B. e L.R.P.).
Access restricted. Please log in or start a trial to view this content.
Materiais
| Name | Company | Catalog Number | Comments |
| Acetonitrile (LC-MS-grade) | Fisher Scientific | A955 | |
| Agarose | ThermoFisher Scientific | R0492 | |
| Ammonium bicarbonate | Fisher Scientific | A643-500 | |
| Analytical Column | Thermo Scientific | 164941 | |
| Analytical microbalance | Mettler-Toledo | XSE105DU | |
| Automatic peptide fractionation platform | Agilent | 1260 Infinity II | |
| Borosilicate Capillaries | Sutter Instruments Co. | B100-50-10 | |
| Borosilicate Capillaries (for making Emmitters) | Sutter Instruments | B100-75-10 | |
| C18 spin columns (for desalting) | ThermoFisher Scientific | 89870 | |
| Camera ro monitor electrospray | Edmund Optics Inc. | EO-2018C | |
| Combretastatin A4 | Millipore Sigma | C7744 | |
| Commercial CESI system | AB SCIEX | CESI | |
| (Cyclohexylamino)-1-propanesulfonic acid (CAPS) | VWR | 97061-492 | |
| Cytochalasin D | Millipore Sigma | C8273 | |
| Dextran, Alexa Fluor 488; 10,000 MW, Anionic, Fixable | ThermoFisher Scientific | D22910 | |
| Diothiothreitol | Fisher Scientific | FERR0861 | |
| Dumont #5 Forceps | Fine Science Tools | 11252-30 | |
| EDTA | Fisher Scientific | AAJ62786AP | |
| Epifluorescence light source | Lumencore | AURA III | |
| Eppendorf LoBing microcentrifuge tubes: protein | Fisher Scientific | 13-698-793 | |
| Formic acid (LC-MS-grade) | Fisher Scientific | A117-50 | |
| Freezer (-20 °C) | Fisher Scientific | 97-926-1 | |
| Freezer (-80 °C) | Thermo Scientific | TSX40086A | |
| Fused silica capillary | Molex | 1088150596 | |
| Heat Block | Benchmark | BSH300 | |
| High pressure liquid Chromatography System | ThermoFisher Scientific | Dionex Ultimate 3000 RSLC nanosystem | |
| High voltage power supply | Spellman | CZE1000R | |
| High-resolution Mass Spectrometer | ThermoFisher Scientific | Orbitrap Fusion Lumos Tribrid Mass Spectrometer | |
| HPLC caps | Thermo Scientific | C4013-40A | |
| HPLC Vials | Thermo Scientific | C4013-11 | |
| Illuminator e.g. Goosenecks | Nikon | C-FLED2 | |
| Ingenuity Pathway Analysis | Qiagen | ||
| Iodoacetamide | Fisher Scientific | AC122275000 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456 | |
| Methanol (LC-MS-grade) | Fisher Scientific | A456-4 | |
| Microcapillary puller | Suttor Instruments | P-2000 | |
| Microinjector | Warner Instrument, Handem, CT | PLI-100A | |
| Micropippette puller | Sutter Instruments Co. | P-1000 | |
| MS data analysis software, commercial | ProteomeDiscoverer | ||
| MS data analysis software, opensource | MaxQuant | ||
| non-idet 40 substitute | Millipore Sigma | 11754599001 | |
| Petri dish 60 mm and 80 mm | Fisher Scientific | S08184 | |
| Pierce 10 µL bed Zip-tips (for desalting) | ThermoFisher Scientific | 87782 | |
| Pierce bicinchoninic acid protein assay kit | ThermoFisher Scientific | 23225 | |
| Pierce quantitative colorimetric peptide assay | ThermoFisher Scientific | 23275 | |
| Pierce Trypsin Protease (MS Grade) | Fisher Scientific | PI90058 | |
| Protein LoBind vials | Eppendorf | 0030108434 , 0030108442 | |
| Refrigerated Centrifuge | Eppendorf | 5430R | |
| Refrigerated Incubator | Thermo Scientific | PR505755R/3721 | |
| sodium isethionate | Millipore Sigma | 220078 | |
| sodium pyrophosphate | Sigma Aldrich | 221368-100G | |
| Stainless steel BGE vial | Custom-Built | ||
| Stainless steel sample vials | Custom-Built | ||
| Stereomicroscope (objective 10x) | Nikon | SMZ 1270, SZX18 | |
| Sucrose | VWR | 97063-790 | |
| Syringe pumps (2) | Harvard Apparatus | 704506 | |
| Syringes (gas-tight): 500–1000 µL | Hamilton | 1750TTL | |
| Transfer pipettes (Plastic, disposable) | Fisher Scientific | 13-711-7M | |
| Trap Column | Thermo Scientific | 164750 | |
| Tris-HCl (1 M solution) | Fisher Scientific | AAJ22638AP | |
| Vacuum concentrator capable of operation at 4–10 °C | Labconco | 7310022 | |
| Vortex-mixer | Benchmark | BS-VM-1000 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| Water (LC-MS-grade) | Fisher Scientific | W6 | |
| XYZ translation stage | Thorlabs | PT3 | |
| XYZ translation stage | Custom-Built |
Referências
- Shoemaker, L. D., Kornblum, H. I. Neural Stem Cells (NSCs) and Proteomics. Molecular & Cellular Proteomics. 15 (2), 344-354 (2016).
- Cervenka, J., et al. Proteomic characterization of human neural stem cells and their secretome during in vitro differentiation. Frontiers in Cellular Neuroscience. 14, 612560(2021).
- Christian, J. L. Morphogen gradients in development: From form to function. Wiley Interdisciplinary Reviews. Developmental Biology. 1 (1), 3-15 (2012).
- Gurdon, J. B., Elsdale, T. R., M, F. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature. 182, 64-65 (1958).
- Harland, R. M., Grainger, R. M. Xenopus research: metamorphosed by genetics and genomics. Trends in Genetics. 27 (12), 507-515 (2011).
- Moody, S. A. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Developmental Biology. 119 (2), 560-578 (1987).
- Moody, S. A. Fates of the blastomeres of the 32-cell stage Xenopus embryo. Developmental Biology. 122 (2), 300-319 (1987).
- Dale, L., Slack, J. M. W. Fate map for the 32-cell stage of Xenopus laevis. Development. 99 (4), 527-551 (1987).
- Sun, L. L., et al. Single cell proteomics using frog (Xenopus laevis) blastomeres isolated from early stage embryos, which form a geometric progression in protein content. Analytical Chemistry. 88 (13), 6653-6657 (2016).
- Lombard-Banek, C., Moody, S. A., Nemes, P. Single-cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angewandte Chemie-International Edition. 55 (7), 2454-2458 (2016).
- Zhang, Y. Y., Fonslow, B. R., Shan, B., Baek, M. C., Yates, J. R. Protein analysis by shotgun/bottom-up proteomics. Chemical Reviews. 113 (4), 2343-2394 (2013).
- Sive, H. L., Grainger, R. M., Harland, R. M. Early development of Xenopus laevis: A laboratory manual. , Cold Spring Harbor Laboratory Press. New York. (2000).
- Briggs, J. A., et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 360 (6392), (2018).
- Gupta, M., Sonnett, M., Ryazanova, L., Presler, M., Wuhr, M. Quantitative proteomics of xenopus embryos I, sample preparation. Xenopus. Methods in Molecular Biology. Vleminckx, K. 1865, Humana Press Inc. NY. 175-194 (2018).
- Baxi, A. B., Lombard-Banek, C., Moody, S. A., Nemes, P. Proteomic characterization of the neural ectoderm fated cell clones in the Xenopus laevis embryo by high-resolution mass spectrometry. ACS Chemical Neuroscience. 9 (8), 2064-2073 (2018).
- Moody, S. A. Cell lineage analysis in Xenopus embryos. Methods in Molecular Biology. 135, 331-347 (2000).
- Sater, A. K., Moody, S. A. Using Xenopus to understand human diseases and developmental disorders. Genesis. 55 (1-2), 1-14 (2017).
- Lombard-Banek, C., Choi, S. B., Nemes, P. Enzyme Activity in Single Cells. Methods in Enzymology. Allbritton, N. L., Kovarik, M. L. 628, 263-292 (2019).
- Lombard-Banek, C., Moody, S. A., Nemes, P. High-sensitivity mass spectrometry for probing gene translation in single embryonic cells in the early frog (Xenopus) embryo. Frontiers in Cell and Developmental Biology. 4, 11(2016).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. Microprobe capillary electrophoresis mass spectrometry for single-cell metabolomics in live frog (Xenopus laevis) embryos. Journal of Visualized Experiments: JoVE. (130), e56956(2017).
- Lombard-Banek, C., Moody, S. A., Manzin, M. C., Nemes, P. Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: developing cell clones in live Xenopus laevis and zebrafish embryos. Analytical Chemistry. 91 (7), 4797-4805 (2019).
- Klein, S. L. The first cleavage furrow demarcates the dorsal-ventral axis in Xenopus embryos. Developmental Biology. 120 (1), 299-304 (1987).
- Karimi, K., et al. Xenbase: a genomic, epigenomic and transcriptomic model organism database. Nucleic Acids Research. 46 (1), 861-868 (2018).
- Kakebeen, A. D., Chitsazan, A. D., Wills, A. E. Tissue disaggregation and isolation of specific cell types from transgenic Xenopus appendages for transcriptional analysis by FACS. Developmental Dynamics. 250 (9), 1381-1392 (2021).
- Garcia, B. A. What does the future hold for top down mass spectrometry. Journal of the American Society for Mass Spectrometry. 21 (2), 193-202 (2010).
- Toby, T. K., Fornelli, L., Kelleher, N. L. Progress in top-down proteomics and the analysis of proteoforms. Annual Review of Analytical Chemistry. (Palo Alto Calif). 9 (1), 499-519 (2016).
- Zhang, Z. B., Dubiak, K. M., Huber, P. W., Dovichi, N. J. Miniaturized filter-aided sample preparation (MICRO-FASP) method for high throughput, ultrasensitive proteomics sample preparation reveals proteome asymmetry in Xenopus laevis Embryos. Analytical Chemistry. 92 (7), 5554-5560 (2020).
- Wisniewski, J. R. Microbial Proteomics: Methods and Protocols.Methods in Molecular Biology. Becher, D. 1841, Humana Press Inc. NY. 3-10 (2018).
- Hughes, C. S., et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols. 14 (1), 68-85 (2019).
- Zhu, Y., et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nature Communications. 9, 882(2018).
- Wessel, D., Flugge, U. I. A method for the quantitative recovery of protein in dilute-solution in the presence of detergents and lipids. Analytical Biochemistry. 138 (1), 141-143 (1984).
- Jiang, L., He, L., Fountoulakis, M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. Journal of Chromatography A. 1023 (2), 317-320 (2004).
- Hildonen, S., Halvorsen, T. G., Reubsaet, L. Why less is more when generating tryptic peptides in bottom-up proteomics. Proteomics. 14 (17-18), 2031-2041 (2014).
- Budnik, B., Levy, E., Harmange, G., Slavov, N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology. 19, 161(2018).
- Drouin, N., et al. Capillary electrophoresis-mass spectrometry at trial by metabo-ring: effective electrophoretic mobility for reproducible and robust compound annotation. Analytical Chemistry. 92 (20), 14103-14112 (2020).
- Sun, L. L., Zhu, G. J., Zhang, Z. B., Mou, S., Dovichi, N. J. Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. Journal of Proteome Research. 14 (5), 2312-2321 (2015).
- DeLaney, K., Sauer, C. S., Vu, N. Q., Li, L. J. Recent advances and new perspectives in capillary electrophoresis-mass spectrometry for single cell "omics". Molecules. 24 (1), 21(2019).
- Nemes, P., Rubakhin, S. S., Aerts, J. T., Sweedler, J. V. Qualitative and quantitative metabolomic investigation of single neurons by capillary electrophoresis electrospray ionization mass spectrometry. Nature Protocols. 8 (4), 783-799 (2013).
- Choi, S. B., Zamarbide, M., Manzini, M. C., Nemes, P. Tapered-tip capillary electrophoresis nano-electrospray ionization mass spectrometry for ultrasensitive proteomics: the mouse cortex. Journal of the American Society for Mass Spectrometry. 28 (4), 597-607 (2017).
- Pino, L. K., Rose, J., O'Broin, A., Shah, S., Schilling, B. Emerging mass spectrometry-based proteomics methodologies for novel biomedical applications. Biochemical Society Transactions. 48 (5), 1953-1966 (2020).
- Chen, C., Hou, J., Tanner, J. J., Cheng, J. L. Bioinformatics methods for mass spectrometry-based proteomics data analysis. International Journal of Molecular Sciences. 21 (8), 25(2020).
- Peshkin, L., et al. On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Developmental Cell. 35 (3), 383-394 (2015).
- Cox, J., et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics. 13 (9), 2513-2526 (2014).
- Gygi, S. P., et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature Biotechnology. 17 (10), 994-999 (1999).
- Thompson, A., et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry. 75 (8), 1895-1904 (2003).
- Mi, H. Y., et al. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive api. Nucleic Acids Research. 49, 394-403 (2021).
- Schmidt, E., et al. On the Move Federated Workshops. , Springer, Verlag. Berlin. 710-719 (2006).
- Deutsch, E. W., et al. Trans-Proteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clinical Applications. 9 (7-8), 745-754 (2015).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Demsar, J., et al. Orange: Data mining toolbox in Python. Journal of Machine Learning Research. 14, 2349-2353 (2013).
- Oberg, A. L., Vitek, O. Statistical design of quantitative mass spectrometry-based proteomic experiments. Journal of Proteome Research. 8 (5), 2144-2156 (2009).
- Jensen, L. J., et al. STRING 8 - a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, 412-416 (2009).
- Schweppe, D. K., Huttlin, E. L., Harper, J. W., Gygi, S. P. BioPlex display: an interactive suite for large-scale AP-MS protein-protein interaction data. Journal of Proteome Research. 17 (1), 722-726 (2018).
- Hornbeck, P. V., et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Research. 43, 512-520 (2015).
- Letunic, I., Khedkar, S., Bork, P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Research. 49, 458-460 (2021).
- Lombard-Banek, C., et al. In vivo subcellular mass spectrometry enables proteo-metabolomic single-cell systems biology in a chordate embryo developing to a normally behaving tadpole (X. laevis). Angewandte Chemie-International Edition. 60 (23), 12852-12858 (2021).
- Lombard-Banek, C., Reddy, S., Moody, S. A., Nemes, P. Label-free quantification of proteins in single embryonic cells with neural fate in the cleavage-stage frog (Xenopus laevis) embryo using capillary electrophoresis electrospray ionization high-resolution mass spectrometry (CE-ESI-HRMS). Molecular & Cellular Proteomics. 15 (8), 2756-2768 (2016).
- Saha-Shah, A., et al. Single cell proteomics by data-independent acquisition to study embryonic asymmetry in Xenopus laevis. Analytical Chemistry. 91 (14), 8891-8899 (2019).
- Onjiko, R. M., Portero, E. P., Moody, S. A., Nemes, P. In situ microprobe single-cell capillary electrophoresis mass spectrometry: metabolic reorganization in single differentiating cells in the live vertebrate (Xenopus laevis) embryo. Analytical Chemistry. 89 (13), 7069-7076 (2017).
- Perez-Riverol, Y., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research. 47, 442-450 (2019).
Access restricted. Please log in or start a trial to view this content.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados