
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Imagem de Longo Prazo de Populações Neurais Identificadas Usando Microprismas em Animais em Movimento Livre e Cabeça Fixada
Neste Artigo
Resumo
Quando integrada com uma placa de cabeça e um design óptico compatível com microscópios de um e dois fótons, a lente de microprisma apresenta uma vantagem significativa na medição de respostas neurais em uma coluna vertical sob diversas condições, incluindo experimentos bem controlados em estados fixos na cabeça ou tarefas comportamentais naturais em animais em movimento livre.
Resumo
Com o avanço da microscopia multifóton e das tecnologias moleculares, a imagem por fluorescência está crescendo rapidamente para se tornar uma abordagem poderosa para estudar a estrutura, a função e a plasticidade dos tecidos cerebrais vivos. Em comparação com a eletrofisiologia convencional, a microscopia de fluorescência pode capturar a atividade neural, bem como a morfologia das células, permitindo registros de longo prazo das populações de neurônios identificadas em resolução unicelular ou subcelular. No entanto, imagens de alta resolução normalmente requerem uma configuração estável e fixa na cabeça que restringe o movimento do animal, e a preparação de uma superfície plana de vidro transparente permite a visualização de neurônios em um ou mais planos horizontais, mas é limitada no estudo dos processos verticais que atravessam diferentes profundidades. Aqui, descrevemos um procedimento para combinar uma fixação da placa cefálica e um microprisma que fornece imagens multicamadas e multimodais. Este preparo cirúrgico não apenas dá acesso a toda a coluna do córtex visual do camundongo, mas permite imagens de dois fótons em uma posição fixa da cabeça e imagens de um fóton em um paradigma de movimento livre. Usando essa abordagem, pode-se amostrar populações celulares identificadas em diferentes camadas corticais, registrar suas respostas sob estados de cabeça fixa e movimento livre, e rastrear as mudanças de longo prazo ao longo de meses. Assim, este método fornece um ensaio abrangente dos microcircuitos, permitindo a comparação direta das atividades neurais evocadas por estímulos bem controlados e sob um paradigma comportamental natural.
Introdução
O advento da imagem fluorescente in vivo de dois fótons 1,2, combinando as novas tecnologias em sistemas ópticos e indicadores de fluorescência geneticamente modificados, surgiu como uma técnica poderosa em neurociência para investigar a intrincada estrutura, função e plasticidade no cérebro vivo 3,4. Em particular, esta modalidade de imagem oferece uma vantagem incomparável sobre a eletrofisiologia tradicional por capturar tanto a morfologia quanto as atividades dinâmicas dos neurônios, facilitando o rastreamento em longo prazo dos neurônios identificados5,6,7,8.
Apesar de seus pontos fortes, a aplicação de imagens de fluorescência de alta resolução muitas vezes requer uma configuração estática e fixa na cabeça que restringe a mobilidade do animal 9,10,11. Além disso, o uso de uma superfície de vidro transparente para visualização de neurônios restringe as observações a um ou mais planos horizontais, limitando a exploração da dinâmica de processos verticais que se estendem por diferentes profundidades corticais12.
Abordando essas limitações, o presente estudo delineia um procedimento cirúrgico inovador que integra fixação da placa cefálica, microprisma e miniscópio para criar uma modalidade de imagem com capacidades multicamadas e multimodais. O microprisma permite observar o processamento vertical ao longo da coluna cortical 13,14,15,16, o que é fundamental para entender como a informação é processada e transformada à medida que se move através das diferentes camadas do córtex e como o processamento vertical é alterado durante as mudanças plásticas. Além disso, permite a obtenção de imagens das mesmas populações neurais em um paradigma de cabeça fixa e em um ambiente de movimento livre, englobando os versáteis cenários experimentais 17,18,19: por exemplo, a fixação da cabeça é frequentemente necessária para paradigmas bem controlados, como avaliação da percepção sensorial e registros estáveis sob o paradigma de 2 fótons, enquanto o movimento livre oferece um ambiente mais natural e flexível para estudos comportamentais. Portanto, a capacidade de realizar uma comparação direta em ambos os modos é crucial para aprofundar nossa compreensão dos microcircuitos que permitem respostas funcionais flexíveis.
Em essência, a integração da fixação da placa cefálica, microprisma e miniscópio em imagens de fluorescência oferece uma plataforma promissora para sondar os meandros da estrutura e funcionalidade do cérebro. Os pesquisadores podem amostrar populações celulares identificadas em várias profundidades que abrangem todas as camadas corticais, comparar diretamente suas respostas em paradigmas bem controlados e naturais e monitorar suas alterações de longo prazo ao longo dos meses20. Esta abordagem oferece informações valiosas sobre como essas populações neurais interagem e mudam ao longo do tempo sob diferentes condições experimentais, fornecendo uma janela para a natureza dinâmica dos circuitos neurais.
Protocolo
Todos os experimentos foram conduzidos de acordo com o UK Animals (Scientific Procedures) Act 1986 sob licenças pessoais e de projeto aprovadas e emitidas pelo Ministério do Interior do Reino Unido após revisão ética apropriada. Linhagens transgênicas adultas CaMKII-TTA; GCaMP6S-TRE21 foram criados e seus filhotes utilizados no experimento. Para a segurança dos experimentadores e a manutenção de condições estéreis, todos os procedimentos foram realizados em condições assépticas e com equipamentos de proteção individual completos.
1. Preparo pré-operatório
- Para minimizar o edema, administrar dexametasona (0,2 mg/kg) por via subcutânea, 12-24 h antes da cirurgia.
- Esterilizar todos os instrumentos cirúrgicos em autoclave e esterilizar a área cirúrgica com ácido hipocloroso estabilizado com água destilada e etanol 70% antes da cirurgia. Certifique-se de que todos os equipamentos cirúrgicos estejam ligados.
- Anestesiar o animal (24 semanas de idade, macho pesando 31 g) usando isoflurano com uma dose de indução de 5%, que é reduzida para 1%-2% quando o camundongo está no quadro estereotáxico, com O2 mantido entre 1-2 L/min. Injetar AINEs (Carprofeno, 2,5 mg/kg) por via subcutânea.
- Verificar a ausência de reflexo de pinça dos pés para avaliar a profundidade da anestesia (aumentar a concentração de isoflurano em incrementos de 0,5% se houver reflexo).
- Faça a barba da cabeça do animal, usando um aparador, de trás das orelhas até um pouco acima dos olhos. Limpe esta área com um lenço de álcool e solução de iodopovidona, garantindo evitar o contato com os olhos do animal.
- Monte o animal na almofada de aquecimento homeotérmica e na estrutura estereotáxica equipada com barras de orelha e dentes e prenda a cabeça. Certifique-se de que a cabeça esteja estável, pois isso é crucial para que o procedimento a seguir seja bem-sucedido (Figura 1).
- Aplique pomada oftálmica sobre os olhos do animal para evitar que eles sequem durante a cirurgia e cubra-os com papel alumínio para protegê-los da luz. Cubra o animal com uma capa cirúrgica estéril.

Figura 1: Preparação pré-operação. O mouse é colocado sobre a estrutura estereotáxica, preso por um pedaço de nariz e barras de ouvido. O mouse é colocado em uma almofada aquecida com temperatura regulada. Os olhos têm pomada oftálmica sobre eles e são cobertos por papel alumínio. A cabeça é raspada e o crânio é exposto. Uma capa estéril é colocada sobre o animal. Clique aqui para ver uma versão maior desta figura.
{kind=link}
2. Craniotomia
- Usando tesoura cirúrgica, incida a pele ao longo da linha média da área raspada da cabeça para expor o crânio.
- Limpar o crânio com um cotonete estéril e peróxido de hidrogênio diluído (3% p/v de 35% H2O2 em 97% dH2O) por 1-3 s para remover qualquer tecido conjuntivo (Figura 2A). Secar o crânio usando uma gota de etanol 70% e um novo cotonete estéril.
- Alinhar o crânio anterior/posteriormente (AP) e medial/lateralmente (ML) para garantir um local de implantação preciso. Para fazer isso, meça a profundidade dorsoventral (DV) do crânio em ambos bregma e lambda e certifique-se de que a diferença entre os dois é de <0,03 mm. Para o alinhamento médio-lateral, medir pontos equidistantes em ambos os ossos parietais a partir da linha média e novamente garantir que a diferença de VD seja de <0,03 mm.
- Usando bregma como origem, encontre e marque a área cortical desejada; aqui, estes são córtex visual primário monocular (V1), AP: -3,5 mm, ML: -2,5 mm.
- Use uma broca de trefina (1,8 mm de diâmetro) e uma broca dental (velocidade de 10.000 rpm) para expor o córtex, garantindo que a marca da área cortical desejada (V1 monocular) esteja localizada no terço inferior da janela da broca.
- Certifique-se de que o ângulo da broca esteja perpendicular à curvatura do crânio. Isso garantirá uma craniotomia uniforme e evitará danos à dura-máter ou córtex.
- Perfurar até que haja diminuição da resistência e então parar (Figura 2B). Remover cuidadosamente o fragmento ósseo descolado com ponta de agulha 23G (Figura 2C).
- Limpe o córtex exposto com espuma cirúrgica saturada em líquido cefalorraquidiano artificial frio (ACSF) para remover quaisquer detritos e parar qualquer sangramento que possa ocorrer.
- Mantenha sempre o córtex exposto hidratado, utilizando ACSF frio, durante toda a cirurgia.

Figura 2: Craniotomia. (A) Incisão cutânea entre bregma e lambda é mostrada. O tecido conjuntivo foi removido da superfície exposta. (B) Craniotomia por broca de trefina antes da retirada do fragmento ósseo. (C) Craniotomia após remoção do fragmento ósseo, mostrando dura-máter e córtex íntegros (barra de escala representa 0,5 mm). Clique aqui para ver uma versão maior desta figura.
{kind=link}
3. Incisão pré-corte
OBS: Para ser considerada na realização da incisão pré-corte, a incisão e o implante do microprisma precisarão ser anteriores à região de imagem de interesse (ROI). Isso é para permitir um campo de visão completo e preciso. No contexto deste protocolo, a incisão será realizada ao longo do eixo médio-lateral, e o microprisma está orientado voltado para a região posterior (Figura 3B).
- Para ajudar na inserção e aliviar a pressão no córtex durante a inserção do microprisma, faça uma incisão.
- Fixe a faca cirúrgica ao suporte do braço estereotáxico e oriente a lâmina ou o braço estereotáxico de modo a que este corte ao longo do eixo ML.
- Mova a faca para a coordenada AP desejada (AP: -3,4mm); o prisma deve estar à frente da ROI, portanto, faça a incisão 100 μm anterior da coordenada AP da ROI da imagem (-3,5mm).
- Agora movendo a faca para a borda medial da craniotomia, onde encontra o crânio, abaixe lentamente a faca até chegar ao osso e depois pare. Como a espessura do osso é de 200 μm, incorpore este valor na profundidade total de inserção (ver cálculo do passo 5.1).
- A imagem ideal está no centro do prisma, ou seja, 500 μm; portanto, certifique-se de que essa profundidade esteja alinhada com a profundidade da coluna cortical (ROI DV: - 0,35 mm).
- Incorporando a espessura do crânio no cálculo da profundidade, use a equação abaixo, que determina a profundidade que a incisão pré-corte precisa ter da superfície do crânio. Para este protocolo, a profundidade de implantação é calculada como:
Espessura óssea (200 μm) + ROI de imagem (por exemplo, 350 μm) + profundidade do microprisma restante (500 μm) = 1.050 μm
- Incorporando a espessura do crânio no cálculo da profundidade, use a equação abaixo, que determina a profundidade que a incisão pré-corte precisa ter da superfície do crânio. Para este protocolo, a profundidade de implantação é calculada como:
- Certifique-se de que o comprimento da incisão seja superior a 1 mm, mas não excessivo; portanto, uma distância de 1,2 mm é ideal, com a coordenada monocular ML no meio dessa distância.
- Quando estiver pronto para realizar a incisão, remova o excesso de FRAC para que a visão não fique obscurecida (Figura 3A).
- Mover o bisturi da borda medial da craniotomia para a coordenada medial inicial da incisão. Lentamente (10 μm/s) abaixe a faca para o córtex.
- Uma vez que a dura-máter é perfurada e a faca entrou no córtex, aplique uma gota de ACSF frio no córtex para manter o tecido lubrificado e hidratado durante a incisão.
- Uma vez atingida a profundidade final, comece a mover a faca ao longo do eixo ML (a uma taxa de 10 μm/s).
- Continue observando o tecido circundante enquanto a incisão está sendo feita. Se o tecido estiver arrastando junto com a faca, mova a faca para cima e para baixo algumas vezes para garantir que o tecido esteja sendo cortado e, em seguida, continue lateralmente, lembrando-se de colocar a faca de volta à sua profundidade final.
- Quando terminar, levante lentamente a faca. Se o sangue surgir durante a incisão, use esse tempo para limpar o local da incisão com espuma cirúrgica embebida em ACSF para diluir o sangue e empurrar qualquer sangue dentro da incisão para fora. Lembre-se de deixar uma espuma cirúrgica fresca e saturada sobre o córtex exposto até que esteja pronta para inserir o microprisma.

Figura 3: Implantação do microprisma. (A) Incisão pré-cortada. (B) Esquema da lente de microprisma integrada demonstrando sua posição dentro do córtex (C) Lente de microprisma integrada na orientação correta para incisão pré-cortada antes da inserção no córtex (barra de escala representa 0,5 mm). (D) Exemplo de acúmulo de cimento ao redor da lente integrada para fixar sua fixação ao crânio. Clique aqui para ver uma versão maior desta figura.
{kind=link}
4. Inserção de microprisma e implantação da placa cefálica
- A lente de microprisma é composta por uma lente índice de gradiente acoplada a um prisma na extremidade distal da lente, que é integrada a uma placa de base. Conecte o microprisma ao kit de implante.
- Verifique se o lado da imagem do prisma é oposto ao parafuso da placa de base. Para auxiliar na inserção e colocação do microprisma, fixe-o ao quadro estereotáxico e oriente o prisma de forma que se alinhe com a incisão (Figura 3C).
- Abaixe lentamente o microprisma para o local da incisão (10 μm/s). Lembre-se de remover ACSF ao inserir inicialmente o prisma, mas uma vez no córtex, lave com ACSF frio para lubrificar a inserção.
- O córtex deve permanecer estável enquanto o prisma é abaixado para a incisão; se não, adicione ACSF extra e agite o córtex movendo o prisma para cima e para baixo para soltar o córtex do prisma.
- Uma vez atingida a profundidade final, secar a superfície cortical exposta com tecido estéril, tomando cuidado para não tocar o prisma.
- Cubra a área cortical exposta ao redor do prisma, bem como a lente, com uma camada protetora de adesivo de silicone, minimizando o excesso de adesivo no crânio circundante e no simulador de ótica.
- Uma vez curado (5-10 min), prenda a placa cefálica ao crânio para estabilizar a cabeça durante imagens fixadas na cabeça.
- Certifique-se de que a placa frontal seja posterior o suficiente para não interferir com a colocação do implante e permitir a aplicação adequada de cimento para fixar adequadamente o implante.
- Certifique-se de que a linha média da placa de cabeça esteja ligeiramente à direita da lente implantada para garantir que ambos os lados da placa de cabeça possam ser fixados ao palco da cabeça ao realizar experimentos fixos na cabeça
- Aplicar cimento adesivo na placa de cabeça e crânio.
- Prepare o cimento dental adesivo misturando 1 colher de pó de cimento opaco com 4 gotas de meio misturador e aplicando uma gota do catalisador.
- Coloque cimento na placa da cabeça e no crânio e segure a placa na cabeça no lugar até cicatrizar, garantindo que ela esteja paralela com as barras da orelha (através do exame visual, inspecione tanto de cima quanto atrás da cabeça do animal).
- Aplicar cimento adesivo para cobrir o restante do crânio e tecido expostos, incorporando o microprisma (até a base da placa de base) e a placa cefálica.
- Não obtenha cimento na placa de base, microscópio simulado ou qualquer um de seus componentes. Continue aplicando o cimento adesivo até que o microprisma e a placa frontal estejam cobertos e estáveis (Figura 3D).
- Quando o cimento estiver curado, desprenda o microscópio simulado movendo o braço estereotáxico para cima lentamente enquanto estabiliza o microprisma com pinças (elas são conectadas através de ímãs, portanto, alguma resistência durante a separação pode ser sentida).
- Insira a capa protetora na lente e aperte o parafuso para prendê-lo no lugar.
- Retirar o animal do quadro estereotáxico, permitir que ele se recupere em uma caixa de recuperação morna e administrar soro fisiológico 0,9% estéril aquecido por via subcutânea (3% do peso corporal).
- Quando o animal estiver acordado e em movimento, coloque-o de volta em uma gaiola limpa e alojada individualmente. Monitorar o animal e administrar analgesia pós-operatória adicional de acordo com a política de analgesia da instituição local.
- Aguarde 4 semanas após a cirurgia, o animal deve estar pronto para exames de imagem.
5. Imagem de cálcio de um fóton de camadas corticais em camundongos que se movem livremente
NOTA: É essencial utilizar imagens capturadas da sessão de imagem original cada vez para garantir a aquisição precisa do plano de imagem pretendido. Esses pontos de referência identificados, juntamente com os neurônios, desempenham um papel crítico no processo de alinhamento descrito em detalhes na etapa 9 do protocolo. Ao adquirir dados de um fóton, o miniscópio é o sistema de imagem e a fonte de laser. A excitação usa LED com uma faixa de potência de 0-2 mW/mm2 na superfície frontal objetiva. O laser usa um comprimento de onda de excitação de 455 ± 8nm (luz azul) para a sinalização GCaMP. O controle deslizante de foco da lente pode ser usado para ajustar o foco (eixo Z), que é representado na interface como 0-1000, onde 0 representa uma distância de trabalho de 0μm e 1000 representa a distância máxima de trabalho de 300μm.
- Antes da aquisição dos dados, deixe o animal se aclimatar à sala e à arena aberta por 1 h antes da sessão de gravação.
- Antes de fazer a imagem, desinfete e limpe tudo com desinfetantes apropriados (por exemplo, ácido hipocloroso estabilizado com água destilada e etanol 70%).
- Configure a caixa DAQ conectando-a a um computador e iniciando o software de aquisição de dados. Estabeleça uma conexão direta através de um cabo ethernet para minimizar a queda de quadros; No entanto, o modo de conexão sem fio pode ser suficiente, dependendo da intensidade da conexão sem fio.
- Fixe o miniscópio à placa de base do animal sob um suave scruff.
- Primeiro, remova a tampa protetora da placa de base desaparafusando o parafuso ajustado. Segure a tampa pela abertura com pinças. Em seguida, prenda o miniscópio à placa de base, onde fica a tampa.
- Verifique a orientação do miniscópio em relação à placa de base antes de instalá-lo para que o lado com a marcação do parafuso, fique voltado para o parafuso.
- Assim que o miniscópio estiver conectado, aperte o parafuso ajustado para estabilizá-lo. Apenas avance o parafuso ajustado até que alguma resistência possa ser sentida. O aperto excessivo do parafuso ajustado potencialmente danificaria o miniscópio e, portanto, deve ser evitado.
- Conecte o miniscópio à caixa DAQ e prepare o software para gravação.
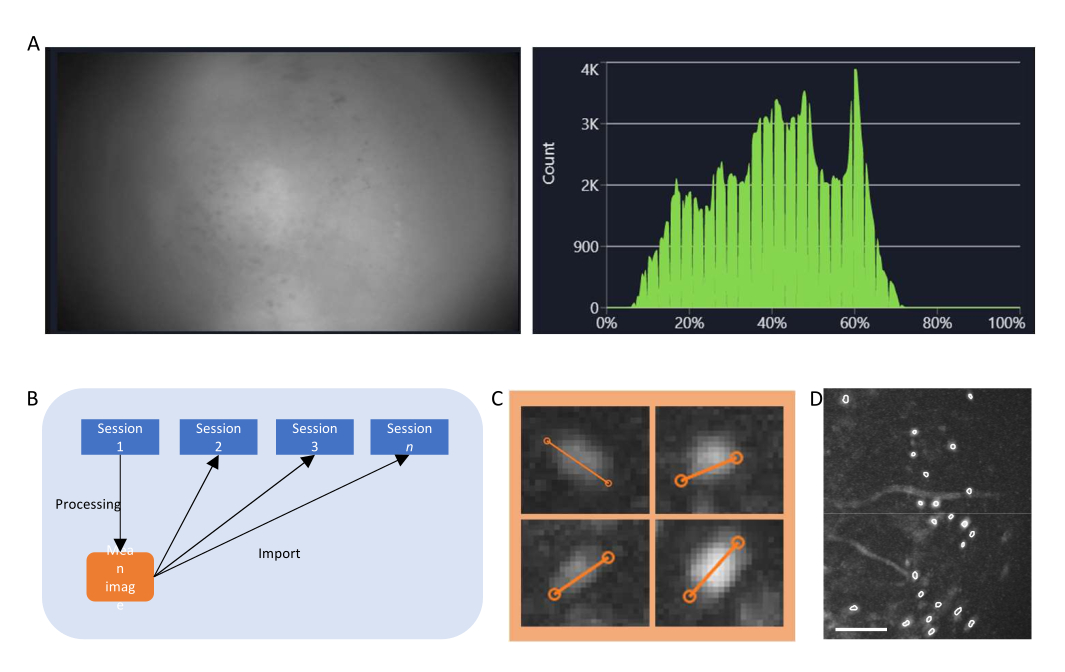
- No software de aquisição de dados, ligue o fluxo para o miniscópio e ajuste os parâmetros de gravação (taxa de quadros de imagem, ganho, potência do LED, valor de efocus) para obter um campo de visão claro (Figura 4A).
- Ligue a janela do histograma e ajuste o ganho e a potência do LED para que a intensidade registrada fique entre 35% a 70%.
- Se estiver realizando um estudo longitudinal, consulte as gravações ou instantâneos tirados em uma sessão anterior e ajuste o valor de efocus para que o mesmo plano de imagem seja claramente visto.
- Inicie o experimento e a aquisição de dados.
- Após a conclusão do experimento, retirar o animal do aparato comportamental.
- Sob um scruff suave, solte o parafuso ajustado e retire o miniscópio da placa de base do animal.
- Devolva a tampa protetora para a placa de base e estabilize-a com o parafuso ajustado.
- Devolva o animal à sua gaiola de origem (prossiga para o passo 7 se quiser processar dados de um fóton).

Figura 4: Aquisição e processamento dos dados com software. (A) Uma imagem mostrando o fluxo em tempo real do miniscópio. Recomenda-se ajustar o valor de foco da lente, para que uma visão clara seja vista na janela de streaming, juntamente com o ganho e a potência do laser de imagem (B) Gráfico esquemático ilustrando o fluxo de trabalho de alinhamento recomendado para sessões gravadas em diferentes pontos de tempo. Recomenda-se gerar uma imagem média a partir da primeira sessão, seguindo as instruções do software de processamento de dados. Esta imagem deve ser usada como imagem de referência durante a correção de movimento para as sessões seguintes. (C) Exemplos de quatro células da mesma imagem ΔF/F projetada no máximo. Uma linha laranja é desenhada em cada célula para medir seu diâmetro celular em pixels, cuja média é tomada como um argumento de entrada para o algoritmo de identificação da célula (canto superior esquerdo: 13, superior direito: 11, inferior esquerdo: 12, inferior direito: 13). (D) Saída do algoritmo de identificação celular após curadoria manual (imagem recortada). Os contornos brancos representam as células identificadas (a barra de escala representa 100 μm). Clique aqui para ver uma versão maior desta figura.
{kind=link}
6. Imagem de cálcio de dois fótons de camadas corticais em camundongos fixados na cabeça
NOTA: Para microscopia de varredura a laser de dois fótons, a fonte de luz é um laser ultrarrápido ajustável com um comprimento de onda de excitação de 920 nm. A potência de excitação, medida na objetiva, foi tipicamente entre 100-150 mW e ajustada em cada sessão para atingir níveis semelhantes de fluorescência. A luz de emissão foi filtrada por um filtro de emissão (525/70 nm) e medida por um tubo fotomultiplicador independente (PMT), denominado canal verde. As imagens foram adquiridas com objetiva de imersão em ar de 20x (NA = 0,45, distância de trabalho de 6,9-8,2mm).
- Antes de adquirir dados, habitue o animal ao aparelho nos dias anteriores (para que o animal se sinta confortável com a configuração comportamental). Permita que o animal passe de 15 a 30 minutos por dia, por 2 a 3 dias, explorando a configuração ou até que exiba comportamento naturalista antes de iniciar a aquisição de dados.
- Ligue o sistema de imagem de dois fótons, inicie o software de aquisição e ligue o laser. Certifique-se de que os lasers estão fechados e os PMTs estão desligados antes de continuar.
- Aparelho de imagem de dois fótons limpo com ácido hipocloroso estabilizado com água destilada e etanol 70%.
- Certifique-se de ajustar o aparelho para se adequar ao tamanho do animal. Fixe suavemente o rato e a sua placa de cabeça à configuração do head-stage e aparafuse-o no lugar para estabilizar a cabeça do rato.
- Uma vez fixada, remova a tampa da lente (consulte o passo 5.4.1) e alinhe a objetiva de modo que fique sobre a placa de base.
- Use os controles de estágio de epifluorescência e XYZ para colocar o tecido cortical em foco.
- Quando as camadas corticais estiverem visíveis, troque o microscópio para permitir a obtenção de imagens de dois fótons (troque o espelho pelo espelho dicroico, feche o obturador fluorescente e desligue o laser e o monitor de epifluorescência). Certifique-se de desligar as luzes principais para proteger os PMTs durante a imagem.
- Configure parâmetros para otimizar os arquivos de imagem adquiridos.
- Use o modo de aquisição ressonante para imagens de cálcio, pois ele pode capturar o disparo rápido de sinais GCaMP.
- Ajuste a potência do laser, o ganho de PMT, o zoom e as tabelas de pesquisa (LUTs) para obter uma imagem ideal e consulte a imagem de um fóton para garantir que o plano focal correto esteja sendo fotografado.
- Iniciar a obtenção de imagens das camadas corticais com o sistema de dois fótons com monitoramento comportamental sincronizado e entradas de estímulo (se aplicável).
- Certifique-se de salvar esses parâmetros de aquisição e distâncias XYZ se quiser reproduzir imagens idênticas ao longo do tempo.
- Para capturar uma pilha z das camadas corticais, siga as etapas descritas abaixo.
- Encontre o plano a partir do qual iniciar a pilha z, ajuste os parâmetros de aquisição para otimizar a imagem e marque isso como o ponto de partida no software.
- Em seguida, usando o controle Z, mova a pilha para baixo, apenas ajustando a potência do laser para manter uma luminância constante da pilha e marque o final da pilha no software.
- Etapa crítica: usando a opção Gradiente Exponencial Relativo na guia Gradiente de Potência do Laser , permita que o software calcule o aumento da potência do laser à medida que ele se move através da pilha z. Certifique-se de marcar os valores de potência do laser de ponto final na tabela fornecida pelo software para permitir que ele calcule o gradiente.
- Uma vez que os parâmetros z-stack estejam definidos, ajuste o tamanho do passo (μm).
Observação : o tamanho da etapa determinará o tempo tomado, o número de fatias e a qualidade dos detalhes da pilha. Tamanhos de passo menores resultarão em maior tempo de aquisição, um aumento no número de fatias e melhores detalhes em comparação com o tamanho de passo maior. Pilhas Z são usadas para auxiliar no registro de imagens de um fóton e dois fótons, pois destacarão quaisquer pontos ou características anatômicas.
- Para adquirir uma série temporal (série T) de alterações de cálcio nos neurônios, encontre um plano focal ideal usando os controles de estágio XYZ e ajuste a potência do laser, o ganho de PMT, o zoom e as LUTs.
- Na guia série T do software de aquisição, defina os parâmetros de frequência de aquisição para corresponder aos dados adquiridos usando o sistema de imagem de um fóton.
NOTA: A correspondência da frequência tornará os dados 1P e 2P comparáveis, e é mais adequado para o algoritmo de identificação celular usado nas etapas de processamento de dados. Múltiplos gatilhos e outros modos de aquisição podem ser incorporados à aquisição da série T.
- Na guia série T do software de aquisição, defina os parâmetros de frequência de aquisição para corresponder aos dados adquiridos usando o sistema de imagem de um fóton.
- Início da aquisição da série T.
7. Processamento de dados de imagens de cálcio de um fóton
- Para filmes de gravação de um fóton, use o software de processamento de dados que acompanha o sistema de miniscópio.
- Primeiro, pré-processe o filme por amostragem espacial e temporal. Geralmente, a redução espacial da amostra do filme por um fator de dois reduziria significativamente o tempo de processamento sem comprometer severamente a precisão da identificação celular.
- Defina o fator de amostragem temporal para que a taxa de quadros do filme seja reduzida para cerca de 10 Hz, o que é mais adequado para o algoritmo de identificação de células usado nas etapas a seguir.
- Se adquirir vários filmes no mesmo dia de imagem, combine os filmes em uma série temporal antes da etapa de pré-processamento para processá-los juntos.
- Opcional: aplique um filtro passa-banda espacial ao filme para remover as frequências espaciais mais baixas e mais altas, resultando em um filme mais suave e com maior contraste.
- Registre o filme usando a funcionalidade de correção de movimento do software. Isso registra o filme e corrige artefatos de movimento causados pelo movimento do miniscópio em relação à superfície da imagem.
- Etapa crítica: Se for realizado um estudo longitudinal, registre os filmes no mesmo campo de visão, por exemplo, a imagem média do filme tirada no primeiro dia de aquisição de imagens (Figura 4B).
- Calcule o ΔF/F do filme usando a guia correspondente e projete o filme para gerar uma imagem de projeção máxima do filme ΔF/F. Esta imagem mostrará regiões que apresentam mudanças nos níveis de fluorescência, potencialmente neurônios individuais, e poderá ser usada para medir o diâmetro médio dos neurônios (Figura 4C).
- Alternativamente, meça o diâmetro da célula no filme corrigido por movimento, onde os neurônios mostram fluorescência clara.
- Identificar células usando os algoritmos do software.
- Embora duas opções (PCA-ICA e CNMF-E) estejam disponíveis nesta etapa, use CNMF-E para este estudo. Insira o diâmetro médio da célula em pixels e execute o algoritmo para gerar um conjunto de células contendo regiões de interesse (ROIs) que demonstram atividades semelhantes às da célula.
- Selecione manualmente as ROIs que são células (têm uma morfologia semelhante à célula e atividade22,23 e estão dentro do FOV) daquelas que não são, e valide o conjunto de células curadas (Figura 4D).
- Exporte os traços de cálcio de cada ROI para análise posterior.
8. Processamento de dados de imagens de cálcio de dois fótons
- Para filmes de gravação de dois fótons, use um pacote python projetado para processar dados de análise de cálcio de dois fótons.
- Primeiro, combine as imagens tiradas em uma série T em uma pilha de .tiff, conforme descrito abaixo.
- Na interface Opções de execução , ajuste os parâmetros, incluindo o valor tau e a taxa de quadros, para que ele corresponda ao GCaMP usado e à taxa de quadros da gravação.
- Opcional: defina o parâmetro do_registration como 1 para registrar o filme. Isso é equivalente à etapa de correção de movimento descrita acima.
- Opcional: defina o parâmetro anatomical_only como 1 para detectar ROIs usando os recursos anatômicos, além da dinâmica de fluorescência. Isso requer a entrada do diâmetro da célula, então faça medições usando o software de processamento de imagem. Isso geralmente é recomendado, pois gera ROIs com formas mais naturais (Figura 5A-C).
- Depois que todos os parâmetros estiverem definidos, execute o algoritmo para que ele execute todos os cálculos juntos. Consulte a interface gráfica do usuário (GUI) para verificar o progresso.
- Feito isso, retorne à interface de seleção de células para curadoria manual dos resultados de identificação da célula.
- Salve uma imagem da célula selecionada definida sobre a projeção máxima do filme. Esta será usada posteriormente como imagem de referência para registrar dados de registro de um fóton.
- Em seguida, o algoritmo salva os resultados automaticamente. formato npy, que pode ser acessado posteriormente com Python. Como alternativa, salve os resultados em outros arquivos formatados para análise adicional em outro software.

Figura 5: Identificação celular utilizando software de processamento de dois fótons. (A) Imagem representativa da identificação celular obtida do software de processamento de dois fótons. Definindo Anatomical_only parâmetro como 0, mas mantendo todos os outros parâmetros iguais, várias não-células estão presentes na área entre linhas tracejadas que interferem na curadoria manual de células reais. (B) Exemplos de medidas de diâmetro celular obtidas de (A), utilizando um software de processamento de imagens (canto superior esquerdo; 7,5 pixels, canto superior direito; 9, canto inferior esquerdo; 6,5, canto inferior direito; 7,5). (C) Imagem representativa da identificação celular. Ao definir Anatomical_only parâmetro como 1 e inserir o diâmetro médio da célula retirado de (B) no algoritmo de diâmetro celular, nenhuma célula está presente na área entre linhas tracejadas (barras de escala representam 200 μm). Clique aqui para ver uma versão maior desta figura.
{kind=link}
9. Registro de conjuntos celulares identificados em modalidades de imagem
- Realizar o registro de células identificadas a partir de registros de um fóton e dois fótons com o algoritmo multimodal de registro e análise de imagens (MIRA), que está disponível através da interface Python do software de imagem de um fóton.
- Este algoritmo alinha os dados de um fóton e dois fótons via registro não rígido. Veja o conjunto de cadernos online demonstrativos encontrados no site e utilizados para este estudo.
NOTA: Os cadernos foram escritos para que todo o processamento fosse concluído no software 1P e, portanto, não são compatíveis com o software de processamento 2P. Assim, siga apenas alguns dos passos dos cadernos para este estudo.
- Este algoritmo alinha os dados de um fóton e dois fótons via registro não rígido. Veja o conjunto de cadernos online demonstrativos encontrados no site e utilizados para este estudo.
- Siga as etapas descritas nos cadernos demonstrativos, que envolvem a geração de uma imagem estrutural para cada modalidade de imagem. Por padrão, isso envolve gerar uma projeção máxima da pilha z de dois fótons e uma imagem média da gravação de um fóton. Alternativamente, use uma imagem média do registro de dois fótons.
- Quando solicitado, o passe de banda espacial filtra as imagens para visualizar melhor os pontos de referência e reorientá-los para que correspondam.
- Selecione pontos de referência correspondentes nas duas imagens (Figura 6A).
- Use-os para calcular a deformação necessária para alinhar as duas imagens. Em geral, 3 a 5 pontos de referência devem ser suficientes.
- O algoritmo calcula a urdidura com base em uma combinação de pontos de referência e similaridade de imagem. Otimizar para o peso relativo dado aos dois fatores até que resultados satisfatórios sejam alcançados.
- Deformar o mapa celular adquirido em uma sessão de um fóton para gerar um novo mapa de célula alinhado aos dados de dois fótons.
- Em seguida, importe esse mapa de célula distorcida para o software de processamento 1P para gerar uma imagem com a imagem de projeção máxima do filme de dois fótons em segundo plano.
- Exporte esta imagem para fins de registro.
- No software de programação, alinhe as duas imagens geradas até o momento (mapa de células de dois fótons em cima de imagem de projeção máxima de dois fótons) e deforme o mapa de células de um fóton em cima de imagem de projeção máxima de dois fótons (Figura 6B,C).
- Para isso, para este estudo, utiliza-se um aplicativo estimador de registro que permite ao usuário comparar os resultados de diferentes técnicas de registro. Como as duas imagens têm o mesmo fundo, a técnica de correlação de fases, com registro rígido, foi suficiente.
- Quando o registro estiver concluído, verifique a imagem agora registrada em busca de ROIs sobrepostas. Essas são ROIs que estão ativas em ambas as sessões de gravação, que poderiam ser usadas em análises posteriores.

Figura 6: Registro de célula entre modalidades usando o fluxo de trabalho MIRA. (A) Imagem representativa do fluxo de trabalho de alinhamento de célula. A imagem média dos dados de um fóton é mostrada à esquerda, e a dos dados de dois fótons é mostrada à direita. Os pontos de referência correspondentes de ambas as imagens são selecionados e rotulados no software por um esquema de cores aleatório (círculos vermelhos). (B) Imagens alinhadas por exemplo mostrando os dois conjuntos de células identificados, um fóton (roxo) e dois fótons (verde), são sobrepostas à imagem média dos dados de dois fótons. (C) Imagem da região marcada com a caixa branca em (B), as células alinhadas são representadas aqui como contornos verdes e roxos sobrepostos. Em todos os painéis, a barra de escala representa 200 μm. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Resultados
O método de conduzir imagens crônicas de cálcio in vivo multicamadas da mesma população neuronal durante um período de várias semanas, usando modalidades de imagem de um e dois fótons, sob condições de movimento livre e cabeça fixada foi mostrado. Aqui, a capacidade de identificar populações neuronais correspondentes sob imagens de um fóton enquanto o animal explorava uma arena aberta no escuro foi demonstrada (Figura 7A). Traços de cálcio foram extraídos dos neurô...
Discussão
Aqui, mostramos a capacidade de observar e comparar diretamente neurônios em condições de cabeça fixa e movimento livre nas mesmas populações neurais. Embora tenhamos demonstrado a aplicação no córtex visual, este protocolo pode ser adaptado a uma infinidade de outras áreas cerebrais, tanto áreas corticais quanto núcleos profundos24,25,26,27,28, bem como outras configurações comportamentais e de
Divulgações
Os autores declaram não haver conflito de interesses ou interesses financeiros concorrentes.
Agradecimentos
Agradecemos à Sra. Charu Reddy e ao Professor Matteo Carandini (Cortex Lab) por seus conselhos sobre protocolo cirúrgico e compartilhamento de cepa de camundongo transgênico. Agradecemos ao Dr. Norbert Hogrefe (Inscopix) pela orientação e auxílio no desenvolvimento da cirurgia. Agradecemos à Sra. Andreea Aldea (Sun Lab) por sua assistência com a configuração cirúrgica e processamento de dados. Este trabalho foi apoiado pela Moorfields Eye Charity.
Materiais
| Name | Company | Catalog Number | Comments |
| 0.9% Sodium Chloride solution for infusion (Vetivex 11) 250ml | Dechra | 20091607 | Saline for hydration and drug reconsitution |
| 18004-1 Trephine 1.8mm diameter bur | FST | 18004-18 | Drill bit |
| 1ml syringe | Terumo | MDSS01SE | 1ml syringe |
| 23G x 5/8 inch 6% LUER needle | Terumo | NN-2316R | 23G needle |
| 71000 Automated stereotaxic apparatus w/ built-in software | RWD | - | RWD |
| Absorbable Haemostatic Gelatin Sponge (10x10x10mm) | Surgispon | SSP-101010 | gel-foam |
| Alcohol pads 70% isopropyl alcohol | Braun | 9160612 | Alcohol pads |
| Aluminium foil | Any retailer | - | Foil to cover eyes during surgery |
| Articifical Cerebrospinal Fluid | Tocris Bioscience a Bio-Techne Brand | 3525/25ML | ACSF |
| Automated microinjection pump | WPI | 8091 | |
| Betadine solution (10% iodinated Povidone) 500ml | Videne/Ecolab | 3030440 | Betadine |
| Bruker Ultime 2Pplus (customised) | Bruker | - | Two-photon imaging system |
| Cardiff Aldasorber | Vet-Tech | AN006 | Anaesthesia absorber |
| CFI S Plan Fluor ELWD ADM 20XC | Nikon | MRH48230 | 20x objective lens |
| Compact Anaesthesia system - single gas - isoflurane K/F, with oxygen concentrator model: ZY-5AC and scavenging unit | Vet-Tech | AN001 | Compact anaesthesia system |
| Contec Prochlor | Aston Pharma | AP2111L1 | Disinfectant (hypochlorous acid) |
| Dexamethasone Sodium Phosphate Injection, USP, 4mg/ml, NDC: 0641-6145-25 | Hikma | Covetrus:70789 | Dexamethasone |
| Dissecting Knife, cutting edge 4mm, thickness 0.5mm, stainless steel | Fine Science Tools | 10055-12 | Knife for incisino of cortex |
| Dual-Sided, Non-Puncture Mouse & Neonatal Rat Ear Bars | Stoelting | 51649 | Ear bar |
| Dummy microscope | Inscopix | Dummy microscope | To help with implantation |
| Ethanol (100%) | VWR | 40-1712-25 | Used to make 70% ethanol |
| Fisherbrand Nitrile Indigo Disposable Gloves PPE Cat III | FischerScientific | 17182182 | Gloves |
| Homeothermic Monitor 50-7222-F | Harvard Apparatus | 50-7222-F | Homeothermic monitoring system/heating pad |
| Image processing software | ImageJ | - | Image processing software |
| Inscopix Data Processing Software (IDPS) | Inscopix | - | One-photon calcium imaging processing software |
| Insight Duals-232, S/N 2043 | InSight | Insight Spectra X3 | Two-photon imaging laser |
| IsoFlo 250ml 100% w/w inhalation | Zoetis | WM 42058/4195 | Isoflurane |
| Kwik-Sil Low Toxicity Silicone Adhesive | World Precision Intruments (WPI) | KWIK-SIL | Silicone adhesive |
| MICROMOT mains adapter NG 2/S, w/ Drill unit 60/E | PROXXON | NO 28 515 | Handheld drill |
| nVoke Integrated Imaging and Optogenetics System package | Inscopix | - | One-photon Imaging system and software |
| ProView Implant Kit | Inscopix | ProView Implant Kit | Dummy microscope, stereotaxic arm and attachment |
| ProView Prism Probe | Inscopix | 1050-002203 | Microprism lens |
| Rimadyl (50mg/ml) | Zoetis | VM 42058/4123 | Carprofen |
| Stereotaxis Microscope on Articulated arm with table clamp | WPI | PZMTIII-AAC | Microscope |
| Super-Bond Universal kit, SUN Medical | Prestige-Dental | K058E | Adhesive cement |
| Two-photon calcium image software | Suite2P | - | Two-photon calcium imaging processing software |
| Vapouriser | Vet-Tech | - | Isoflurane vapouriser |
| Xailin Lubricating Eye Ointment 5g | Xailin-Night | MLG/28/1551 | Ophthalmic ointment |
Referências
- Denk, W., Strickler, J. H., Webb, W. W. Two-photon laser scanning fluorescence microscopy. Science. 248 (4951), 73-76 (1990).
- Svoboda, K., Yasuda, R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 50 (6), 823-839 (2006).
- Dombeck, D. A., Khabbaz, A. N., Collman, F., Adelman, T. L., Tank, D. W. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 56 (1), 43-57 (2007).
- Vaziri, A., Emiliani, V. Reshaping the optical dimension in optogenetics. Curr Opin Neurobiol. 22 (1), 128-137 (2012).
- Holtmaat, A., et al. high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 4 (8), 1128-1144 (2009).
- Sun, Y. J., Sebastian Espinosa, J., Hoseini, M. S., Stryker, M. P. Experience-dependent structural plasticity at pre- and postsynaptic sites of layer 2/3 cells in developing visual cortex. Proc Natl Acad Sci U S A. 116 (43), 21812-21820 (2019).
- Andermann, M. L., Kerlin, A. M., Reid, R. C. Chronic cellular imaging of mouse visual cortex during operant behavior and passive viewing. Front Cell Neurosci. 4, 3 (2010).
- Sofroniew, N. J., Flickinger, D., King, J., Svoboda, K. A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. Elife. 5, 14472 (2016).
- Puścian, A., Benisty, H., Higley, M. J. NMDAR-dependent emergence of behavioral representation in primary visual cortex. Cell Rep. 32 (4), 107970 (2020).
- Trachtenberg, J. T., et al. Long-term in vivo. imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 420 (6917), 788-794 (2002).
- Seaton, G., et al. Dual-component structural plasticity mediated by αCaMKII autophosphorylation on basal dendrites of cortical layer 2/3 neurones. J Neurosci. 40 (11), 2228-2245 (2020).
- Helmchen, F., Denk, W. Deep tissue two-photon microscopy. Nat Methods. 2 (12), 932-940 (2005).
- Andermann, M. L., et al. Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron. 80 (4), 900-913 (2013).
- Chia, T. H., Levene, M. J. Microprisms for in vivo multilayer cortical imaging. J Neurophysiol. 102 (2), 1310-1314 (2009).
- Low, R. J., Gu, Y., Tank, D. W. Cellular resolution optical access to brain regions in fissures: Imaging medial prefrontal cortex and grid cells in entorhinal cortex. Proc Natl Acad Sci U S A. 111 (52), 18739-18744 (2014).
- Buxhoeveden, D. P., Casanova, M. F. The minicolumn hypothesis in neuroscience. Brain. 125, 935-951 (2002).
- Chen, S., et al. Miniature fluorescence microscopy for imaging brain activity in freely-behaving animals. Neurosci Bull. 36 (10), 1182-1190 (2020).
- Gulati, S., Cao, V. Y., Otte, S. Multi-layer cortical Ca2+ imaging in freely moving mice with prism probes and miniaturized fluorescence microscopy. J Vis Exp. (124), e55579 (2017).
- Resendez, S. L., et al. Visualization of cortical, subcortical, and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat Protoc. 11 (3), 566-597 (2016).
- Guo, Z. V., et al. Procedures for behavioral experiments in head-fixed mice. PLoS One. 9 (2), 88678 (2014).
- Wekselblatt, J. B., Flister, E. D., Piscopo, D. M., Niell, C. M. Large-scale imaging of cortical dynamics during sensory perception and behavior. J Neurophysiol. 115 (6), 2852-2866 (2016).
- Pnevmatikakis, E. A., et al. Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron. 89 (2), 285-299 (2016).
- Zhou, P., et al. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife. 7, 28728 (2018).
- Beckmann, L., et al. Longitudinal deep-brain imaging in mouse using visible-light optical coherence tomography through chronic microprism cranial window. Biomed Opt Express. 10 (10), 5235-5250 (2019).
- Wenzel, M., Hamm, J. P., Peterka, D. S., Yuste, R. Reliable and elastic propagation of cortical seizures in. Cell Rep. 19 (13), 2681-2693 (2017).
- Heys, J. G., Rangarajan, K. V., Dombeck, D. A. The functional micro-organization of grid cells revealed by cellular-resolution imaging. Neuron. 84 (5), 1079-1090 (2014).
- Barson, D., Hamodi, A. S. Simultaneous mesoscopic and two-photon imaging of neuronal activity in cortical circuits. Nat Methods. 17 (1), 107-113 (2020).
- Paquelet, G. E., et al. Single-cell activity and network properties of dorsal raphe nucleus serotonin neurons during emotionally salient behaviors. Neuron. 110 (16), 2664-2679 (2022).
- Yang, Q., et al. Transparent microelectrode arrays integrated with microprisms for electrophysiology and simultaneous two-photon imaging across cortical layers. bioRxiv. , (2022).
- Priestley, J. B., Bowler, J. C., Rolotti, S. V., Fusi, S., Losonczy, A. Signatures of rapid plasticity in hippocampal CA1 representations during novel experiences. Neuron. 110 (12), 1978-1992 (2022).
- Zong, W., et al. Miniature two-photon microscopy for enlarged field-of-view, multi-plane and long-term brain imaging. Nat Methods. 18 (1), 46-49 (2021).
- Engelbrecht, C. J., et al. Ultra-compact fiber-optic two-photon microscope for functional fluorescence imaging in vivo. Opt Express. 16 (8), 5556-5564 (2008).
- Suzuki, M., Aru, J., Larkum, M. E. Double-μ Periscope, a tool for multilayer optical recordings, optogenetic stimulations or both. Elife. 10, 72894 (2021).
- Stibůrek, M., et al. 110 μm thin endo-microscope for deep-brain in vivo observations of neuronal connectivity, activity and blood flow dynamics. Nat Commun. 14 (1), 1897 (2023).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados