
Method Article
Определение и виды Количественное в смесях с помощью масс-спектрометрии MRM пептидов прикладной к мясу аутентификации
В этой статье
Резюме
We present a protocol for identifying and quantifying the components in mixtures of species possessing similar proteins. Mass spectrometry detects peptides for identification, and gives relative quantitation by ratios of peak areas. As a tool food for fraud detection, the method can detect 1% horse in beef.
Аннотация
We describe a simple protocol for identifying and quantifying the two components in binary mixtures of species possessing one or more similar proteins. Central to the method is the identification of 'corresponding proteins' in the species of interest, in other words proteins that are nominally the same but possess species-specific sequence differences. When subject to proteolysis, corresponding proteins will give rise to some peptides which are likewise similar but with species-specific variants. These are 'corresponding peptides'. Species-specific peptides can be used as markers for species determination, while pairs of corresponding peptides permit relative quantitation of two species in a mixture. The peptides are detected using multiple reaction monitoring (MRM) mass spectrometry, a highly specific technique that enables peptide-based species determination even in complex systems. In addition, the ratio of MRM peak areas deriving from corresponding peptides supports relative quantitation. Since corresponding proteins and peptides will, in the main, behave similarly in both processing and in experimental extraction and sample preparation, the relative quantitation should remain comparatively robust. In addition, this approach does not need the standards and calibrations required by absolute quantitation methods. The protocol is described in the context of red meats, which have convenient corresponding proteins in the form of their respective myoglobins. This application is relevant to food fraud detection: the method can detect 1% weight for weight of horse meat in beef. The corresponding protein, corresponding peptide (CPCP) relative quantitation using MRM peak area ratios gives good estimates of the weight for weight composition of a horse plus beef mixture.
Введение
The European horse meat scandal of 2013, in which undeclared horse meat was found in a number of supermarket beef products1, highlights the need for testing methods capable of detecting and measuring food fraud in meat. Several technologies have been explored, especially enzyme-linked immunosorbent assay (ELISA) and DNA-based methods2. An alternative route, based on mass spectrometry, targets species-specific peptides which in turn arise from species-specific proteins. Here we outline one such peptide-based approach that offers both identification and relative quantitation of the adulterant species in a meat mixture3.
The protocol is framed in the context of red meats and the desire to determine the presence of one in another at the level of 1% by weight, the level considered by some to represent fraudulent food adulteration as opposed to contamination4. The method relies in the first instance on identifying a protein which is nominally 'the same' in all target meats. Myoglobin, the protein responsible for the red color of meat, is a good candidate since it is abundant, relatively heat tolerant and water soluble, and has been used for species determination of meat previously5,6. The myoglobins for beef (Bos Taurus), pork (Sus scrofa), horse (Equus caballus) and lamb (Ovis aries)3, for instance, are nominally the same, as required, but their sequences are not identical. Such groups of 'similar but different' proteins, like these four myoglobins, can conveniently be described as 'corresponding proteins'. The sequence differences in these four myoglobins are species-specific: for example, the full myoglobin proteins for beef and horse, P02192 and P68082 respectively, each comprise 154 amino acids with 18 sequence differences between the two. Subject to proteolysis using trypsin these proteins produce two sets of peptides, some of which are identical, and some which show one or more species-specific amino acid differences: corresponding proteins therefore give rise to corresponding peptides.
The CPCP approach, therefore, seeks first to identify proteins from two or more species where these proteins exhibit limited species-specific sequence variants. These are corresponding proteins. Following proteolysis, corresponding proteins give rise to peptides, some of which likewise display species-specific sequence variants inherited from the parent protein. These are corresponding peptides. The CPCP approach can be used to compare levels of two corresponding proteins in a mixed species sample by monitoring the levels of corresponding peptides.
The natural technology for the detection of known peptides is multiple reaction monitoring mass spectrometry, or MRM-MS7. Species-specific peptides yield precursor ions, which along with their mass spectrometry fragment ions, are easily itemized in advance by software tools. These lists are then used to instruct the mass spectrometer to record only specific precursor plus fragment ion pairs, called transitions. A particular target peptide is therefore identified not only by its retention time in the chromatography preceding the mass spectrometer, but also by a set of transitions sharing a common precursor ion. This is a highly selective means of detecting known peptides that makes efficient use of the mass spectrometer resource.
Other authors have used mass spectrometry to test for meat adulteration via peptide markers but from disparate proteins8-14. Using the corresponding proteins, corresponding peptides (CPCP) scheme, however, means experimental conditions can be optimized, aiding identification of the species in the mixture from known species-specific transitions. In addition, corresponding proteins and peptides will generally behave similarly in the extraction, proteolysis and detection stages. Since transition peak areas are quantitative and reproducible, ratios of peak areas arising from pairs of corresponding peptides from different species provide a direct estimate of the relative quantities of two meats in a mixture. In contrast, more traditional quantitation routes exploit calibrations based on reference materials to establish absolute quantitation14,15.
Though the protocol is outlined in the context of myoglobin and meat, proteins other than myoglobin could be used for identification and relative quantitation via the CPCP strategy in meat mixtures, though potentially with modifications to the protocol. In addition the strategy is also applicable to binary mixtures of other species sharing one or more corresponding proteins.
The starting point for the protocol is purified 'reference' myoglobin, which for some species can be purchased but which for others must be prepared by conventional size-exclusion chromatography. The procedure for preparing reference myoglobin is not included in the protocol, but is described elsewhere3. Software tools16 are used to list candidate peptides and transitions arising from myoglobins of interest. Each reference myoglobin is subjected to proteolysis and the resultant peptides analyzed by liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) to discover which of the candidate precursor ions and transitions are most useful, and to determine the matching peptide retention times. The outcome of this stage is a revised list of target peptides with their transitions, suitable for species determination, and a list of CPCP pairs, suitable for relative quantitation. To test real meats, sample extractions are prepared then subjected to proteolysis to generate peptides both from myoglobin and other extraneous proteins. The myoglobin-based peptides are then monitored by LC-ESI-MS/MS based on their listed transitions. The species present in a mixture are identified by the transition peaks associated with marker peptides. Estimates of the relative amounts of two meats in a binary mixture are calculated using ratios of transition peak areas. A set of test mixtures of pairs of meats will allow the ratio of peak areas for a given pair of transitions to be checked and calibrated against actual mixtures.
протокол
1. протеолиза и анализ эталонных Myoglobins
- Протеолиз эталонных myoglobins
- Готовят растворы очищенных эталонных myoglobins (диапазон 0,2 - 0,5 мг / мл в 25 мМ бикарбоната аммония) 3.
- Передача 1 мл аликвоты каждого образца по 2 мл центрифужные пробирки.
- Термически денатурации распакованные белки путем нагрева образца в горячем блоке при 95 ° С в течение 30 мин. Охлаждают образец в течение примерно 15 мин, пока она не достигнет комнатной температуры. Добавить 30 мг мочевины (конечная концентрация 0,5 М) для улучшения пищеварения, а затем перемешать.
- Триптический протеолиз
- Приготовьте 1 мг / мл раствора трипсина в 25 мМ бикарбоната аммония и хранить на льду в соответствии с требованиями. Добавьте достаточный объем трипсина таким образом, что активность фермента является окончательным 420 ВАЕЕ (N - бензоил-L-аргинином эфира гидрохлорида этил) единиц / мг экстрагированного белка, затем перемешать, осторожно вортексе и позволяют proteolyze в течение ночи при 37 ° С.
- Проводят додецилсульфат натрия гель - электрофореза в полиакриламидном сульфат (SDS-PAGE) 17 , чтобы продемонстрировать полноту протеолиза.
- Обессоливание образца после протеолиза
- Разбавьте образец 1: 2: V с водой.
- Активизировать полимерный обращенно-фазной (RP) картридж заполнен 30 мг Р.П. материала путем добавления 1 мл метанола, затем уравновешивают картридж путем добавления 1 мл 1% муравьиной кислоты.
- Загрузить образец в картридж под действием силы тяжести.
- Промыть 1 мл 5% метанол / 1% муравьиной кислоты, под действием силы тяжести.
- Элюируйте пептиды с 1 мл смеси ацетонитрил / вода (90:10 по объему, 0,1% муравьиной кислоты) под действием силы тяжести в 2 мл микропробирок предварительно заполненном с 5 мкл диметилсульфоксида (ДМСО).
- Растворитель удаляют в вакууме при 50 ° С с использованием центробежного испарителя в течение 120 мин, а затем повторно растворить остаток в 250 мкл смеси ацетонитрил / вода (3:97 по объему; 0,1% муравьиной кислоты).
- Переводраствор для флакона автосамплером низкой громкости.
Примечание: Образцы могут храниться при температуре 4 ° С до готовности для жидкостной хроматографии масс-спектрометрии (ЖХ / МС) анализа.
- Генерация списков переходов для управления записями
- Найдите миоглобина последовательности для различных мяса из базы данных UniProt.
- Введите миоглобина последовательности в поле "Target" программного обеспечения прогнозирования пептида и перехода (например, горизонт). При необходимости, наведите курсор мыши на пептид раскрыть свой список фрагментов.
- Нажмите на "Настройки" и выберите "Peptide Settings". Входные предпочтения для переваривания (т.е. трипсина) и количество пропущенных расколы (0). Введите необходимый выбор дополнительных параметров, в частности, длина пептида (6 - 25), N-терминальные исключений (0) и не предполагается модификаций аминокислот (нет).
- Нажмите на "Настройки" и выберите "Настройки перехода». Выберите настройки длятип инструмент, используемый для анализа LC / MS.
- Нажмите на "Экспорт" и выберите "Transition List ', чтобы создать таблицу, содержащую сгенерированные MRM переходов и параметров.
- Анализ с помощью ЖХ / МС
- Настройка системы бинарных градиента (вода (А) и ацетонитрил (В), каждый с 0,1% кислоты V муравьиной: V) Высокоэффективный жидкостный хроматограф (HPLC) с автоматическим пробоотборником, С18 колонки ядро оболочки ВЭЖХ (10 см х 2,1 мм , 2,6 мкм, размер частиц), соединенный с тройной квадрупольный масс-спектрометр, эксплуатируемого в положительном режиме с электрораспылением с детектированием MRM.
- В программном обеспечении сбора данных (например, аналитик), выберите "Файл" и "Новый" и нажмите на кнопку «Приобретение метода» в всплывающем окне нажмите кнопку "OK".
Примечание: Это открывает редактор метода инструмент, который содержит список подключенных устройств, которые позволят установку нового метода LC / MS. - Нажмите на "Binary Pump", и яNput значение скорости потока (300 мкл / мин), а время градиент в таблице, устанавливая бинарный профиль градиента 3% B до 30% B в течение 22 мин, с увеличением до 100% B на 23 мин в течение 5 мин промывать прежде чем вернуться к начальным условиям и повторного установления равновесия в течение 6 мин.
- Нажмите на 'пробоотборник' и вставьте объем впрыска (5 мкл). Включить "Игла цикла стирки 'и введите' Wash Time '(30 сек) и выберите' Flush Port '.
- Нажмите на 'термостатированной контроллера колонке' и в 'Column Oven Свойства' Установить 'Left температуры' и 'правильной температуре' (40 ° C).
- Нажмите на "масс-спектрометром", а затем нажмите на кнопку "Изменить параметры", чтобы ввести условия источника газа. Выберите "Тип сканирования" в качестве "MRM (MRM)» и «Полярность» как «Позитивный». Перейти к 'Период Резюме' и введите '' продолжительность времени, общее время для анализа ЛКd уравновешивания (35 мин).
- В таблице правой кнопкой мыши и выберите "декластеризация потенциал (DP) 'и' энергии столкновения (CP) ', чтобы добавить эти столбцы в таблице. Введите Q1, Q3, время (мс), идентификатор значения DP и CE для всех переходов, для одного вида мяса, созданных в списке переходов (см шаг 1.4.5).
Примечание: Время (мсек) относится ко времени задержки, время масс-спектрометр проводит сканирование каждого перехода, суммирование которых не должна превышать 3 сек. - Сохраните файл метода приобретения (расширение файла .dam).
Примечание: Шаги 1.5.2 - 1.5.8 должны быть повторены для каждого вида мяса. Это позволит создать единый файл метод для каждого вида мяса в экранном режиме в рамках подготовки к анализу ниже. - В программном обеспечении сбора данных, нажмите на кнопку "Acquire" и выберите "уравновешивания". В открывшемся окне выберите нужный метод приобретения, чтобы начать уравновешивание прибора.
- Поместите образец ампул в АреПодтверждено в автоматическом пробоотборнике.
- Нажмите на "Файл" и выберите "New", затем "Batch Acquisition". На вкладке "Образец" выберите "Add Set", затем "Добавить образцы". Вставьте число анализируемых образцов и нажмите на кнопку "OK". В поле "Acquisition" выберите файл метод, который будет использоваться для анализа из выпадающего меню.
- В таблице, выберите 'Plate код "и выберите нужную конфигурацию лотка из выпадающего меню. Щелкните левой кнопкой мыши в 'Тарелка Code' заголовок столбца затем правой кнопкой мыши и выберите "Fill Down". В "Виал" Позиция ввести положение каждого образца в автоматическом пробоотборнике в строках.
- В "файл данных" введите имя файла для приобретения, а затем щелкните левой кнопкой мыши в заголовке столбца с последующим правой кнопкой мыши и выберите "Fill Down". В 'Sample Name "вставить личность каждого из анализируемых образцов. Сохранить как пакетного файла приобретения (файл электроннойXTension .dab).
- Нажмите на вкладку "Отправить" затем выделите образцы, которые должны быть проанализированы на LC / MS. Нажмите на кнопку "Отправить". Нажмите на ссылку "Приобретите" и "Старт образца", чтобы начать анализ.
Примечание: Каждый метод сбора просканирует для MRM переходов по всей длине хроматографа для одного вида мяса. Массовые настройки спектрометра для приобретения MRM варьироваться в зависимости от типа прибора и пептида. - Просмотр созданные файлы данных, используя программное обеспечение просмотра данных. Нажмите на Xic (извлекаемых ионов) и в выпадающем списке выделить все фрагменты (значения Q3) для одного предшественника (Q1). Новая панель откроется, которая показывает только выбранные переходы.
- Регистрируют время удерживания (R Т) для групп параллельных переходов , так как они соответствуют одному пептиду.
- Повторите предыдущие два шага для каждого набора переходов, чтобы назначить пики их соответствующих пептидов для каждогоиз видов мяса.
- Запись маркеров пептиды , которые пригодны для обеспечения идентификации видов (например, пептид HPGDFGADAQGAMTK, предшественник m / z = 752, R T = 12,0 мин, для лошади), вместе с их временем удерживания, и заметьте которые образуют соответствующие пары , подходящие для относительного количественного ,
Примечание: Например, лошадь маркерного пептида (предшественник м / Z = 752) имеет соответствующую говяжий пептид, HPSDFGADAQAAMSK (прекурсор m / z = 767, R T = 13,2 мин). - Для того, чтобы создать единый динамический метод, охватывающей все виды мяса, в программном обеспечении просмотра данных, для каждого вида мяса, в свою очередь, открыть данные XIC перехода для каждого предшественника (назначенного конкретного пептида 1.5.8).
- Нажмите на пик кластера в заданное время удержания левой кнопкой мыши и перетаскивая курсор под кластера. Определить наиболее интенсивные переходы (щелкнув правой кнопкой мыши на пик этикетке).
- Вручную записать переходыи время удерживания в электронную таблицу.
- Для ввода параметров в качестве нового метода динамического на LC программного обеспечения / MS, нажмите на кнопку "Масс-спектрометры", а затем нажмите на ссылку "Изменить параметры", чтобы ввести условия источника газа. Выберите "Тип сканирования" в качестве "MRM (MRM)» и «Полярность» как «Позитивный».
- Перейти к "Период" Резюме и введите продолжительность времени (устанавливается как общее время для анализа LC и уравновешивания). В таблице правой кнопкой мыши и выберите "декластеризация потенциал (DP) 'и' энергии столкновения (CP) ', чтобы добавить эти столбцы в таблице.
Примечание: В столбце "Время" в настоящее время относится к ожидаемому времени удерживания (мин) для каждого перехода. - В разделе "изменения параметров" программного обеспечения для сбора данных / MS LC, установите флажок "Назначенные MRM" окно. Введите Q1, Q3, Время (мин), ID, DP и CE значения для переходов, созданных в электронной таблице (1.5.21) и сохраните метод приобретения (филе расширение .dam).
Примечание: Этот метод обычно снижает количество MRM переходов наиболее интенсивным 4 для каждого пептида и сканирует только через окно времени удерживания для каждого пептида пика, что дает повышенную чувствительность и качество данных. «Динамический» метод является методом 'руководствовались время удерживания оконная', который иногда называют планированием.
2. Получение и анализ образцов градуировочного
- Добыча мясных смесей
- Используя мясо ранее замороженным затем измельчают в порошок, готовят ассортимент мясных смесей путем взвешивания соответствующих количеств мяса (общей массой около 300 мг) в 15 мл пластиковые центрифужные пробирки.
- Добавляют 4 мл буфера для экстракции (0,15 М хлорида калия + 0,15 М фосфатном буфере при рН 6,5). Vortex в течение 30 сек. Выдержка на лабораторном шейкере при комнатной температуре в течение 2 ч при 250 циклов / мин.
Примечание: Циклы / мин относится к колебательное движение. - Передача 2 млэкстракта в пробирку микроцентрифужных 2 мл. Центрифуга в течение 5 мин при температуре 4 ° С при 17000 х г.
- Передача 200 мкл аликвоты супернатанта (резервируя небольшое количество для анализа белков, см 2.2) в 2 мл центрифужные пробирки и сухой с использованием центробежного испарителя (предварительно заданную программу: 50 ° C, без каких-либо вентилирования и 120 продолжительности мин).
- анализ белка
- Передача 7 мкл аликвоты надосадочной жидкости зарезервированной (см 2.1.4) в трех экземплярах, в лунки 96-луночного планшета.
- Передача 7 мкл аликвоты ряда белковых стандартов в трех экземплярах, диапазон 0 - 1,0 мг / мл бычьего сывороточного альбумина (БСА), к тому же 96-луночного планшета.
- Добавить 200 мкл Кумасси плюс анализа белков реагента в каждую лунку.
- Визуально сравнить цвет образца скважин со стандартами белка, чтобы проверить образцы находятся в диапазоне калибровочных стандартов. При необходимости повторить с разбавленной пробы таким образом это становится в пределах досягаемости.
- Оставьте пластину на ули в течение 3 мин.
- Взрыв все пузырьки, которые сформировались с иглой для подкожных инъекций.
- Анализируют пластину на планшет-ридере использованием стандартного протокола конечной точки при длине волны 595 нм.
- Определить концентрацию белка в образцах с использованием калибровочных данных из белковых стандартов.
Примечание: Это необходимо для расчета количества трипсина, используемого в трипсинизированном дайджеста.
- Протеолиз мясных смесей
- Растворяться высушенный остаток, полученного на стадии 2.1.4 в 1 мл 25 мМ раствора бикарбоната аммония. Хорошо перемешайте на rotamixer.
- Следуйте протоколу, начиная с шага 1.1.3 до 1.3.7.
- Анализ с помощью ЖХ / МС
- Настройка LC / MS, как ранее (этап 1.5.1).
- Создать новую партию приобретения, как это описано ранее (шаги 1.5.9 - 1.5.14), при выборе метода приобретения, созданного на этапе в 1.5.24, который использует / MS метод динамического LC сочетающую всех видов мяса, а также приобретать данные для в DigesTed образцы мяса.
- Покажите полный хроматограмму в программном обеспечении просмотра данных. Отображение Xic для каждого перехода, установленного в свою очередь. Визуально подтвердить каждый кластер содержит необходимое количество колпаковых пиков в ожидаемое время удерживания, тем самым подтверждая существование выбранного пептида.
- Выполнение количественного, используя данные программы просмотра для интеграции площадей пиков для каждого из переходов, представляющих интерес с помощью двойного щелчка на 'Build Количественное определение Метод' в панели навигации.
- В 'Select Sample' панели выберите "Файл данных" и "образец", чтобы быть проанализированы, чтобы сформировать таблицу 'аналитов.
- Нажмите на вкладку "интеграция", чтобы отобразить первый из переходов (аналитов) должны быть интегрированы.
- Нажмите на поле "аналит", чтобы отобразить выпадающий список переходов. Выберите каждый переход в свою очередь, чтобы отобразить его и визуально подтвердить правильный пик выбран для интеграции. Для мodify или форсировать интеграцию, щелкните левой кнопкой мыши и перетащите курсор над целевой пик (это будет выделен зеленым цветом). Нажмите на кнопку "Select Peak" и нажмите кнопку "Применить".
- Сохранение рабочего пространства в качестве метода файла (.qmf).
Примечание: Это создает файл Количественный метод для последующего расчета площадей пиков образцов. - Двойной щелчок 'Количественное Wizard' в панели навигации. В окне "выбрать образцы" создать "Количественное определение Set", выбрав одну "файл данных", а затем один или более "Доступные Образцы". Выберите 'Next' для отображения "выберите Настройки и Query" окно. Оставьте со значениями по умолчанию, выберите "Далее" для отображения "Select Method". Из выпадающего списка '' метод выбора файла в «интеграции» метод, созданный на шаге 2.4.8, а затем выберите "Готово".
Примечание: Это создает 'Таблица результатов, в том числе площади пиков переходных вытекающих из мясных смесей. - Сохранить "Таблица результатов" (расширение файла .rdb), экспорт в виде текстового файла (.txt) и открыть его в таблицу, чтобы просмотреть данные.
- Построить графики процентной доли (переходными площади пика) одного мяса в другой по отношению к измеренному процентах (вес / вес) от двух мяса для выбранного MRM для выбранных переходов из соответствующих пептидов, сосредоточив внимание на тех случаях, когда два фрагмента содержат такое же количество аминокислот, считая от с-терминального конца.
Примечание: Идентичные фрагменты с одинаковыми сайтов фрагментации дают оптимальные результаты. - Изучите сюжеты из 2.4.11 выше. Либо визуально, либо с помощью инструмента линии тренда в пакете черчения, определить группу участков, которые являются как линейные, так и аналогичного градиента. Используйте одну или несколько из этих СРСР плюс фрагментов комбинаций для калибровки в реальных образцах мяса.
Примечание: график, показывающий необычный градиент может указывать либо пептид или подавление фрагмента с последующим уменьшением сигнала Strength. Нелинейные участки могут свидетельствовать о плохом обнаружения пика или другие проблемы.
3. Мясо Образцы
- Выделение белков из образцов целевого мяса
- Там, где это применимо, акцизный посторонний немясных материал из образца с помощью шпателя. Например, соскрести соус и пасту из охлажденной лазаньи.
- Взвешивают 20 г мяса в металлический стакан.
- Добавляют 100 мл 0,15 М хлорида калия / 0,15 М монофосфат буфера калия при рН 6,5.
- Извлечение белков путем смешивания мяса в высокоскоростном гомогенизаторе в течение 1 мин.
- Следуйте протоколу, начиная с шага 2.1.4 - 2.3.2.
- Анализ образцов с помощью ЖХ / МС
- Повторите шаг 2.4.2 для получения данных с использованием динамического метода LC / MS.
- Определить пептиды из каждого мяса миоглобина, как выполняется на этапе 2.4.3.
- Для количественному, используйте программное обеспечение для интеграции количественный площадей пиков для каждого перехода интересов,как показано на этапе 2.4.9.
- Для идентификации видов в смеси, запись этих маркеров пептиды, удовлетворяющие согласованным критериям для чисел переходов и сигнала к шуму для этих переходов.
- Для количественной оценки, использование комплексных переходных пиков, согласованных с шага 2.4.12 и, используя процент по площади пика перехода, вычислить процент миоглобина из двух видов в смеси.
- Использование предварительного знания из литературы 18 возможных уровней миоглобина в мясе для оценки относительных вес / вес количество двух мяса , присутствующих в образце.
Результаты
В одном динамическом режиме MRM эксперимента каждый запрограммированный переход записывается отдельно (как отсчетов детектора в секунду, сП) в течение определенного интервала времени удержания. Таким образом, из всех данных, собранных в одном эксперименте, пик интенсивности для каждого перехода, можно вынимать. Тогда только конечный сигнал для окна времени удерживания, установленного для этого перехода. За пределами окна, сигнал равен нулю по определению. Сигнал для любого одного перехода, например, 752 → 1269 от лошади (пептид одноизотопные масс 1,501.66 дальтон, ион - предшественник m / z 751.84 дальтон, зарядовое состояние = 2, ионный фрагмент у 13) , как правило , должен конкурировать только с шумом измерения и не от других пиков переходных, которые могли бы, возможно, быть от других видов. Таким образом, выход представляет собой набор чистых пиков, по одному на переходном этапе, в общем времени удерживания для тех переходов, разделяющих общий ион-предшественник.
На рисунке 1 показан вывод для набора из четырех переходов 752 → (1269, 706, 248, 1366) для смеси 1% вес / вес лошадь в говядине. Поскольку четыре перехода, отображаемые связаны с лошади, и отсутствуют в образцах чистого говядины, баранины или свинины, эти пики означают наличие лошади. В зависимости от критериев надежности, набор из двух или более переходов каждый превышающий некоторый заданный уровень сигнал-шум устанавливает идентификацию. Таким образом, этот показатель устанавливает наличие лошади в смеси 1% вес / вес лошади в говядине.
Иногда обнаруживается один изолированный переход. Это указывает на случайное совпадение иона-предшественника и один фрагмент, возможно, от постороннего белка, с теми, которые ожидаются от системы и программируются в масс-спектрометр. Особая природа пика, и его появление в неожиданное время удержания, является сиgnature случайного перехода, который может быть проигнорировано.
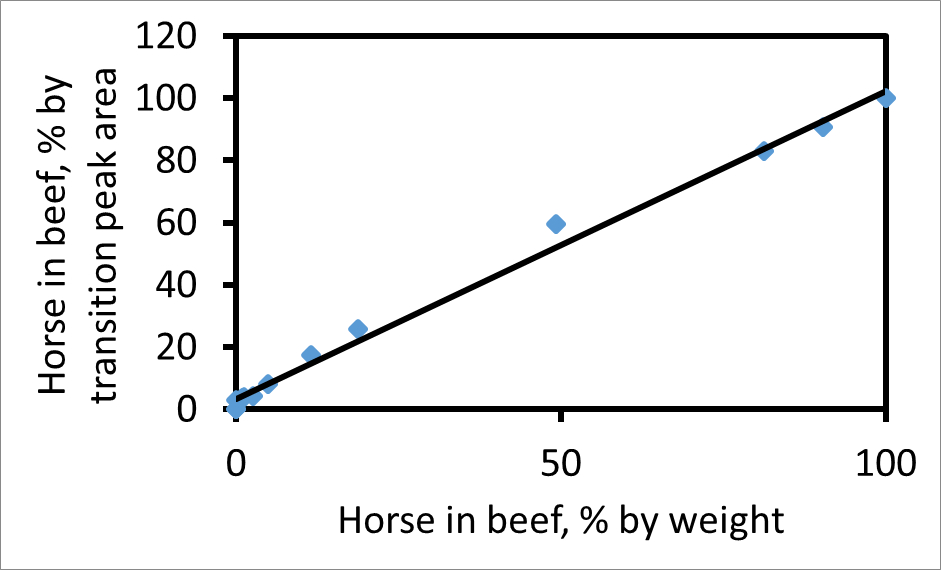
Площадь под каждым пиком перехода можно рассчитать по отдельности. На основе подходящего фрагмента, отношение лошади к площадям пиков переходных говядины, например, 752 → 1269 (лошади) до 767 → 1299 (говядина), будет пропорционально отношению фактического мяса в смеси. На рисунке 2 показана участок процент по площади пика для этих двух переходов к проценту веса для веса лошади в смеси с лошади говядины. Если площади пиков в процентах переходов соответствует процент веса для веса мяса, то наклон равен 1. Наклон в этом участке составляет 1,03, что свидетельствует о том, что для этих переходов и СРСР пары, переход площади пиков дают надежное измерение относительных количеств из двух мяса в смеси. Если мясо лошади в образце был вдвое богат миоглобина, как говядина, то, с другими факторами неизменными, наклон линия будет больше единицы.

Рисунок 1. интенсивности переходов MRM в зависимости от времени удерживания на 1% вес / вес лошади в говядине. Переходы 752 → (1269, 706, 248, 1366), показано в оранжевый, черный, синий и зеленый, соответственно. Маркер пептид HPGDFGADAQGAMTK. Четыре фрагмента перехода можно обозначать Y 13, Y 7, Y 2 и Y 14 соответственно, где у п обозначает подсчета в п аминокислот от пептида C-терминального конца. Сигнал шума варьируется от 23 до 53 в течение четырех переходов. Дополнительная красная линия обозначает переход на 752 → 1269 для 0% лошади, 100% говядины для сравнения. Отображается только ненулевая область времени удерживания. Эта цифра была изменена с Уотсоном и др. 3.эс / ftp_upload / 54420 / 54420fig1large.jpg "целевых =" _blank "> Пожалуйста, нажмите здесь, чтобы посмотреть увеличенную версию этой фигуры.

Рисунок 2. Участок лошади в говядине, как процент веса для веса, по сравнению с лошади в говядине как процент перехода площади пика. Участок использует пару пептидов говядины (767) и лошади (752) и фрагмент иона у 13 для обоих. Если А обозначает площадь пика , то по оси ординат 100 А Н / (А + Н А Б). Наклон лучшей подгонки линии (R 2 = 0,99) составляет 1,03. Эта цифра была изменена с Уотсоном и др. 3. Пожалуйста , нажмите здесь , чтобы посмотреть увеличенную версию этой фигуры.
{kind=link}
Обсуждение
Выбор подходящего целевого белка имеет важное значение. Хороший целевой белок должен иметь соответствующие формы в видов, представляющих интерес, достаточный зависит от вида вариации последовательности, видовой специфичности, и существуют в доступных количествах в организмах. Для оценки смесей, которые подверглись обработке (например, термообработка), белок, имеющий последовательность, относительно невосприимчивы к этой обработки желательно. Миоглобин является хорошим кандидатом для красного мяса, в том числе приготовленные из красного мяса, но это не единственная возможность. После того, как белок-мишень принимается решение, наиболее важной частью протокола является белок протеолиз. Белок отличается от миоглобина также может потребовать альтернативный протокол протеолиза.
Протокол, как описано включает в себя сегмент на основе эталонного очищенного белка. Это имеет целью открыть окна времени удерживания и подходящие ионы-предшественники и фрагментов. Этот сегмент является очень полезным, но не обязательно.
Несмотря на то, соответствующие пептидные пары из двух видов, представляющих интерес могут быть перечислены даже без опыта, иногда бывает, что разница последовательность имеет драматические последствия на профиле пищеварения. Например, пептид пара VLGFHG (говядина) и ELGFQG (лошадь) дают аномальный результат количественный (проявляется в виде градиента меньше , чем один на рисунке 2). Это происходит потому, что последний пептид возникает из относительно подавленной расщепления К.Э., что приводит к недооценке уровня лошади в смеси. Соответствующие пептиды, которые начинаются с разных аминокислот, поэтому лучше избегать. Часто фрагменты из двух соответствующих пептидов имеют идентичные аминокислотные последовательности, и хорошо себя, но это не всегда так, и должно быть проверено в процессе разработки метода. идентификация видов гораздо менее чувствительны к этим вопросам, чем относительное количественному.Протокол был продемонстрирован на четыре красного мясаs 3. Дополнительные виды мяса могут быть включены, хотя качество пик формы перехода может ухудшиться, если слишком много маркерные пептиды коэлировать, эффективно уменьшая время задержки и, в конечном счете деградирует относительных оценок Количественное определение. Улучшение измерительных приборов, уже доступны, улучшит это. Связанный с этим вопрос заключается в том, что не все виды мяса имеют разные myoglobins. Например, лошади, ослы и зебры myoglobins идентичны и, таким образом, строго говоря, метод способен только обнаруживать лошадь или осла или зебру в говядине. В некоторых случаях, даже если myoglobins не идентичны, некоторые ключевые пептиды могут быть. Например, некоторые бараньи миоглобина происхождения маркерные пептиды также появляются в козу.
Усложнение перед этим и любой другой на основе белка Количественный способ является то, что уровень содержания белка должен считать постоянным во всех видов, если уровни белка или пептида являются приравнять тривиальным к уровням мяса в смеси. Для миоглобина и четыре красных мест это не всегда верно. Уровни в целом виды, зависящие, со свининой, проявляющие низкий уровень четырех. Кроме того, уровень миоглобина изменяется в зависимости от мяса вырезать и возраста животных. Таким образом, хотя отношения площадей пиков переходных достоверно сопоставить соотношения миоглобина, отображение соотношение фактического мяса является оценкой рисунок на предположениях относительно вероятных источников на мясо в смеси.
Подход, изложенный в данной работе отличается несколькими способами от других опубликованных вкладов. Более типичный путь заключается в использовании протеомические методы для идентификации различных несопоставимых зависит от вида маркерные пептиды, причем в этом случае маркеры для различных видов не обладают особого отношения друг с другом 8-12,14,19. В отличие от этого , мы отобрали белки , общие для всех видов , представляющих интерес до видов-зависимых вариантов последовательностей 3. Помимо того, что центральное место в нашей относительной стратегии количественного, это имеет то преимущество, что образецСтратегии препарат может быть оптимизировано. Кроме того, такие соответствующие белки можно было бы ожидать, ведут себя подобно, например, при добыче или в коммерческой обработке образцов, таких как приготовление пищи или консервирования. идентификация видов, то обычно протекает через обнаружение несопоставимых пептидов маркерные, в то время как в СРСР подход определения видов протекает через обнаружение близко родственных пептидов, обладающих, как правило, один или два различия последовательности. И, наконец, определение количества белков , чтобы оценить процент по массе одного вида в другой может условно протекать по абсолютному количественному каждого белка в отдельности , на основе известных стандартов 7,14,15. Однако с помощью метода СРСР нет никакой необходимости в методах калибровки. Вместо того, чтобы относительные уровни оцениваются путем сравнения уровней сигналов двух соответствующих пептидов из двух видов, минуя стадию абсолютного измерения в целом. Поскольку конечной целью является процент по весу одного вида в ANOTher, родственник Количественное, то СРСР является одновременно более прямым и проще, чем сравнения двух абсолютных измерений Количественное определение. Эти функции перевода в короткие экспериментальные времена, как ожидается, будет примерно в два ч с использованием уточненных протоколов, что делает этот метод полезен в качестве быстрого средства наблюдения в области выявления случаев мошенничества пищи.
Раскрытие информации
The authors have nothing to disclose.
Благодарности
We acknowledge financial support from Institute of Food research BBSRC Core Strategic Grant funds, BBSRC Project BB/J004545/1.
Материалы
| Name | Company | Catalog Number | Comments |
| Uniprot database | www.uniprot.org | Freely accessible database of protein sequences | |
| Skyline software | www.skyline.gs.washington.edu | Free software to download that enables the creation of targeted methods for proteomic studies, peptide and fragment prediction | |
| Ammonium bicarbonate | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | O9830 | |
| Methanol, HPLC grade | Fisher Scientific, Loughoborough, UK www. fisher.co.uk | 10674922 | |
| Acetonitrile, HPLC grade | Fisher Scientific, Loughoborough, UK www. fisher.co.uk | 10010010 | |
| Urea | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | U5378 | |
| Trypsin(from bovine pancreas treated with TPCK) | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | T1426 | |
| Formic acid | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | F0507 | |
| Coomassie Plus Protein Assay Reagent | Thermo Fisher Scientific www.thermofisher.com | 1856210 | |
| Protein standard | Sigma-Aldrich Co Ltd, Gillingham, UK www.sigmaaldrich.com | P0914 | |
| Ultra Turrax homogeniser T25 | Fisher Scientific, Loughoborough, UK www. fisher.co.uk | 13190693 | |
| Edmund and Buhler KS10 lab shaker | |||
| Heraeus Fresco 17 Centrifuge | Thermo Fisher Scientific www.thermoscientific.com | 75002420 | |
| Vacuum centrifuge RC 1022 | Jouan | ||
| Plate Reader | |||
| Strata-X 33u polymeric reversed-phase cartridges 60 mg/3 ml tubes | Phenomenex, Macclesfield, UK | 8B-S100-UBJ | |
| 4000 QTrap triple-quadrupole mass spectrometer | AB Sciex, Warrington, UK www.sciex.com | ||
| 1200 rapid resolution LC system | Agilent, Stockport, UK | ||
| XB C18 reversed-phase capillary column (100 mm x 2.1 mm, 2.6 µm particle size) | Phenomenex, Macclesfield, UK www.phenomenex.com | ||
| Analyst 1.6.2 software | AB Sciex, Warrington, UK www.sciex.com | QTrap data acquisition and analysis, including peak area integration | |
| Autosampler vials |
Ссылки
- O'Mahony, P. J. Finding horse meat in beef products-a global problem. QJM-An Int. J. Med. 106, 595-597 (2013).
- Sentandreu, M. A., Sentandreu, E. Authenticity of meat products: Tools against fraud. Food Res. Int. 60, 19-29 (2014).
- Watson, A. D., Gunning, Y., Rigby, N. M., Philo, M., Kemsley, E. K. Meat Authentication via Multiple Reaction Monitoring Mass Spectrometry of Myoglobin Peptides. Anal. Chem. 87, 10315-10322 (2015).
- Food Standards Agency. . Report of the investigation by the Food Standards Agency into incidents of adulteration of comminuted beef products with horse meat and DNA. , (2013).
- Taylor, A. J., Linforth, R., Weir, O., Hutton, T., Green, B. Potential of electrospray mass-spectrometry for meat pigment identification. Meat Science. 33, 75-83 (1993).
- Ponce-Alquicira, E., Taylor, A. J. Extraction and ESI-CID-MS/MS analysis of myoglobins from different meat species. Food Chem. 69, 81-86 (2000).
- Gallien, S., Duriez, E., Domon, B. Selected reaction monitoring applied to proteomics. J. Mass Spectrom. 46, 298-312 (2011).
- Orduna, A. R., Husby, E., Yang, C. T., Ghosh, D., Beaudry, F. Assessment of meat authenticity using bioinformatics, targeted peptide biomarkers and high-resolution mass spectrometry. Food Addit. Contam. Part A-Chem. 32, 1709-1717 (2015).
- Claydon, A. J., Grundy, H. H., Charlton, A. J., Romero, M. R. Identification of novel peptides for horse meat speciation in highly processed foodstuffs. Food Addit. Contam. Part A-Chem. 32, 1718-1729 (2015).
- von Bargen, C., Brockmeyer, J., Humpf, H. U. Meat Authentication: A New HPLC-MS/MS Based Method for the Fast and Sensitive Detection of Horse and Pork in Highly Processed Food. J. Agric. Food Chem. 62, 9428-9435 (2014).
- von Bargen, C., Dojahn, J., Waidelich, D., Humpf, H. U., Brockmeyer, J. New Sensitive High-Performance Liquid Chromatography Tandem Mass Spectrometry Method for the Detection of Horse and Pork in Halal Beef. J. Agric. Food Chem. 61, 11986-11994 (2013).
- Montowska, M., Alexander, M. R., Tucker, G. A., Barrett, D. A. Authentication of processed meat products by peptidomic analysis using rapid ambient mass spectrometry. Food Chem. 187, 297-304 (2015).
- Montowska, M., Alexander, M. R., Tucker, G. A., Barrett, D. A. Rapid detection of Peptide markers for authentication purposes in raw and cooked meat using ambient liquid extraction surface analysis mass spectrometry. Anal. Chem. 86, 10257-10265 (2014).
- Sentandreu, M. A., Fraser, P. D., Halket, J., Patel, R., Bramley, P. M. A Proteomic-Based Approach for Detection of Chicken in Meat Mixes. J. Proteome Res. 9, 3374-3383 (2010).
- Elliott, M. H., Smith, D. S., Parker, C. E., Borchers, C. Current trends in quantitative proteomics. J. Mass Spectrom. 44, 1637-1660 (2009).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26, 966-968 (2010).
- Keeton, J. T., Ellerbeck, S. M., Nunez de Gonzalez, M. T., Devine, C., Dikeman, M. . Encyclopedia of Meat Sciences. 1, 235-243 (2014).
- Montowska, M., Alexander, M. R., Tucker, G. A., Barrett, D. A. Rapid Detection of Peptide Markers for Authentication Purposes in Raw and Cooked Meat Using Ambient Liquid Extraction Surface Analysis Mass Spectrometry. Anal. Chem. 86, 10257-10265 (2014).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены