
Method Article
Крыса метил Seq платформа для выявления эпигеномные изменения, связанные с воздействием стресса
В этой статье
Резюме
Здесь мы описываем протокола и осуществления метил-Seq, epigenomic платформы, с помощью мышиной модели для идентификации эпигеномные изменения, связанные с воздействием хронического стресса. Результаты показывают, что метил-Seq платформа крыса способен обнаруживать метилирования различия, которые возникают от воздействия стресса у крыс.
Аннотация
Как геномов более широкий спектр животных становятся доступными, существует растущая потребность для инструментов, которые могут захватить динамические эпигеномные изменения в этих моделях животных. Крыса является одной конкретной модели животных, где эпигеномные инструмент может дополнять многих фармакологических и поведенческих исследований обеспечить глубокий механистический информации. С этой целью мы адаптировали SureSelect целевой захвата системы (упоминаемый как метил-Seq) для крысы, которые могут оценить уровень метилирования ДНК через геном крысы. Крыса дизайн ориентированных промоутеров, острова CpG, Остров Шорс и GC-богатые регионы от всех RefSeq генов.
Для осуществления платформы на эксперимент крыса, крысах Sprague-Dawley были подвержены хронический стресс переменной за 3 недели, после чего были собраны образцы крови для экстракции геномной ДНК. Метил-Seq библиотеки были построены из образцов ДНК крыса стрижка, адаптер перевязки, цели обогащения, преобразование бисульфита и мультиплексирования. Библиотеки были виртуализировать на платформе виртуализации нового поколения и последовательного читает были проанализированы для определения ЗВД между ДНК безударных и подчеркнул крыс. Топ кандидата ЗВД были независимо проверены бисульфита пиросеквенирования для подтверждения надежности платформы.
Результаты показывают, что крыса метил-Seq платформа является полезным эпигеномные инструментом, который может захватить метилирования изменений, вызванных воздействием подчеркнуть.
Введение
Достижения в высокопроизводительного секвенирования привели к богатству genomic последовательностей для модели и номера модели организмов. Наличие таких последовательностей способствовала исследованиям в области генетики, сравнительной геномики и transcriptomics. К примеру, доступные genomic последовательностей являются весьма полезными для выравнивания последовательности данных от чип-Seq экспериментов, которые обогащают ДНК, на основе своей ассоциации с гистонов модификации1, или гидросульфит последовательности, которая измеряет метилирования дна путем Обнаружение урацила формируется из преобразование бисульфита unmethylated cytosines2. Однако имели место задержки в осуществлении epigenomic платформ, которые включают имеющиеся геномные последовательности данных в их конструкции из-за отсутствия аннотированный данных вегетационных регулирования последовательностей, которые могут влиять на функции гена.
В частности метилирование ДНК является одним из наиболее широко изучены эпигеномные изменения на ДНК, которая может использовать имеющиеся геномных данных для создания methylomic платформы. Одним из таких примеров является платформой на основе массива для человека methylome3, которая широко используется в различных дисциплинах по психиатрии4,5от онкологии. К сожалению подобные платформы для не человеком животных моделей являются скудными, как есть практически не широко используемых платформ, которые воспользовались геномные последовательности в их первоначальный дизайн.
Общий метод для оценки methylomic пейзаж не человеком животных моделей — уменьшенная бисульфита виртуализации (RRBS)6. Этот подход преодолевает Стоимость секвенирования бисульфита всего генома, что, обеспечивая всеобъемлющую methylomic пейзаж, обеспечивает чтение глубина охвата ниже из-за стоимости и ограниченной функциональной информации в крупных ген бедных районах генома2 . RRBS предполагает ограничение дайджест и выбор размера геномной ДНК для обогащения для высоко GC-богатых последовательностей как острова CpG, которые обычно вблизи генным стимуляторам и думал играть определенную роль в ген регулирование7. В то время как в ряде важных исследований был использован метод RRBS, ее опора на энзимов ограничения — не без известные проблемы и ограничения. К примеру обогащение GC-богатых последовательностей в RRBS полностью зависит от наличия специфических последовательностей, признанные энзима ограничения и последующая размер выбор электрофорезом. Это означает, что любой геномной районы, которые не содержат эти места ограничения исключаются во время выбора размера. Кроме того кросс видов сравнения являются сложными, если же места ограничения нет в же локусов среди различных видов.
Один из подходов к преодолению ограничений RRBS заключается в использовании метод обогащения, который использует опубликованные геномные последовательности в разработке платформы. На основе массива человека платформа использует грунтовка зонды, направленных против конкретных СКП для Аллел Специфически (CG против TG после преобразования бисульфит) целевой отжига и грунтовка расширения. Его дизайн отражает не только доступные человека геномные последовательности, но и экспериментально проверены регулирования регионов приобретены из нескольких строк запроса, таких как кодирование и ENSEMBL8. Несмотря на его широкое применение в человека methylomic исследования подобные платформы для модели животных не существует. Кроме того формат на основе массива места значительные ограничения на площадь поверхности для размещения зонд. В последние несколько лет были предприняты усилия, чтобы объединить цели специфика обеспечиваемой захвата зонд дизайн и высок объём особенность секвенирование нового поколения. Такие усилия привела к системе обогащения на основе последовательности целевого объекта для генома мыши (мыши метил-Seq), которая была использована для выявления мозга конкретные или глюкокортикоидов индуцированной различия в метилирования9,10. Подобных платформ для других моделей и животных-модели необходимы для облегчения epigenomic исследований в этих животных.
Здесь мы демонстрируем осуществления этот роман платформы для проведения methylomic анализа на крысу. Крыса служил важным животной модели в фармакологии, метаболизм, Нейроэндокринологии и поведение. Например существует возрастает необходимость понимания основных механизмов, которые приводят к токсичности препарата, ожирение, стресс ответ или наркомании. Высок объём платформы, способной захвата methylomic изменения, связанные с этими условиями будет увеличить наше понимание механизмов. Поскольку геном крысы по-прежнему не хватает Аннотация для регулирования регионов, мы включены нерезервном промоутеров, острова CpG, Остров Шорс11и ранее определены ГК богатыми последовательности в крыса метил-Seq платформа12.
Чтобы оценить успешной разработки и осуществления SureSelect цели обогащения (обобщенно называют метил-Seq) платформы для геном крысы, мы использовали крыс модель13 переменных хронического стресса (CVS) для выявления дифференциально метилированной регионах между безударных и подчеркнул животных. Наш дизайн платформы, протокол и осуществления могут быть полезны для следователей, которые хотят провести всеобъемлющее и объективное эпигеномные расследование на организм которых геномные последовательности уже доступна, но по-прежнему плохо аннотированный.
протокол
Все эксперименты были завершены в соответствии и соблюдение всех соответствующих нормативных и институциональных руководящих принципов, включая институциональный уход животных и использования Комитетом на Джона Хопкинса Школа медицины.
1. Животные
- На 4-недельного возраста получите подростков крысах Sprague-Dawley. Дом животных в клетках крыс из поликарбоната в температуре- и под контролем влажности номер на 12 ч света, 12 h темные цикла с наступлением света в 0600 h. животных с ad libitum доступ к воде.
- Разрешить крысы, чтобы акклиматизироваться на 1 неделю, чтобы уменьшить стресс, связанные с транспортом. Пара дом животных (N = 16) исключает изоляции стресс, и в возрасте 5 недель, начать переменной хронический стресс (CVS) режим за 3 недели.
2. хронический стресс переменной
- Администрировать режима CVS однажды утром (9-11 утра) и один раз в день (1-3 вечера) на нерегулярные раз держать рутину непредсказуемым. Включить на ночь мягкий стресс. CVS режим включает в себя: 1) 3 ч в цилиндре сдержанность; 2) 10 мин плавать; 3) 3 h Кейдж наклона 4) 1 ч медленно покачивая платформы; и 5) 1 ч в холодной комнате 4 ° С.
Примечание: Ночь раздражители включают социального вытеснения (5 в клетке), социальной изоляции, мокрой кровати, еда ограничения и огни на. Типичная еженедельный график режима стресс приводится в таблице 1.
3. Эндокринная анализов
-
Определить уровень кортикостерона (CORT) с помощью хвоста отборы проб крови (~ 50 мл) собраны в то же время (9 утра) два раза в неделю на протяжении всего эксперимента перед CVS режим для установления базовых уровней гормонов (день 0), один раз в середине загрузок CVS (дней 4,11 и 18), через каждые 7 дней CVS (7 дней и 14) и в заключение CVS (21 день). Сбор проб крови до режима ежедневного стресса.
- Собирают один заключительный ствола крови во время эвтаназии (день 25) для РИА и экстракции геномной ДНК.
- Центрифуга для всех образцов крови (600 x g, 4 ° C, 10 мин), чтобы отделить плазму от клеток крови. Накапайте из плазмы (супернатант) и хранить образцы-80 ° c.
- Размораживание и использовать плазмы для определения уровней CORT радиоиммуноанализ (РИА). Убедитесь, что 3 недели плазмы CORT уровни повышаются в подчеркнул животных для проверки надежности режима стресса.
4. поведение
- После режима CVS (дней 23 – 24), оценить каждое животное для тревоги подобное поведение на повышенные плюс лабиринт (EPM)14.
- С помощью видеокамеры, запись животных на аппарат EPM для 300 s и оценка, время, проведенное в центре, закрыт оружия и распростертыми объятиями.
5. дизайн метил Seq крыса
- С помощью браузера геноме UCSC, получить нерезервном геномной координаты (Крыса Ноя 2004 rn4 Ассамблеи) для островов CpG и Остров Шорс (± 1 КБ, окаймляющие острова CpG), промоутеры (± 1 КБ каждый ТП) каждого гена RefSeq, и другие последовательности, которые могут быть доступны из соответствующей литературе.
Примечание: Для крысы метил-Seq, дополнительные последовательности GC-богатые люди от предыдущей платформы на основе массива метилирования была добавлена12. Для регионов более чем 5 кбит/с чередуя регионов 500 бод были взяты пробы следуют 1 кбит/с, которые были пропущены. Окончательный крыса метил-Seq конструкция состоит из 111 Мбит/с, 2.3 миллионов СКП; и регион средний размер 594 bps. Он нацелен на 228,800 уникальный локусов. - Введите составлен список геномной координат в коммерчески доступных целевых захвата дизайн программного обеспечения для соответствующего зонд дизайн.
6. строительство библиотеки метил Seq крысы от дна Genomic
Примечание: Для ликвидации партии эффекты, обработать несколько образцов в то же время и наращивать мастер смеси соответственно. Извлечь ДНК, используя набор коммерчески доступных для извлечения ДНК. Методы на основе столбцов или осадков дают высокое качество геномной ДНК (отношение ~ 1.8 260/280). Не рекомендуется использовать методы на основе фенола. Элюировать или Ресуспензируйте ДНК в буфере TE низкий (10 мм TE, 0,1 мм ЭДТА, рН 8,0).
- Подготовка образца
Примечание: Для каждого шага с помощью ДНК связывающих магнитные бусы, убедитесь, бусины приспособиться к комнатной температуре по крайней мере 30 минут и хорошо перемешать перед использованием.- Сдвига ДНК
- Используйте флуориметр для определения начальной концентрации двуцепочечной ДНК каждого образца. Разбавить > 1 мкг геномная ДНК до 50 мкл с низким TE буфера (10 мм TE, 0,1 мм ЭДТА, рН 8.0) в низких ДНК связывающих microcentrifuge трубы.
- Сдвига образцы с использованием изотермических sonicator (10% ПВ, 5 интенсивности, 200 циклов в лопнул, 6 циклов 60 s, частота подметать, 4 ° C).
- Оцените качество ДНК с помощью электрофореза-систему на основе меры ДНК размер и количество.
Примечание: Рекомендованная сумма ДНК-1 мкг, или 3 мкг. Если есть ограниченный исходный материал, низкий входной сумма должна быть > 500 нг, как нижнюю отрицательно скажется на количество и качество созданных библиотек.
- Заканчивается ремонт ДНК.
- Используйте крыса метил-Seq комплект для подготовки мастер ремонтно конец микс на льду. Мкл 52 смеси для каждой выборки и инкубировать в тепловая велосипедист без крышки с подогревом (20 ° C за 30 мин., владение 4 ° C).
Конец ремонт Мастер микс (на сэмпл):
35.2 мкл воды
10 мкл буфера конце ремонта (10 x)
1,6 мкл dNTP микс
1 мкл T4 ДНК полимеразы
2 мкл фрагменты ДНК полимеразы
2.2 мкл T4 Polynucleotide киназы - Очищайте образцы с использованием 180 мкл ДНК связывающих магнитные бусы и 400 мкл свежеприготовленные 70% этанола на сэмпл. 180 мкл бусы для каждой выборки и инкубировать в течение 5 мин при комнатной температуре. Пелле бусы, удалить супернатант и Ресуспензируйте Пелле в 200 мкл на 70% спирте. Удаление этанола и повторите мыть раз.
- Используйте магнитные пластины Пелле бусины и удалить столько этанол как можно скорее. Сухой в 37 ° C теплоблока для 3-5 мин до тех пор, пока шарик Пелле совершенно сухой. Ресуспензируйте в 44 мкл нуклеиназы свободной воды и собирать приблизительно 42 мкл супернатант.
Остановочный пункт: После окончания ремонта ДНК образцов может быть опечатаны и хранятся при температуре-20 ° C.
- Используйте крыса метил-Seq комплект для подготовки мастер ремонтно конец микс на льду. Мкл 52 смеси для каждой выборки и инкубировать в тепловая велосипедист без крышки с подогревом (20 ° C за 30 мин., владение 4 ° C).
- Adenylate 3' заканчивается.
- Подготовка мастер Adenylation микс на льду. Добавить 9 мкл смесь для каждой выборки и инкубировать в тепловая велосипедист без крышки с подогревом (37 ° C за 30 мин., владение 4 ° C).
Мастер Adenylation микс (на сэмпл):
5 мкл буфера фрагменты
1 мкл dATP
3 мкл фрагменты ДНК полимеразы - Очищайте образцы с использованием 90 мкл ДНК связывающих магнитные бусы и 400 мкл свежеприготовленные 70% этанола на сэмпл. Мкл 90 бисера для каждой выборки и инкубировать в течение 5 мин при комнатной температуре. Пелле бусы, удалить супернатант и Ресуспензируйте Пелле в 200 мкл на 70% спирте. Удаление этанола и повторите мыть раз.
- Используйте магнитные пластины Пелле бусины и удалить столько этанол как можно скорее. Сухой в 37 ° C теплоблока для 3-5 мин до тех пор, пока шарик Пелле совершенно сухой. Ресуспензируйте в 35 мкл нуклеиназы свободной воды и собирать около 33,5 мкл супернатант.
- Подготовка мастер Adenylation микс на льду. Добавить 9 мкл смесь для каждой выборки и инкубировать в тепловая велосипедист без крышки с подогревом (37 ° C за 30 мин., владение 4 ° C).
- Перевязать метилированной адаптер.
- Подготовить лигирование Мастер микс на льду и 16,5 мкл смеси для каждой выборки. Инкубируйте в тепловая велосипедист без крышки с подогревом (20 ° C на 15 мин, удерживайте 4 ° C).
Перешнуровка Мастер микс (на сэмпл):
2.5 мкл воды
2.5 мкл метил Seq метилированию адаптер
10 мкл T4 ДНК лигаза буфера (5 x)
1.5 мкл T4 ДНК лигаза - Очищайте образцы с использованием 90 мкл ДНК связывающих магнитные бусы и 400 мкл свежеприготовленные 70% этанола на сэмпл. Мкл 90 бисера для каждой выборки и инкубировать в течение 5 мин при комнатной температуре. Пелле бусы, удалить супернатант и Ресуспензируйте Пелле в 200 мкл на 70% спирте. Удаление этанола и повторите мыть раз.
- Используйте магнитные пластины Пелле бусины и удалить столько этанол как можно скорее. Сухой в 37 ° C теплоблока для 3-5 мин до тех пор, пока шарик Пелле совершенно сухой. Ресуспензируйте в 22 мкл нуклеиназы свободной воды и собирать примерно 22 мкл супернатант. Оцените качество с помощью bioanalyzer.
Примечание: Если общее количество ДНК является менее 500 нг, сдвига и процесс дополнительные ДНК перед выполнением последующих шагов. Если средний размер ДНК не увеличится более чем 30 bps, убедитесь, что реагенты являются новыми, как T4 ДНК-полимеразы, фрагменты, и/или T4 лигаза могут быть старые.
Остановочный пункт: После безлигатурные метилированной адаптер, образцы могут быть опечатаны и хранятся при температуре-20 ° C.
- Подготовить лигирование Мастер микс на льду и 16,5 мкл смеси для каждой выборки. Инкубируйте в тепловая велосипедист без крышки с подогревом (20 ° C на 15 мин, удерживайте 4 ° C).
- Сдвига ДНК
- Гибридизация
- Передать низкий трубы microcentrifuge ДНК связывающих образцы и использовать с подогревом вакуумные концентраторы для уменьшения объема образца менее 3,4 мкл. воссоздать образцов 3.4 мкл.
Примечание: Концентрат образцов примерно ~ 3 мкл чтобы убедиться, что образцы удаляются из вакуума концентратор до все жидкости испаряется. - Подготовьте гибридизации буфера при комнатной температуре и метил-Seq блок микс на льду. 5.6 мкл метил-Seq блок микс для каждой выборки и инкубировать в тепловой циклователь (95 ° C за 5 мин, 65 ° C для 2 мин, удержание 65 ° C).
Гибридизация буфер (на сэмпл):
6.63 мкл метил Seq Гибнер 1
0.27 мкл метил Seq Гибнер 2
2.65 мкл метил Seq Гибнер 3
3.45 мкл метил Seq Гибнер 4
Метил-Seq блок микс (на сэмпл):
2.5 мкл метил Seq индексации блока 1
2.5 мкл метил Seq блока 2
0,6 мкл метил Seq блока 3 - Подготовьте РНКазы блок смесь и перемешать гибридизации захватить библиотеки. 20 мкл захвата библиотека гибридизации смеси для каждой выборки и Инкубируйте на 65 ° C для по крайней мере 16 h.
РНКазы блок микс (на сэмпл):
0.5 мкл РНКазы блока
1.5 мкл воды
Захват библиотека гибридизации Mix (на сэмпл):
13 мкл буфера гибридизации
2 мкл РНКазы блок микс
5 мкл крыса метил Seq захватить Библиотека
Примечание: держать реакции при 65 ° C при добавлении гибридизации микс для предотвращения неспецифической привязки. - Аликвота 50 мкл стрептавидина магнитные бусы на сэмпл в новой полосы 8-Ну трубку. Вымойте бусины с 200 мкл буфера метил-Seq привязки. Пелле бусины и удалить супернатант между каждой стирки для в общей сложности 3 смывки используйте магнитные пластины. После окончательной стирки Ресуспензируйте стрептавидина бусины в 200 мкл буфера метил-Seq привязки.
- Добавьте образцы 200 мкл промывают стрептавидина магнитные бусы и инкубации при комнатной температуре за 30 минут с помощью вращающегося смеситель. Во время смешивания, аликвота 200 мкл метил-Seq мыть буфера 2 в трех экземплярах скважины 96-луночных плиты каждого образца и место в тепловой cycler для предварительно нагреть до 65 ° C.
- После инкубации, Пелле стрептавидина магнитные бусы, используя магнитные пластины и Ресуспензируйте бисер в 200 мкл метил-Seq мыть буфера 1. Инкубируйте 15 мин при комнатной температуре. Используйте магнитные пластины для гранул и удалить супернатант.
- Вымойте бусины 3 раза с метил-Seq мыть буфер 2: Ресуспензируйте шарик Пелле в 200 мкл мыть буфера 2 (предварительно подогреть в шаг 6.2.5.), инкубировать бусины в тепловой циклователь (65 ° C, 10 мин) и Пелле бусины. Выбросите надосадке после каждого мытья, используя магнитные пластины.
Примечание: Сохранить реакций гибридизации при 65 ° C при добавлении 2 буфера мытья для предотвращения привязки неспецифические. - 20 мкл метил-Seq Элюирующий буфер промывают бисером и инкубации при комнатной температуре 20 минут использования магнитные пластины Пелле бусины и передачи супернатанта новой трубки газа. Выбросите бисер.
Примечание: Время инкубации, подготовьте бисульфита преобразования реагента.
- Передать низкий трубы microcentrifuge ДНК связывающих образцы и использовать с подогревом вакуумные концентраторы для уменьшения объема образца менее 3,4 мкл. воссоздать образцов 3.4 мкл.
- Преобразование бисульфита
Примечание: Выполните преобразование бисульфита eluted ssDNA, используя соответствующие реагенты и инструкции от коммерчески доступных бисульфита конверсирования.- Добавьте 130 мкл подготовленных бисульфита преобразования реагента в надосадке из предыдущего шага. Разделите каждый из 150 мкл реакций одинаково в две скважины. Инкубируйте в тепловой циклователь (64 ° C для 2,5 ч, удерживайте 4 ° C).
Примечание: 150 мкл реакции делится поровну на два отдельных скважин для обеспечения однородной температуры. После инкубации в течение 2,5 ч, сразу переходите к следующему шагу. - Привяжите образцы спина столбцов путем добавления 600 мкл буфера привязки и мыть раз с 100 мкл буфера мытья. Центрифуга столбцы (15000 x g, 1 мин) между все этапы преобразования бисульфит и отбросить потока через.
- Desulphonate образцы, добавив столбцы 200 мкл буфера Desulphonation. Инкубации при комнатной температуре 15-20 мин повторить центрифугирования и отбросить потока через.
- Вымойте столбцы дважды с 200 мкл буфера мыть. Элюировать каждый пример, добавив 10 мкл буфера к столбцу, инкубации на 3 мин при комнатной температуре и центрифугирования (15000 x g, 1 мин). Повторите шаг элюции для в общей сложности 20 мкл.
- Подготовьте реакции PCR 1 Mix Master на льду. Мкл 82 смеси для каждой выборки. Инкубируйте в тепловая велосипедист с следующей программы.
ПЦР реакции Master Mix 1 (на сэмпл):
30 мкл воды
50 мкл метил Seq ПЦР Мастер микс
1 мкл метил Seq PCR1 праймера F
1 мкл метил Seq PCR1 грунтовка R
Тепловая велосипедист программа:
Этап 1, 1 цикл: 95 ° C 2 мин.
Этап 2, 8 циклов: 95 ° C 30 s, s 60 ° C 30, 72 ° C 30 s
Этап 3, 1 цикл: 72 ° C 7 мин.
Этап 4, 1 цикл: 4 ° C Hold - Очищайте образцы с использованием 180 мкл ДНК связывающих магнитные бусы и 400 мкл свежеприготовленные 70% этанола на сэмпл. 180 мкл бусы для каждой выборки и инкубировать в течение 5 мин при комнатной температуре. Пелле бусы, удалить супернатант и Ресуспензируйте Пелле в 200 мкл на 70% спирте. Удаление этанола и повторите мыть раз.
- Используйте магнитные пластины Пелле бусины и удалить столько этанол как можно скорее. Сухой в 37 ° C теплоблока для 3-5 мин до тех пор, пока шарик Пелле совершенно сухой. Ресуспензируйте в 21 мкл нуклеиназы свободной воды и собирать около 19,5 мкл супернатант.
- Добавьте 130 мкл подготовленных бисульфита преобразования реагента в надосадке из предыдущего шага. Разделите каждый из 150 мкл реакций одинаково в две скважины. Инкубируйте в тепловой циклователь (64 ° C для 2,5 ч, удерживайте 4 ° C).
- Индексирование
- Подготовьте реакции PCR Master Mix 2 на льду. 25.5 мкл Master Mix 2 для каждого образца. 5 мкл. коммерческие индексации Праймеры для отдельных образцов и инкубировать в тепловая велосипедист.
ПЦР реакции Master Mix 2 (на сэмпл):
25 мкл метил Seq ПЦР Мастер микс
0.5 мкл метил Seq общих индексации грунт
Тепловая велосипедист программа:
Этап 1, 1 цикл: 95 ° C 2 мин.
Этап 2 — 6 циклов: 95 ° C 30 s, s 60 ° C 30, 72 ° C 30 s
Этап 3, 1 цикл: 72 ° C 7 мин.
Этап 4, 1 цикл: 4 ° C Hold
Примечание: Дополнительные циклы (2-3) может быть необходимым, если Начальная концентрация ДНК ниже рекомендованных значений. - Очищайте образцы с использованием 90 мкл ДНК связывающих магнитные бусы и 400 мкл свежеприготовленные 70% этанола на сэмпл. Мкл 90 бисера для каждой выборки и инкубировать в течение 5 мин при комнатной температуре. Пелле бусы, удалить супернатант и Ресуспензируйте Пелле в 200 мкл на 70% спирте. Удаление этанола и повторите мыть раз.
- Используйте магнитные пластины Пелле бусины и удалить столько этанол как можно скорее. Сухой в 37 ° C теплоблока для 3-5 мин до тех пор, пока шарик Пелле совершенно сухой. Ресуспензируйте в 24 мкл нуклеиназы свободной воды и собирать примерно 24 мкл супернатант.
- Оценка концентрации и bp размер с помощью реагентов обнаружения ДНК высок чувствительности на bioanalyzer.
Примечание: Если bioanalyzer не удается обнаружить присутствие библиотеки ДНК, повторите шаги подготовки с дополнительные ДНК.
Остановочный пункт: после очистки, индексированные образцы могут быть опечатаны и хранятся при температуре-20 ° C. - Объединение образцов для используемой платформы соответствующей последовательности следующего поколения.
- Концентрации с использованием данных bioanalyzer, которая определяет Молярность ДНК, основанный на библиотеке размер и количество в данном объеме, развести с низкой TE буфера (6.1.1.1) и объединить всех образцов до конечной концентрации 15 pM.
Примечание: Более чувствительным методом количественного определения библиотеки является Количественная ПЦР в реальном времени, используя праймеры которые нацелены перевязаны адаптеры. - Запуск пула образцы на число полос движения, которые являются достаточно для 4 выборок на полосе на секвенсор следующего поколения.
Примечание: например, если 16 библиотеки образцов были однозначно индексируются и комбинированные, запустите библиотеки свыше 4 полосы, эквивалентна 4 выборок на пер.
- Концентрации с использованием данных bioanalyzer, которая определяет Молярность ДНК, основанный на библиотеке размер и количество в данном объеме, развести с низкой TE буфера (6.1.1.1) и объединить всех образцов до конечной концентрации 15 pM.
- Подготовьте реакции PCR Master Mix 2 на льду. 25.5 мкл Master Mix 2 для каждого образца. 5 мкл. коммерческие индексации Праймеры для отдельных образцов и инкубировать в тепловая велосипедист.
7. виртуализации программа Sequencer следующего поколения
- Отправьте образцы в основной институциональной последовательности для кластеризации метил-Seq библиотеки, следуют виртуализации на компьютере виртуализации нового поколения.
8. анализ для выявления ЗВД
- Реализуйте Bismark15, который вызывает Bowtie 2.0 как внутренней последовательности выравниватель16,17, для выравнивания сырой ввода чтение преобразование бисульфита, плюс стренги генома. После выравнивания используйте Bismark_methylation_extractor для контроля качества и присвоить значение оценкам метилирования CpG каждый.
- Создайте список ЗВД с BS-Seq пакет18 в Bioconductor. Фильтр ЗВД основан на наличие более чем 3 последовательных стандартов клинической практики и P-значение < 0,05.
Примечание: Генерировать DMR список, который включает в себя геномной координаты, расстояние до ближайшего гена RefSeq, количество СКП в пределах каждой DMR, средний % значение метилирования CpG через DMR для сравнения двух групп (например, подчеркнул против безударных), P-значение, и Рузвельт (ложные обнаружения ставка) значение. Используйте список DMR, т.е., геномных координаты, для разработки грунты пиросеквенирования для проверки.
9. Проверка бисульфита пиросеквенирования
-
Конструкция праймера
- Дизайн праймеров PCR бисульфита и пиросеквенирования. Таким образом, чтобы вложенные ПЦР усилится 150 – 400 bps DMR дизайн два набора праймеры PCR (снаружи и вложенных).
Примечание: В общем, разработанный грунтовки являются по меньшей мере 24 баз долго с по крайней мере 4-5 подряд G (C для обратного грунтовки) счет сокращены отжига температура от потери последовательность сложности. Один из вложенных праймеры будет биотин меченых и ВЭЖХ очищенная. Однако стандартный грунтовки должны быть заказаны первым, чтобы оптимизировать ПЦР шаг путем разрешения реакции на геле агарозы.- Дизайн пиросеквенирования пробирного грунт, так, что он нацелен на взаимодополняющие биотинилированным нити основы всего 1 – 2 вверх по течению СКП для быть assayed. Дизайн несколько пиросеквенирования грунтовки в случае необходимости, как каждый пиросеквенирования грунт можно надежно проба 30 bps вниз по течению.
- Для Rt1-m4 используйте следующую команду:
rRT1M4 вне-F TGTAYGATTTTGGTTATYGTAAAT
rRT1M4 вне-R AACTTACAAATTTCACCAACTCA
rRT1M4 Nested – F GTGGGTTAYGTGGATAATATATAG
Вложенная rRT1M4 – R AATCACTTACCATTCTCTCTCTAACTA
rRT1M4 Pyro1 TAYGTGGATAATATATAGAT
rRT1M4 Pyro2 GATAGTTATTTGGYGAGTTAG
rRT1M4 Pyro3 GAGTATTTGGAGGAGTTGAT
rRT1M4 Pyro4 GGATTTTAATATTTGGT
- Дизайн праймеров PCR бисульфита и пиросеквенирования. Таким образом, чтобы вложенные ПЦР усилится 150 – 400 bps DMR дизайн два набора праймеры PCR (снаружи и вложенных).
-
Используйте имеющиеся комплект для преобразование бисульфита геномная ДНК крови крыс.
Примечание: Преобразование бисульфита шаги были адаптированы из коммерчески доступных комплект со следующими изменениями: В шаге 1, добавьте 50 – 100 нг крови геномная ДНК и разбавляют водой до 20 мкл. В шаге 9 элюировать 20 мкл на сэмпл.- Подготовка реагента преобразование бисульфита согласно протоколу производителя и объединить с разбавленным геномная ДНК. Инкубируйте в тепловой циклователь (64 ° C для 2,5 ч, удерживайте 4 ° C).
- Добавьте привязки буфера преобразованы геномная ДНК в столбцах спин и центрифуги (15000 x g, 1 мин). Мыть столбцы один раз, затем добавить Desulphonation буфера столбцы и Инкубируйте 15 мин при комнатной температуре. Центрифуга (15000 x g, 1 мин).
- Промойте колонку с мыть буфер и центрифуги (15000 x g, 1 мин). Повторите шаг мыть с центрифугированием (15000 x g, 2 мин). 20 мкл Элюирующий буфер и центрифуги (15000 x g, 1 мин) элюировать.
-
Амплификации PCR
- Подготовка внешних ПЦР Мастер микс. 21.5 мкл Мастер микс для 3,5 мкл бисульфит преобразованы геномная ДНК и запустите программу тепловая велосипедист.
За пределами ПЦР Мастер микс:
16.25 мкл воды
2.5 мкл буфера полимеразы [10 x]
0.5 мкл dNTP [10 мм]
1 мкл вперед грунт [0.1 мкм]
1 мкл обратного грунт [0.1 мкм]
0,25 мкл полимеразы дна Taq [5000 ед/мл].
Тепловая велосипедист программа:
Этап 1, 1 цикл: 94 ° C 4 мин
Этап 2, 47 циклов: 94 ° C 1 мин, 53 ° C 30 сек, 72 ° C 1 мин
Этап 3, 1 цикл: 72 ° C 8 мин., 4 ° C Hold - Подготовка вложенных ПЦР Мастер микс. 23 мкл Мастер микс по 2 мкл пример за пределами ПЦР и повторите вне программы тепловая велосипедист ПЦР. Оцените качество продукта ПЦР через гель-электрофорез (1 x TAE буфера, гель агарозы 1%).
Вложенные ПЦР Master Mix:
17.75 мкл воды
2.5 мкл буфера полимеразы [10 x]
0.5 мкл dNTP [10 мм]
1 мкл вперед грунт [0.1 мкм]
1 мкл обратного грунт [0.1 мкм]
0,25 мкл полимеразы дна Taq [5000 ед/мл]
Примечание: Для вложенных PCR, либо вперед или обратный грунтовки должны быть биотинилированным.
- Подготовка внешних ПЦР Мастер микс. 21.5 мкл Мастер микс для 3,5 мкл бисульфит преобразованы геномная ДНК и запустите программу тепловая велосипедист.
-
Пиросеквенирование
- Сделайте мастер смеси, содержащие 38 мкл буфера привязки, 35 мкл воды и 2 мкл стрептавидина покрытием sepharose бусы на сэмпл. В 96-луночных тарелку добавьте 75 мкл мастер смеси и 5 мкл вложенных продукта PCR. Встряхните в шейкере пластины для 15-60 мин.
- При встряхивании, мкл 12 праймера (0,5 мкм, разбавленных в отжига буфер) в скважины пиросеквенирования пробирного плиты.
- После встряхивания, выполните мыть с помощью привязки реакции мыть буферов. Поместите вакуумный инструмент в корыто с водой, а затем собирать образцы от пластины. Опускайте вакуумные инструмент в наполовину заполненный желоба, содержащие 70% этанол, NaOH (0,2 М) и буфер Tris ацетат (10 мм, рН 7,4). Отсоедините от вакуума и место вакуумные инструмент в HS пробирного пластины для передачи бусины.
- Установите пластину на теплового блока и Инкубируйте на 80 ° C на 2 мин разрешить пластины остыть в течение 5 минут, а затем начать программу Пиро.
Результаты
Успешное осуществление платформы метил-Seq крыс зависит от нескольких критериев. Рисунок 1 показывает общий рабочий процесс исследования и освещаются конкретные контроля качества (КК) шаги, которые необходимы прежде чем двигаться вперед. Одним из первых факторов, чтобы рассмотреть является надежность модели на животных и режим стресс, которые определяют масштабы эпигеномные изменения, которые происходят через methylome. Поскольку наша животных работа основывается на наших предыдущих наблюдение, что воздействие кортикостерона (CORT) может привести к изменениям в19,метилирование ДНК20, нашего режима хронический стресс переменной (CVS) должна быть достаточной строгости производить подчеркнул крыс с повышенными плазмы CORT уровнями. Типичный загрузок CVS режим показано в таблице 1 и состояла из ежедневного стресса в утром, днем, и на ночь, постоянно изменяются, чтобы не допустить привыкания и уменьшилась реакции на стресс. На протяжении 3-х недельный режим, подчеркнул животных выставлены значительно повышенные уровни среднего плазмы CORT [дней 4 – 21, контроль: 32,7 3.7 нг/мл, стресс: 103.0 11,9 нг/мл (средний SEM), P = 2,2 x 10-4, рисунок 2A] над теми из безударных, Управление животных. Последовательно эти животные также показал больше тревоги подобное поведение на повышенные плюс лабиринт (EPM), как указано значительно больше времени в закрытом оружия EPM и меньше времени в объятия (Рисунок 2Б). Эти результаты показывают, что CVS воздействия привели к значительным эндокринной и поведенческие изменения, ведущие нас расследовать ли эти изменения были связаны с определенными сигнатурами метилирование ДНК.
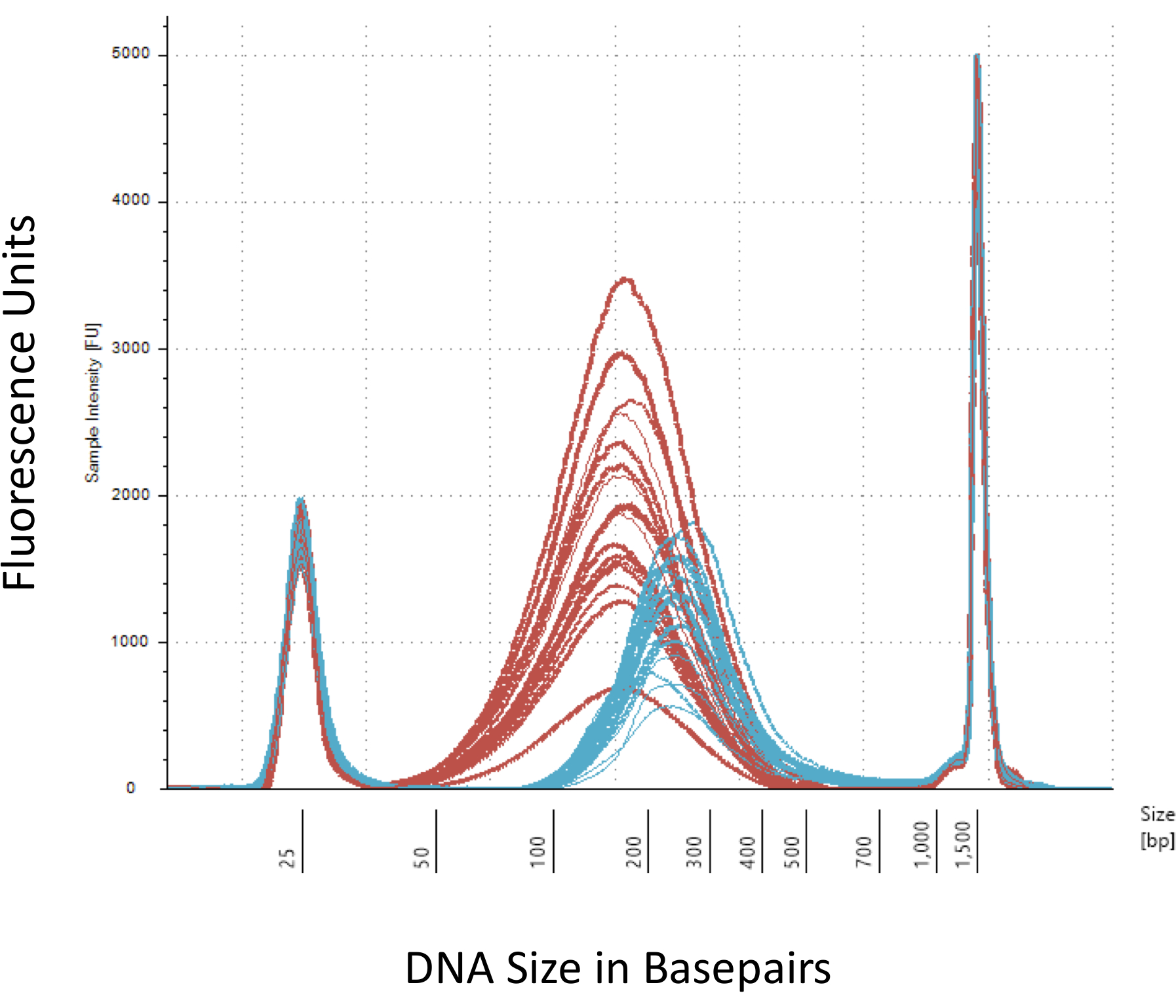
Мы подчеркиваем несколько контрольно-пропускных пунктов, которые имеют решающее значение для успешного строительства метил-Seq библиотеки. Начиная с достаточное количество ДНК является необходимой, как sonication, умывальник/очистки, цели обогащения, и преобразование бисульфита шаги последовательно сокращать количество ДНК в готовой библиотеке. Хотя некоторые шаги амплификации PCR смягчить потери шаблона дна, чрезмерное число цикла PCR может ввести выше повторяющиеся читает. Для текущего исследования крыс метил-Seq был использован 2 g геномная ДНК крови на крысу. Мы отмечаем, что метил-Seq библиотеки могут быть сделаны с начиная сумма ДНК как низко как 500 нг. Меньше исходного материала позволяет пользователям создания библиотек из ДНК, изолированные СУИМ (сортировки активирован флуоресценции клеток) или иглы штампов, хотя есть повышенный риск производит недостаточное количество библиотек для последующего последовательности. QC производится электрофорез 1 Л образца на bioanalyzer, который обеспечивает молекулярный вес, количество и Молярность ДНК. Три важные шаги, которые требуют использования bioanalyzer являются: 1) после sonication шаг для обеспечения достаточной стрижка ДНК (~ 170 bp, красный, рис. 3); 2) после адаптер лигирование шага свидетельствует сдвиг в средний размер стриженый ДНК (~ 200 bp, синий, рис. 3) для обеспечения их последующего усиления методом ПЦР; и 3) после шага очистки окончательный библиотека для обеспечения количество и размер библиотеки для виртуализации.
R-пакеты для анализа бисульфита, Виртуализация данных18использовались BSSeq и BSmooth в Bioconductor. Они включают в себя инструменты и методы для выравнивания считывает последовательность, осуществляющие контроль качества, и выявления дифференциально метилированию регионов (ЗВД). Программное обеспечение BSmooth вызывает Боути 2.016,17 как внутренней последовательности выравниватель для получения резюме CpG уровня измерения, путем выравнивания сырой ввода чтение бисульфит преобразованы genomic последовательностей. Соответствие читает затем фильтруют через строгий контроль качества процедур, которые стремятся для выявления систематической последовательности и базы вызова ошибок, которые может привести к искажению течению анализы. Ряд участков создаются визуально помощи в этом процессе фильтрации. Секвенирование метрики также создаются для документа соответствующей информации, например число унифицированных гласит, % целевой и за освещение CpG, среди прочих (Таблица 2). После фильтрации данных, алгоритм сглаживания/нормализация выполняется, где каждый CpG назначается значение оценкам метилирования, основанный на всех КК считывает из каждой выборки и оценки из соседних СКП для обеспечения более точного вызова метилирования статус даже в тех случаях, когда низкий охват последовательности. Это значение обеспечивает сглаженного оценку вероятности метилирование на каждом сайте ВПУ. Сравнивая среднее сглаженного метилирования оценки каждого образца между двумя группами лечения и ранжирования геномной регионов от наиболее значительно отличается от наименее, создается список ЗВД (Таблица 3).
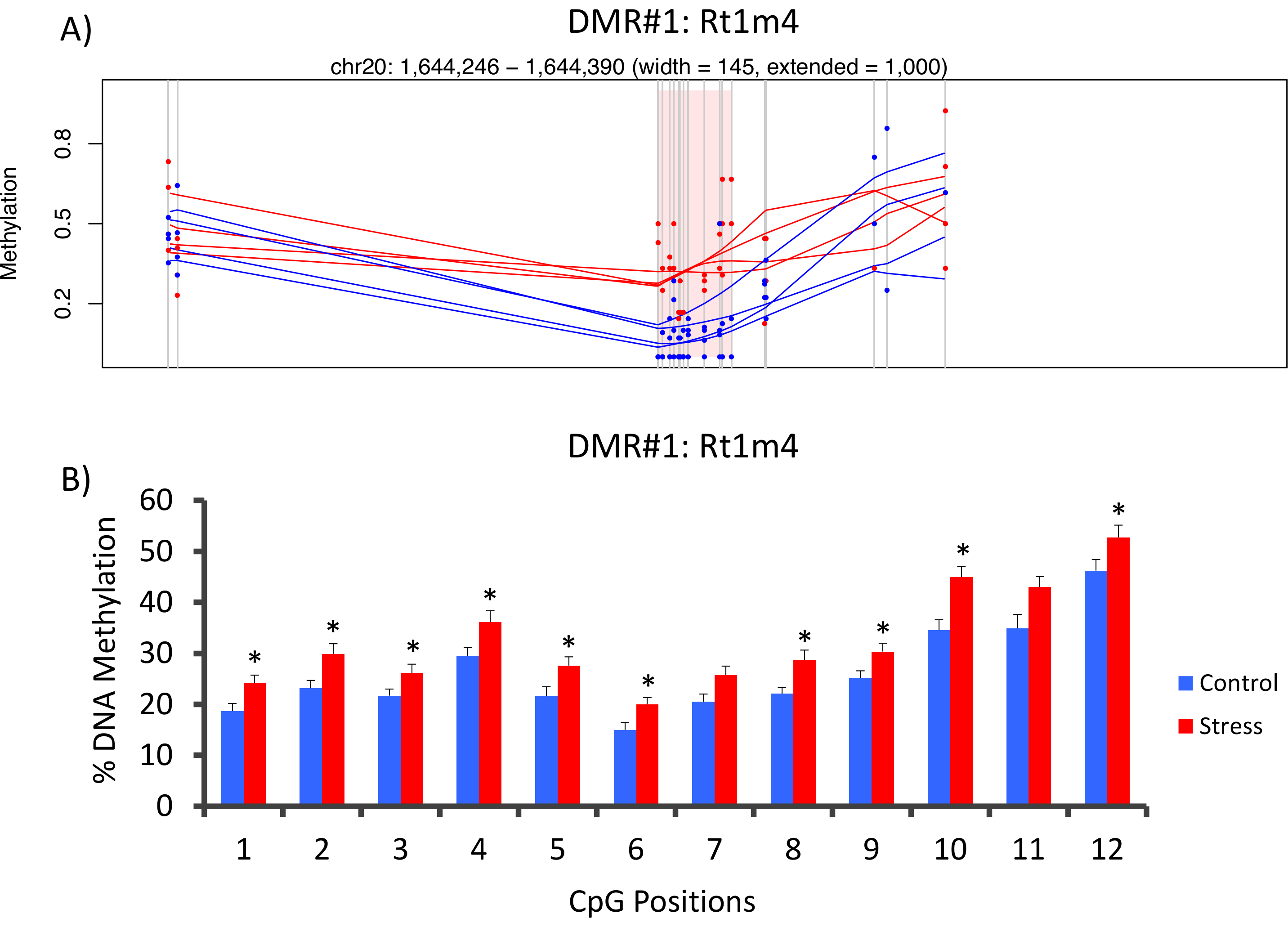
DMR между безударных и подчеркнул группами был расположен в промоутер крыса гистосовместимости гена Rt1-m4, с верхней подчеркнул животных экспонируется более высокий уровень метилирования во всех СКП чем безударные животных (рис. 4A). Чтобы подтвердить успешное осуществление платформы метил-Seq и анализа данных, грунтовки были разработаны против DMR и уровень метилирования ДНК крови в всей когорте безударных и подчеркнул животных (8 Виртуализирован с метил-Seq и 8 не виртуализированных) были оценены бисульфита пиросеквенирования. Результаты демонстрируют значительное увеличение метилирование ДНК через 10 из 12 СКП assayed (изменение 5.1 – 10,4% метилирования, P < 0,037, рис. 4B). KEGG путь анализ проводился на все номинально значительные ЗВД для выявления путей, связанные со стрессом. Последовательно DMR-связанные пути замешан заболеваний, связанных с воздействием хронического стресса, таких как диабет, сердечно-сосудистых заболеваний и рака (Таблица 4). 21 , 22 , 23 продемонстрировать связь между эпигеномные данных и степень воздействия подчеркнуть, уровень метилирования CpG-10 были по сравнению с средние уровни CORT 3 недели для каждого животного. Результаты показали скромный корреляции между эндокринной и метилирования данных (R2= 0,54, P = 0,001, рис. 5).

Рисунок 1: Общая схема рабочего процесса для платформы крыса метил-Seq. Один g геномной ДНК, извлеченные из крови подчеркнул и крысы управления сначала обрабатывается для построения библиотек метил-Seq последовательность, анализа и идентификации цели. Еще 100 нг ДНК используется для независимой проверки выявленных эпигеномные целей бисульфита пиросеквенирования. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 2: воздействие переменной хронический стресс (CVS) приводит к эндокринной и поведенческих изменений в крысах. (A) несколько выборок кортикостерона (CORT) продемонстрировать надежность 3 недели CVS режима. Утром до ежедневного стресса режима были собраны пробы крови. (B) подчеркнул животных больше времени в закрытом оружия и меньше времени в объятия повышенный плюс лабиринт (EPM). Показываются Boxplots с точки данных для каждого животного. T-критерия Стьюдента была выполнена на статистическую значимость. * P < 0,05, ** P < 0.01, и *** P < 0,001. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 3: количественный стриженый и адаптера лигируют крыса ДНК на bioanalyzer. Красные и синие кривые показывают количество и размер геномной ДНК (красный) после стрижки в изотермических sonicator и адаптер перевязки, соответственно. Каждая строка представляет один образец и красный и синий кривых отражать оба потери ДНК в ходе нескольких шагов (конец ремонт, 3'-adenylation и очистка образца) и увеличение размера bp вследствие перешнуровка адаптеры. Острые пики на 25 bp и 1500 bp являются стандартных маркеров, которые были добавлены в буфер загрузки. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 4: CVS-индуцированной эпигеномные изменения обнаруживаются крыса метил-след (A) анализ крысы метил-Seq данных причастны промоутер гена Rt1m4 как дифференциально метилированной региона (DMR) между подчеркнул (красный) и управления (синий) крыс. Графический вывод для Rt1m4 DMR (розовые тени район) отображает каждый CpG (Вертикальная серая линия), четырех образцов в каждой группе (красные или синие линии) и уровень метилирования % для каждого животного (красная или синяя точка). (B) двенадцать СКП в пределах РРЛ были подтверждены бисульфита пиросеквенирования. Столбчатые диаграммы представлены как означает SEM, и студент T-тест был проведен для статистической значимости. * P < 0,05. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 5: Линейный регрессионный анализ показал скромный корреляции между % ДНК метилирования CpG-10 Rt1m4 и 3 недели означает плазмы CORT уровни обоих подчеркнул и контроля животных (N = 16). Данные из подчеркнул животных представлены красными кругами. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}
| Неделя | 1 день | День 2 | День 3 | День 4 | День 5 | День 6 | День 7 |
| AM | Сдержанность | Плавать | Холодный номер | Плавать | Сдержанность | Шейкер | Плавать |
| ВЕЧЕРА | Шейкер | Кейдж наклона | Сдержанность | Шейкер | Холодный номер | Сдержанность | Холодный номер |
| Ночлег | Ограничить питание | Мокрый постельных принадлежностей | Изоляция | Свет на | Вытеснения | Свет на | Мокрый постельных принадлежностей |
Таблица 1: Типичный еженедельный график режима хронический стресс переменной (CVS).
| Секвенирование метрики | Стресс-1 | Элемент управления1 |
| (n = 4) | (n = 4) | |
| Паре конец читает (за год) | 89,290,397 | 80,165,674 |
| Однозначно сопоставленных парных конец читает (UMPER) | 39,200,255 | 35,013,406 |
| Выравнивание ставка/сопоставления эффективности (UMPER / PER) | 44% | 44% |
| Считывает повторяющиеся (% UMPER) | 73% | 65% |
| Подвергнутые дедупликации UMPER | 10,481,031 | 12,306,018 |
| Средняя глубина охвата чтения (x) (ARDC) | 6 x | 6 x |
| СКП (N) | 12,056,878 | 12,056,878 |
| ARDC (x) стандартов клинической практики | 2 x | 2 x |
| СКП с по крайней мере 10 гласит (N) | 481,383 | 595,850 |
| ARDC (X) СКП с по крайней мере 10 чтений | 19 | 19 |
| На целевых СКП (полного перекрытия с зонд целевых регионах) | 1,923,872 | 2,007,638 |
| Створ ARDC (x) стандартов клинической практики | 7 x | 8 x |
| На целевых СКП с по крайней мере 10 гласит (N) | 428,249 | 531,419 |
| На целевых ARDC (x) из СКП с по крайней мере 10 считывает | 18 x | 18 x |
| На цели (% с 1 или более пары совпадения с зонд целевых регионов) (UMPER) | 8,277,715 | 9,369,523 |
| % На цель (всего дедуплицированного UMPER) | 78% | 77% |
| На цели (всего баз сопоставления) Mb | 125 МБ | 128 МБ |
| На целевых средний чтения глубина охвата (x) (ARDC) | 9 x | 10 x |
| 1 Секвенирование метрики, основанные на средние показатели по предметам в каждой группе |
Таблица 2: Последовательность показателей, полученных из крыса метил-Seq платформы.
| Chr | Начало | конец | Джин | Расстояние | areaStat | meanDiff | стресс | управления | направление |
| chr20 | 1,644,246 | 1,644,390 | RT1-M4 | in_gene | 93.03 | 0,22 | 0,33 | 0,11 | получить |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | -0.19 | 0,72 | 0.91 | потеря |
| chr3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0,21 | 0,94 | 0,72 | получить |
| chr2 | 143,064,811 | 143,065,010 | Ufm1 | 8569 | -59.48 | -0.11 | 0,13 | 0,24 | потеря |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57.04 | 0,21 | 0,94 | 0,73 | получить |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0.13 | 0.74 | 0.88 | потеря |
| chr7 | 47,101,725 | 47,101,930 | Pawr | in_gene | -50.54 | -0,12 | 0,64 | 0,76 | потеря |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0.11 | 0,85 | 0,96 | потеря |
| chr11 | 80,640,132 | 80,640,356 | Dgkg | in_gene | -50.07 | -0.16 | 0,73 | 0,89 | потеря |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0.17 | 0,58 | 0.75 | потеря |
Таблица 3: Топ 10 дифференциально метилированию регионов. Для каждого DMR, выходная таблица показывает от левой к правой колонке: хромосомных местоположение (КПЧ), координирует (начало/конец), расстояние от транскрипции гена имя запуска сайта, разностной области статистики между подчеркнул и контрольных групп (areaStat), означает, Дифференциальный метилирования (meanDiff), среднее метилирования уровней через каждый DMR для подчеркнул и контрольных групп (стресс/управления) и направление метилирования изменения от элементов управления.
| KEGG путь термины | Ген игр | % | P-значение | Benjamini |
| Диабет | ||||
| Сахарный диабет II типа | 12 | 0.1 | 3.6 x 10-4 | 9,8 x 10-3 |
| Сердечно-сосудистые заболевания | ||||
| Сосудистые гладких мышц | 18 | 0.1 | 1.6 x 10-3 | 3.6 x 10-2 |
| Аритмогенная кардиомиопатия и правого желудочка (ARVC) | 13 | 0.1 | 4.0 x 10-3 | 7.1 x 10-2 |
| Расширенная кардиомиопатия | 14 | 0.1 | 7.6 x 10-3 | 1,2 х 10-1 |
| Нейрон функция | ||||
| Долгосрочный потенцирование | 11 | 0.1 | 1,5 x 10-2 | 1.4 x 10-1 |
| Сигнализации | ||||
| Сигнальный путь MAPK | 35 | 0.2 | 2.4 x 10-4 | 9,9 x 10-3 |
| Сигнальный путь кальция | 22 | 0.1 | 1.2 x 10-2 | 1.4 x 10-1 |
| Сигнальный путь хемокиновых | 21 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
| Рак | ||||
| Пути в раке | 42 | 0,3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Глиомы | 15 | 0.1 | 4.4 x 10-5 | 2.4 x 10-3 |
| Non-немелкоклеточного рака легкого | 10 | 0.1 | 7,9 x 10-3 | 1.1 x 10-1 |
| Колоректальный рак | 13 | 0.1 | 8.4 x 10-3 | 1.1 x 10-1 |
| Хронический миелолейкоз | 12 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
Таблица 4: KEGG путь анализ ЗВД определены от крысы метил-Seq.
Обсуждение
В этом исследовании мы разработаны и осуществлены метил-Seq платформы геном крысы. Демонстрируя его полезность с мышиной модели стресса, мы продемонстрировали, что экспериментальные и аналитических трубопровода может обеспечить дифференциально метилированной регионов между двумя группами сравнения.
Для обеспечения успешного осуществления платформы, несколько критических шагов необходимо соблюдать. Во-первых, первоначальный ДНК качество и количество имеет значительное влияние на качество и количество окончательного метил-Seq библиотеки. Мы использовали флуориметр, вместо того чтобы спектрофотометр, для обеспечения отражения количество двуцепочечной ДНК настоящее нашей ДНК измерения. Bioanalyzer был использован для измерения молекулярного размера и количества ДНК после стрижки и после перевязки адаптер. Проверка молекулярного размера «сдвиг» между этими шагами важно подтвердить наличие адаптеров на концах каждого фрагмента ДНК, которая будет проходить адаптер опосредованной ПЦР в последующих шагах. Количество ДНК, оставшиеся в конце шага лигирование адаптер также имеет важное значение, поскольку по крайней мере 100 нг библиотека продукта необходима на этом этапе для обеспечения достаточного количества доступен после целевой обогащения и бисульфит Преобразование шаги. Окончательный расчет высок чувствительности была проведена сконструированный метил-Seq библиотеки так, что библиотека может быть разбавлен должным образом для последующего кластеризации по sequencer следующего поколения. Наконец бисульфит пиросеквенирования работал как высоко количественных, независимый метод для оценки точности аналитических трубопровода. Окончательной проверки, используя оригинальные образцы и репликации с использованием дополнительных животных являются важнейшие шаги, чтобы обеспечить, что эксперимент может обнаруживать биологически значительные изменения в метилирование ДНК.
Мы также включать ряд руководящих принципов в случае отклонения от протокола или если возникают проблемы. Во-первых это можно потерять слишком много ДНК при конце ремонт, адаптер перевязки или магнитный шарик очистки шагов. Кроме того, начиная количество ДНК может быть небольшим (< 200 нг) из-за наличия ограниченных ткани/ДНК или осуществление различных методов обогащения, например сортировки клеток активированных флуоресцированием. Увеличение числа цикла в течение двух библиотека амплификации шаги могут быть способны компенсировать чрезмерное потерю ДНК или низкий начиная сумма ДНК во всем протокол строительство библиотеки. Однако не более чем еще 2-3 циклов рекомендуется, как чрезмерное шаблон усиления может привести к увеличению числа дубликатов читает время виртуализации. Эти дубликаты исключаются во время шага выравнивания для предотвращения смещения в расчетах процент метилирования. Во-вторых если средний размер ДНК не увеличится более чем 30 bps, убедитесь, что реагенты являются новыми, как T4 ДНК-полимеразы, фрагменты, и/или T4 лигаза могут быть старые. Может быть использован коммерчески доступных замена реагентов.
Кроме того вполне возможно, что предсказал ЗВД может не пройти проверку на пиросеквенирования, где различия метилирование ДНК не существуют или значительно меньше, чем те, которые предсказывали анализа. Плохой проверки кандидата регионов проблема слишком общие для многих генома общесистемного анализа, например когда пиросеквенирования результаты не подтверждают дифференциального метилирования или размер эффекта намного меньше, чем предсказано анализа. BSmooth это один аналитический пакет «сглаживает» уровень метилирования через окно несколько стандартов клинической практики. Для текущего эксперимента BSmooth замешан DMR, чей уровень метилирования были подтверждены бисульфита пиросеквенирования. Однако скорее всего будет расхождения между уровень метилирования предсказано BSmooth и те подтверждены пиросеквенирования. Эти расхождения обусловлены функцию сглаживания, которая оценкам средняя метилирования значения во всех СКП в пределах РРЛ, в том числе последовательных стандартов клинической практики, которая в метилирование ДНК могут отличаться более чем на 50% или СКП, значения которых метилирования были исключены из-за подпороговые читать глубины. R-пакеты, такие как MethylKit24 может использоваться для выявления небольших окон СКП или даже одной СКП, чей уровень метилирования сильно коррелируют с те подтверждены пиросеквенирования. Реализация различных пакетов и тестирования их предсказал регионов или СКП дифференциальной метилирования пиросеквенирования обеспечит надежность данных. Кроме того оригинальные метил-Seq библиотеки можно resequenced и чтения файлов для увеличения глубины чтения. Поскольку определение уровней метилирования полуколичественного и продиктовано на количество прочтений [(# of CpGs) / (# ТПГС + СКП)], увеличивая чтения глубины для данного CpG будет увеличить точность его значения процентов метилирования. В этом исследовании мы рассматривали только СКП метилирования, значения которых были определены по крайней мере десять читает и достичь общего чтения освещение 19 x для каждого CpG.
Метил-Seq платформа крыса — не без его ограничения. Хотя это более экономически эффективным, чем последовательность всего генома бисульфита, это значительно дороже, чем другие методы. Тем не менее большая часть стоимости был для покупки полос на программы sequencer, а не для захвата системы. В зависимости от чтения глубины, необходимых, с кросс ткани сравнения, требующие меньше из-за большой (25-70%) различия12 в метилирование ДНК стоимость может быть уменьшена мультиплексирования более выборок на полосу и с использованием платформы большей емкости. Кроме того Подготовка образца является более длительным, чем другие методы. Похож на другие пулдаун подходы, которые включают секвенирование нового поколения, добавлен бисульфита преобразования и очистки шагов добавить рабочую нагрузку. В целом метил-Seq платформа является экономически эффективные альтернативы всего генома и обеспечивает разрешение базы пара на более 2,3 миллионов СКП, который значительно больше, чем те Анализ microarray дна-платформ. На сегодняшний день, коммерчески доступных человека и мыши метил-Seq платформ были использованы для документирования алкогольной зависимостью изменений в макаки мозга25,26, психомоторного развития генов в мыши мозга9и кровоснабжение мозга цели глюкокортикоидов10. Кроме того способность целевым конкретных регионов, независимо от последовательности признание энзимов ограничения делает его идеальной платформой для кросс видов сравнений. Для этого исследования мы разработали платформу метил-Seq для крыс, для которого многих фармакологических, метаболизм и поведенческих эксперименты выполняются без использования инструмента генома всей methylomic. Наши данные показывают, что он может использоваться для обнаружения ЗВД в мышиной модели стресса и коррелируют других физиологических параметров, таких как общий плазменный CORT уровнях.
Метил-Seq платформа идеально подходит для эпигенетических экспериментов в животных с последовательности геномов, которые не могут иметь достаточно экспериментальных доказательств, документирование регулирования регионов. Когда такие регионы доступны, дополнительные регионы могут специально и прилагается к текущей версии. Кроме того платформа идеально подходит для сравнительной геномики, поскольку цели обогащения не ограничивается энзима ограничения распознавания. Например промоутер региона любой ген может быть захвачен независимо от ли он укрывает сайта конкретного ограничения. Аналогично любые нормативные регионах, таких как те, которые определены в мыши или людей, которые сохраняются в геноме интерес может быть захвачен.
Раскрытие информации
Рукопись является частью конкурса приз от Agilent Technologies.
Благодарности
Это исследование финансировалось NIH Грант MH101392 (RSL) и поддержка со стороны следующих награды и фонды: NARSAD молодой следователь премии, Маргарет Энн Цена следователь фонд, Фонд Джеймса Wah настроение расстройств ученый через Фонд Чарльз т. Бауэр, Бейкер Фонд и фонд проекта матч (RSL).
Материалы
| Name | Company | Catalog Number | Comments |
| Radioimmuno assay (RIA) | MP Biomedicals | 7120126 | Corticosterone, 125I labeled |
| Master Pure DNA Purification Kit | Epicentre/Illumina | MC85200 | |
| Thermal-LOK 2-Position Dry Heat Bath | USA Scientific | 2510-1102 | Used with 1.5 mL tubes |
| Vortex Genie 2 | Fisher | 12-812 | Vortex Mixer |
| Ethyl alcohol, Pure | Sigma-Aldrich | E7023 | 100% Ethanol, molecular grade |
| Centrifuge 5424 R | Eppendorf | - | Must be capable of 20,000 x g |
| Qubit 2.0 | ThermoFisher Scientific | Q32866 | Fluorometer |
| Qubit dsDNA BR Assay Kit | ThermoFisher Scientific | Q32850 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Q32851 | High sensitivity DNA detection reagents |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| SureSelectXT Rat Methyl-Seq Reagent Kit | Agilent Technologies | G9651A | Reagents for preparing the Methyl-Seq library |
| SureSelect Rat Methyl-Seq Capture Library | Agilent Technologies | 931143 | RNA baits for enrichment of rat targets |
| IDTE, pH 8.0 | IDT DNA | 11-05-01-09 | 10 mM TE, 0.1 mM EDTA |
| DNA LoBind Tube 1.5 mL | Eppendorf | 22431021 | |
| Covaris E-series or S-series | Covaris | - | Isothermal sonicator |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm (25) | Covaris | 520045 | |
| Water, Ultra Pure (Molecular Biology Grade) | Quality Biological | 351-029-721 | |
| Veriti 96 Well-Thermal Cycler | Applied Biosystems | 4375786 | |
| AMPure XP Beads | Beckman Coulter | A63880 | DNA-Binding magnetic beads |
| 96S Super Magnet | ALPAQUA | A001322 | Magnetic plate for purification steps |
| 2200 TapeStation | Agilent Technologies | G2965AA | Electrophresis-based bioanalyzer |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 | |
| D1000 ScreenTape High Sensitivity | Agilent Technologies | 5067-5584 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 Reagents High Sensitivity | Agilent Technologies | 5067-5585 | |
| DNA110 SpeedVac | ThermoFisher Scientific | - | Vacuum Concentrator |
| Dynabeads MyOne Streptavidin T1 magnetic beads | Invitrogen | 65601 | Streptavidin magnetic beads |
| Labquake Tube Rotator | ThermoFisher Scientific | 415110Q | Nutator Mixer is also acceptable |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5006 | Bisulfite conversion kit. Contains Binding, Wash, Desulphonation, and Elution buffers |
| Illumina Hi-Seq 2500 | Illumina | - | Next-generation sequencing machine |
| PCR and Pyrosequencing Primers | IDT DNA | Variable | |
| Taq DNA Polymerase with ThermoPol Buffer - 2,000 units | New England BioLabs | M0267L | |
| Deoxynucleotide (dNTP) Solution Set | New England BioLabs | N0446S | |
| Pyromark MD96 | QIAGEN | - | Pyrosequencing machine |
| Ethyl Alcohol 200 Proof | Pharmco-Aaper | 111000200 | 70% Ethanol solution |
| Sodium Hydroxide Pellets | Sigma-Aldrich | 221465 | 0.2 M NaOH denature buffer solution |
| Tris (Base) from J.T. Baker | Fisher Scientific | 02-004-508 | 10 mM Tris Acetate Buffer wash buffer solution |
| PyroMark Gold Q96 Reagents (50x96) | QIAGEN | 972807 | Reagents required for pyrosequencing |
| PyroMark Annealing Buffer | QIAGEN | 979009 | |
| PyroMark Binding Buffer (200 mL) | QIAGEN | 979006 | |
| Streptavidin Sepharose High Performance Beads | GE Healthcare | 17-5113-01 | Streptavidin-coated sepharose beads |
| PyroMark Q96 HS Plate | QIAGEN | 979101 | Pyrosequencing assay plate |
| Eppendorf Thermomixer R | Fisher Scientific | 05-400-205 | Plate mixer. 96-well block sold separately (cat. No 05-400-207) |
| SureDesign Website | Agilent Technologies | - | Target capture design software (https://earray.chem.agilent.com/suredesign/) |
| UCSC Genome Browser | University of California Santa Cruz | - | rat Nov 2004 rn4 assembly |
| Agilent Methyl-Seq Protocol | Agilent Technologies | - | https://www.agilent.com/cs/library/usermanuals/public/G7530-90002.pdf |
Ссылки
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Meissner, A., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454 (7205), 766-770 (2008).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Naumov, V. A., et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 8 (9), 921-934 (2013).
- Wockner, L. F., et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational Psychiatry. 4, e339 (2014).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research. 33 (18), 5868-5877 (2005).
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A., Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48 (3), 226-232 (2009).
- Slieker, R. C., et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 6 (1), 26 (2013).
- Hing, B., et al. Adaptation of the targeted capture Methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 10 (7), 581-596 (2015).
- Seifuddin, F., et al. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 12 (8), 637-652 (2017).
- Irizarry, R. A., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 41 (2), 178-186 (2009).
- Lee, R. S., et al. Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics. 6 (11), 1378-1390 (2011).
- Jankord, R., et al. Stress vulnerability during adolescent development in rats. Endocrinology. 152 (2), 629-638 (2011).
- Pellow, S., Chopin, P., File, S. E., Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 14 (3), 149-167 (1985).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 10 (3), R25 (2009).
- Hansen, K. D., Langmead, B., Irizarry, R. A. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology. 13 (10), R83 (2012).
- Lee, R. S., et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 151 (9), 4332-4343 (2010).
- Lee, R. S., et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. , (2011).
- Bose, M., Olivan, B., Laferrere, B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Current Opinion in Endocrinology, Diabetes, and Obesity. 16 (5), 340-346 (2009).
- Brydon, L., Magid, K., Steptoe, A. Platelets, coronary heart disease, and stress. Brain, Behavior, and Immunity. 20 (2), 113-119 (2006).
- McKlveen, J. M., et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry. 80 (10), 754-764 (2016).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology. 13 (10), R87 (2012).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Translational Psychiatry. 7 (1), e994 (2017).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 60, 103-113 (2017).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены