
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Высокопроизводительная ДНК плазменный мультиплексирование и трансфекция с использованием акустической технологии нанодозирования
В этой статье
Резюме
Этот протокол описывает высокопроизводительную плазмидную трансфекцию клеток млекопитающих в 384-хорошую пластину с использованием акустической технологии выброса капель. Трудоемкий, подверженный ошибкам раздавливание ДНК и мультиплексирование, а также дозирование реагентов трансфекционного реагента, являются программным обеспечением и выполняются нанодайзером. Клетки затем посеяны в этих предварительно заполненных скважин.
Аннотация
Трансфекция клеток, незаменимая для многих биологических исследований, требует контроля многих параметров для точного и успешного достижения. Чаще всего выполняется при низкой пропускной их, это к тому же трудоемкие и подверженные ошибкам, тем более при мультиплексации нескольких плазмидов. Мы разработали простой, быстрый и точный метод для выполнения трансфекции клеток в 384-хорошо макет пластины с использованием акустических капель катапульты (ADE) технологии. Устройство nanodispenser используемое в этом изучении основано на этой технологии и позволяет точно поставку nanovolume на высокой скорости от плиты скважины источника к назначению одному. Он может обойтись и мультиплекс ДНК и трансфекционного реагента в соответствии с заранее разработанной таблицы. Здесь мы представляем оптимальный протокол для выполнения ADE основе высокой пропускной способностью плазмидтранса, что позволяет достичь эффективности до 90% и почти 100% котрансфекции в котрансфекционных экспериментов. Мы расширяем первоначальную работу, предлагая удобный для пользователя макрос на основе электронных таблиц, способный управлять до четырех плазмидов/колодцев из библиотеки, содержащей до 1536 различных плазмидов, и приложения для планшетного нахлыстое направляющее приложение. Макрос разрабатывает необходимый шаблон (ы) исходной пластины (ы) и генерирует готовые к использованию файлы для нанодайзера и планшетного приложения. Четырехэтапный протокол трансфекции включает в себя i) разбавитель обойтись с классическим жидким обработчиком, ii) плазмидное распределение и мультиплексирование, iii) трансфекционный реагент распределяется нанодсером, и iv) клеточное покрытие на предварительно заполненных скважинах. Описанный программно-основанный контроль мультиплексирования и трансфекции Плазмидных плазмидных aDE позволяет даже неспециалистам в полевых условиях быстро и безопасно выполнять надежную трансфекцию клеток. Этот метод позволяет быстро определить оптимальные настройки для данного типа ячейки и может быть перенесен на более высокие и ручные подходы. Протокол облегчает применение, такие как человеческий белок ORFeome (набор открытых кадров чтения в геноме) или CRISPR-Cas9 на основе генной функции проверки, в непулированных стратегий скрининга.
Введение
Представленный здесь метод подробно описывает, как выполнять мультиплексирование и трансфекцию ДНК в клетках млекопитающих при высокой пропускной состоянии с помощью жидкого нанодайзера на основе акустической системы в 384-хорошей пластине, даже для неспециалистов в этой области. Этот недавно опубликованный метод1 позволяет выполнять столько, сколько 384 независимых плазмидных мультиплексирования ДНК и трансфекции условиях в одном эксперименте, менее чем за 1 ч. Одиночные или котрансфекционные эксперименты были успешными, достигнув почти 100% котрансфекция в популяции трансинфицированных клеток. Этот протокол упрощает трансфекцию, поскольку большинство утомительных, трудоемких и подверженных ошибкам шагов теперь ориентированы на программное обеспечение (см. рисунок 1 для общего обзора). Были предприняты дальнейшие усилия по разработке специализированных инструментов для повышения простоты использования при одновременном предотвращении человеческих ошибок в ходе общего процесса и содействия успешному трансфекции даже для неспециалистов в этой области. Описанный протокол включает в себя "удобную" макротаблицу, которую мы разработали для управления 384 независимыми трансфекционными условиями с мультиплексированием возможностей до четырех плазмидов в каждой скважине. Макрос автоматически генерирует шаблоны исходной пластины (ы) для загрузки ожидаемого объема плазмидной плазмы ДНК от стартовых фондовых решений и файлов, необходимых для привода программного обеспечения нанодайзера на экспериментальной конструкции, которая была введена. Поскольку ручная раздача ДНК в 384-хорошо источник пластины утомительно и подвержены ошибкам, мы также разработали специальное приложение на основе таблетки для руководства пользователя при распределении решения ДНК в соответствии с шаблоном.

Рисунок 1: Экспериментальный рабочий процесс. Схематическое представление оптимального автоматизированного протокола обратного трансфекции высокой пропускной связи (от экспериментального проектирования до пользовательского биологического асссе). Ручные шаги указаны символом руки, а приблизительное время для каждого шага написано в красной коробке. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
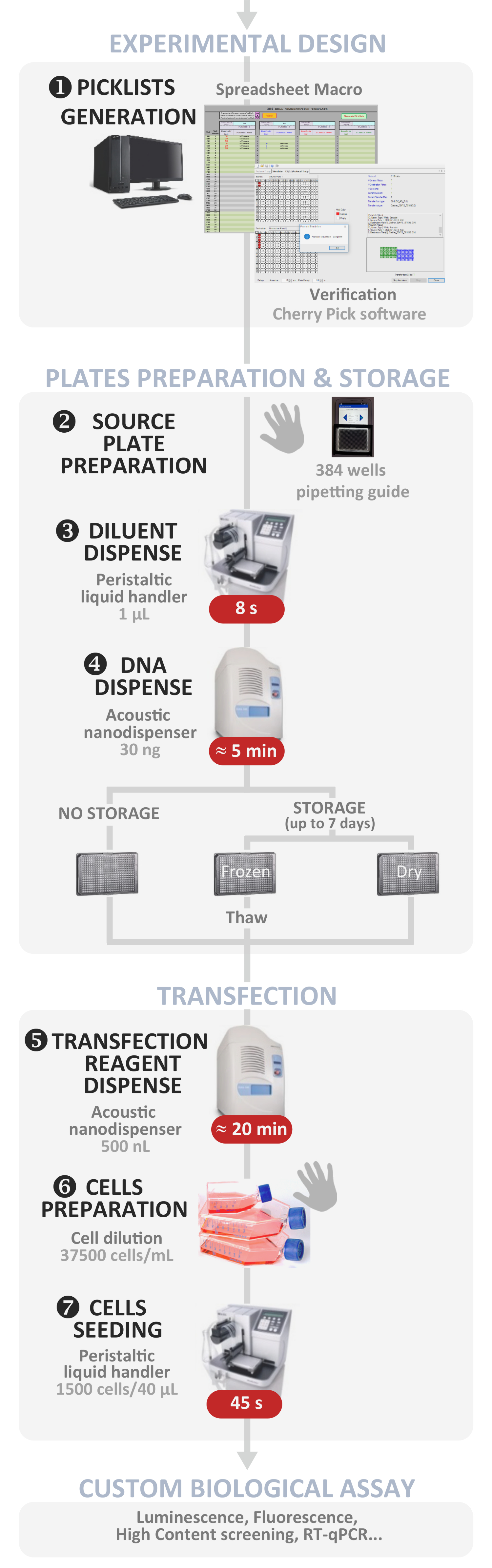{kind=link}
Многие клеточные эксперименты начинаются с плазмидной трансфекции ДНК, и даже если многие выделенные реагенты были и все еще разрабатываются для повышения эффективности трансфекции и / или облегчить процедуру, многое еще предстоит сделать2,3 , 4. Трансфекция плазмидных клеток ДНК включает в себя несколько шагов для достижения высокой эффективности, таких как начальное комплексное поглощение, эндосомальный побег и цитоплазмический перенос к ядру5,6. В дополнение к выпасам кальция или физическим методам, таким как электропорация или микроинъекция с использованием специальных устройств7, современные химические методы были сосредоточены на повышении доставки клеток ДНК при снижении цитоксичности клеток8, 9. Использование липидов или катионных полимеров, образующих липосомы-подобные комплексы, а в последнее время нелипсоомальные полимерные химические системы сделали трансфекцию проще и эффективнее10. Несмотря на эти изменения, трансфекция клеток по-прежнему требует конкретных навыков, которые должны быть точно выполнены, поскольку большинство из этих физических или химических протоколов трансфекции требуют, чтобы ученые вручную подготовить каждое состояние реакции трансфекции ДНК, таким образом ухудшает пропускную выливку. Чтобы обойти эту проблему, обратные протоколы трансфекции были разработаны с использованием химических реагентов трансфекции11,12,13, что позволяет пользователю проверить или объединить несколько плазмидов в более быстрый способ. В этих протоколах перед посевом клеток на комплексах образуются нуклеиновые кислотные комплексы с трансфекционными реагентами. Однако эти обратные протоколы по-прежнему ограничены ручным управлением решениями ДНК и сочетанием каждого из независимых условий. Хотя это возможно, чтобы выполнить их в 96-ну хорошо пластины формате, подготовка ДНК и дозы будет утомительным, и там, вероятно, будут ошибки. Когда требуется различное количество нескольких плазмидов ДНК и мультиплексируется друг с другом, трансфекция клеток становится еще более трудной и более трудоемкой, и человеческие ошибки становятся совершенно неизбежными. Масштабирование до 384-колодцевого формата пластин в обратном подходе трансфекции, несмотря на несколько мультиплексированных условий трансфекции ДНК, становится невозможной задачей из-за следующих причин. i) Объемы ДНК, трансфективный реагент или объемы реакционной смеси для управления ниже, чем 1 КЛ для каждой скважины. ii) Мультиплексирование плазмидов для 384 независимых условий становится чрезвычайно сложным. Поставка в каждой из 384 скважин также iii) очень трудоемкие и iv) ошибка подвержена. Действительно, выдать правильное решение в ожидаемых скважинах трудно управлять, потому что низкие объемы уже обойтись не позволяют визуального мониторинга между пустыми и уже заполненными скважинами. v) Наконец, существует высокий риск высыхания смеси путем испарения до добавления клеток в связи со временем, необходимым для выполнения необходимых мер по дозированию. Таким образом, ограничивающим фактором для создания высокой пропускной днк плазмидных трансфекционных анализов, как представляется, миниатюризация анализа, который подразумевает малообъемный мультиплексирование и управление, которые не могут быть обработаны вручную больше, но также вряд ли достижимы в надежным способом классическими перистатическими жидкими обработчиками.
В качестве доказательства сложности автоматизировать такие assays и получить высокую пропускную стоимость, только несколько попыток автоматизировать трансфекции были опубликованы до сих пор: 96-ну хорошо пластины формат с использованием коммерческого устройства обработки жидкости и фосфата кальция осадков14 и, совсем недавно, липоплексрей, и микрофлюидный чип, позволяющий 280 независимых трансфекций15, но требующих специальных навыков в этой области. Другой метод, акоутофорез, позволяющий жидкой левитации и ведущих к жидкости манипуляции и смешивания, был использован для выполнения трансфекции ДНК в 24-96-ну хорошо пластины форматов16. Хотя этот подход является осуществимым, он страдает от крайне низкой пропускной всей перевалки, поскольку смешивание клеток с трансфекционной смесью ДНК требует инкубации 60 с для каждой точки перед посевом. Это означает продолжительность не менее 96 мин для полной 96-колодца пластины. Кроме того, этот протокол далек от того, чтобы поддаваться общей аудитории биологов, как эта работа была сделана с в доме разработан и изготовлены устройства, которые в настоящее время не доступны на рынке. Напротив, в последние несколько лет с помощью нанообъемных диспенсеров появилась простая в использовании технология акустического дозирования на основе программного обеспечения. Используя сфокусированную акустическую энергию, эти устройства позволяют жестко контролируемый выброс небольших объемов жидкости от 2,5 нл до 500 нл от исходной пластины до пункта назначения17. Эта технология, называемая акустической выкатывания капель (ADE), имеет множество преимуществ: она полностью автоматизирована, бесконтактная, без кончиков, точная, точная и высоко воспроизводимая, и имеет высокую пропускную связь18. Первый посвященный доставке диметилсульксида (DMSO) решения, настройки были расширены, чтобы обойтись aqueous буферов19. Таким образом, акустические нанодятелы, кажется, подходят для протоколов трансфекции обратных клеток и могут обойти большинство вышеупомянутых ручных ограничений. Поскольку с помощью этой технологии не было описано никаких попыток трансфекции плазмидных трансфекций, мы недавно оценили пригодность акустической системы дозирования для выполнения обратного трансфекции клеток.
Воспользовавшись пропускной стоимостью нанодчебирования и простотой использования, мы оптимизировали обратный протокол трансфекции для клеток HeLa путем перекрестного тестирования нескольких параметров, которые могут влиять на трансфекцию ДНК на 384-ну, одной пластине, а именно, общее количество ДНК и источник концентрации ДНК, разбавитель, трансфекционный реагент, и количество спреда клеток. Разработанный протокол обходит вышеописанные ручные ограничения трансфекции клеток и предоставляет ряд преимуществ по сравнению с другими автоматизированными попытками трансфекции. Во-первых, он миниатюризирован, что позволяет экономически эффективным трансфекционным реагентом, экономя препараты плазмида ДНК и трансфекционный реагент. Во-вторых, он гораздо более высокопроизводительен и воспроизводим, чем ручной протокол (даже для начинающих), так как трансфекция всей 384-й пластины может быть достигнута менее чем за 1 ч. Наконец, это программное обеспечение, что позволяет контролировать дозированных количество ДНК и мультиплексирования нескольких плазмидов. Действительно, благодаря программному обеспечению nanodispenser(Таблица материалов), пользователь может разработать план исследования для управления объемами, которые будут освобождены от определенного источника хорошо пластины назначения один.
Представленный здесь протокол предназначен в основном для тех, кто имеет доступ к нанодционизму и хотел бы создать трансфекционные эксперименты с высокой пропускной их пропускной вот и доступной, но также и для тех, кто хочет быстро оптимизировать свои параметры трансфекции для данного типа клеток применение этого протокола для перекрестного тестирования нескольких параметров при высокой пропускной состоянии. Действительно, мы показали, что оптимизированные параметры, идентифицированные с этим наномасштабным протоколом, могут быть перенесены в более масштабные и ручные трансфекционные эксперименты. Наконец, поскольку трансфекционный реагент, используемый в настоящем протоколе, позволяет трансфекцию ДНК или siRNA в соответствии с производителем, протокол также представляет интерес для тех, кто стремится к выполнению комплексных подходов к переэкспрессии генов или нокдауну. Назначения пластины предварительно заполнены ДНК могут быть сохранены до 7 дней до использования в трансфекционном анализе без потери эффективности, которая является еще одним преимуществом следующего протокола для такого рода применения.
протокол
1. Предварительная подготовка
- Подготовка программ перистальтических жидкостных обработок
ПРИМЕЧАНИЕ: Для разбавитель ныхивательных и клеточных этапов протокола должна быть подготовлена специальная программа с учетом высоты дозирующей головки к используемой пластине и намерения шага.- Для шага разбавителя с 1 злицы установите кассету номиналу 1 л и подготовьте программу с настройками, описанными в шагах 1.1.1.1 и 1.1.1.2.
- Отрегулируйте параметр скорости потока до максимума для лучшей пропускной связи, так как на этом этапе не ожидается никакого биологического материального ущерба. Отрегулируйте высоту дозировки до 9,6 мм (в соответствии с используемой пластиной культуры клеток, Дополнительная рисунок 1), чтобы 1 л капли коснуться нижней части скважин во время диспенсации.
ПРИМЕЧАНИЕ: Этот шаг имеет решающее значение, чтобы избежать удержания капель на дозирующей головке до достижения достаточного объема, чтобы упасть. - Отрегулируйте прозрачую высоту пластины до 14,4 мм, чтобы после раздавливания каждого ряда можно было свободно перемусливаться головой над плитой. Визуально контролировать надлежащие настройки перистальтической жидкости обработчик головы высоты: убедитесь, что не капли сохраняются на дозирования советы при распределении и убедитесь, что голова достаточно высока, чтобы сделать смещение головы после дозирования каждого ряда.
ПРИМЕЧАНИЕ: Избегая удержания капли является важным параметром, поскольку это ухудшит точность объема диспенсации.
- Отрегулируйте параметр скорости потока до максимума для лучшей пропускной связи, так как на этом этапе не ожидается никакого биологического материального ущерба. Отрегулируйте высоту дозировки до 9,6 мм (в соответствии с используемой пластиной культуры клеток, Дополнительная рисунок 1), чтобы 1 л капли коснуться нижней части скважин во время диспенсации.
- Для раздавливания 40-л суспензии ячейки, смонтировать кассету 10 л и подготовить программу с настройками, как описано в шагах 1.1.1.1.1.2.2.
- Отрегулируйте параметр скорости потока до Low, чтобы обойтись без клеток с низкой скоростью, чтобы избежать потенциального повреждения клеток сдвига стрессом и высоким воздействием на дно скважин. Отрегулируйте высоту диспенсации до 11.43 мм (в соответствии с используемой пластиной культуры клеток, Дополнительная рисунок1), достаточно высокая, чтобы снизить воздействие клеток на дно скважин во время процесса дозирования, но достаточно низко, чтобы избежать удержания капель на дозирования головы. Отрегулируйте плиту ясной высоты до 16 мм, чтобы позволить свободное смещение дозирующей головы над пластиной после дозирования каждого ряда.
- Визуально контролировать надлежащие настройки перистальтической жидкости обработчик головы высоты: убедитесь, что не капли сохраняются на дозирования советы при распределении и убедитесь, что голова достаточно высока, чтобы сделать смещение головы после дозирования каждого ряда.
ПРИМЕЧАНИЕ: Избегая удержания капли является важным параметром, поскольку это приведет к распределению ненадежного номера ячейки.
- Для шага разбавителя с 1 злицы установите кассету номиналу 1 л и подготовьте программу с настройками, описанными в шагах 1.1.1.1 и 1.1.1.2.
- Препарат плазмида ДНК (классический протокол экстракции мини-подготовки)
- Выращивайте преобразуемый штамм бактерий DH5 в среде LB, дополненный 125 мкг/мл антибиотиком выбора ампициллин (ТаблицаМатериалов) на ночь при 37 градусах Цельсия и под нежным возбуждением (200 об/мин) на орбитальном шейкере (Таблицаматериалов).
- Урожай 2 мл культуры, гранулы клетки центрифуги в течение 5 мин на 6000 х г, и отбросить супернатанта.
- Приостановите действие клеточной гранулы с 250 злиц буфера, содержащего RNase A(Таблица материалов). Добавьте 250 л люсис-буфера и инкубировать в течение 5 минут при комнатной температуре, в соответствии с инструкциями производителя.
- Остановите реакцию лизиса, добавив 300 йЛ буфера нейтрализации(Таблицаматериалов) и vortexting в ближайшее время. Центрифуга трубки в течение 5 мин на 11000 х г.
- Поместите новую плазмидную мини-колонну(Таблицаматериалов) в трубку коллекции 2 мл и декантирует супернатант в колонке центрифугией в течение 1 мин при 11 000 х г.
- Отбросьте поток через и поместите мини-колонку обратно в трубку сбора.
- Вымойте плазмидную миниколонку с 500 злителка юаня буфера(таблицаматериалов) и центрифугу в течение 1 мин при 11000 х г, в соответствии с инструкциями производителя.
- Отбросьте поток через и поместите плазмидную миниколонку обратно в трубку коллекции.
- Добавьте 700 л стирального буфера(Таблицаматериалов), дополненного этанолом и центрифугой в течение 1 мин при 11 000 х г, в соответствии с инструкциями производителя.
- Отбросьте поток через и центрифуги плазмид мини-колонны и его сбора трубки 1x больше в течение 2 мин на 11000 х г, чтобы высушить мембрану кремнезема.
- Поместите высушенную плазмидную миниколонку в новую трубку 1,5 мл и добавьте 30 л дистиллированной воды, предварительно разогретой при температуре 60 градусов по Цельсию, нависните на 2 мин при комнатной температуре, а затем центрифугите его в течение 1 мин при 11 000 х г.
- Откажитесь от плазмидной миниколонки и держите элюат, содержащий очищенную плазмиду ДНК.
- Измерьте концентрацию ДНК элутированной ДНК с помощью микротомного спектрофотометра(Таблицаматериалов).
- Включите спектрофотометр и выберите параметры измерения ДНК.
- Поднимите руку выборки спектрофотометра и пипетки 1 л воды на пьедестал измерения для выполнения пустой калибровки.
- Опустите руку выборки, начните пустое измерение и дождитесь завершения.
- Поднимите руку выборки и протрите образец с верхних и нижних пьедесталах.
- Пипетка 1 злициста раствора ДНК на нижний пьедестал для его измерения.
- Опустите руку выборки, начните измерение концентрации ДНК и дождитесь завершения.
- Поднимите руку выборки и протрите образец с верхних и нижних пьедесталах.
- Для дальнейших измерений концентрации ДНК повторите шаги 1.2.13.5-1.2.13.7.
- После завершения измерений храните решения ДНК при температуре 4 градусов по Цельсию до использования.
2. Экспериментальный дизайн и генерация пиклистов для управления диспенсами на основе ADE
ПРИМЕЧАНИЕ: Для управления объемами ДНК и смешивания до четырех плазмидов в формате 384 колодца был разработан специальный макрос электронной таблицы. Основываясь на введенном экспериментальном дизайне, этот макрос генерирует необходимые файлы для управления протоколом трансфекции ДНК на основе ADE с помощью нанодайзера. Для того, чтобы создать эти файлы, несколько полей должны быть заполнены в листе шаблонов, как показано на рисунке 2.

Рисунок 2 : Поколение списков для управления диспенсацией ADE с помощью макроса электронной таблицы. Несколько параметров должны быть заполнены, а именно :1 ) трансфекционный реагент (TR) и минимальные/максимальные объемы, которые будут использоваться в исходной пластине, (2) первоначальные концентрации плазмида, которые должны быть распределены в исходной пластине, и(3 ) конструкция цельной пластины, включая ожидаемые количества плазмидных и мультиплексирования в каждом из 384 скважин. (4) Активация Picklists позволяет проверить различные поля и, после правильного заполнения, пиклисты для диспенсации ДНК и TR и необходимый шаблон исходной пластины автоматически генерируются. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
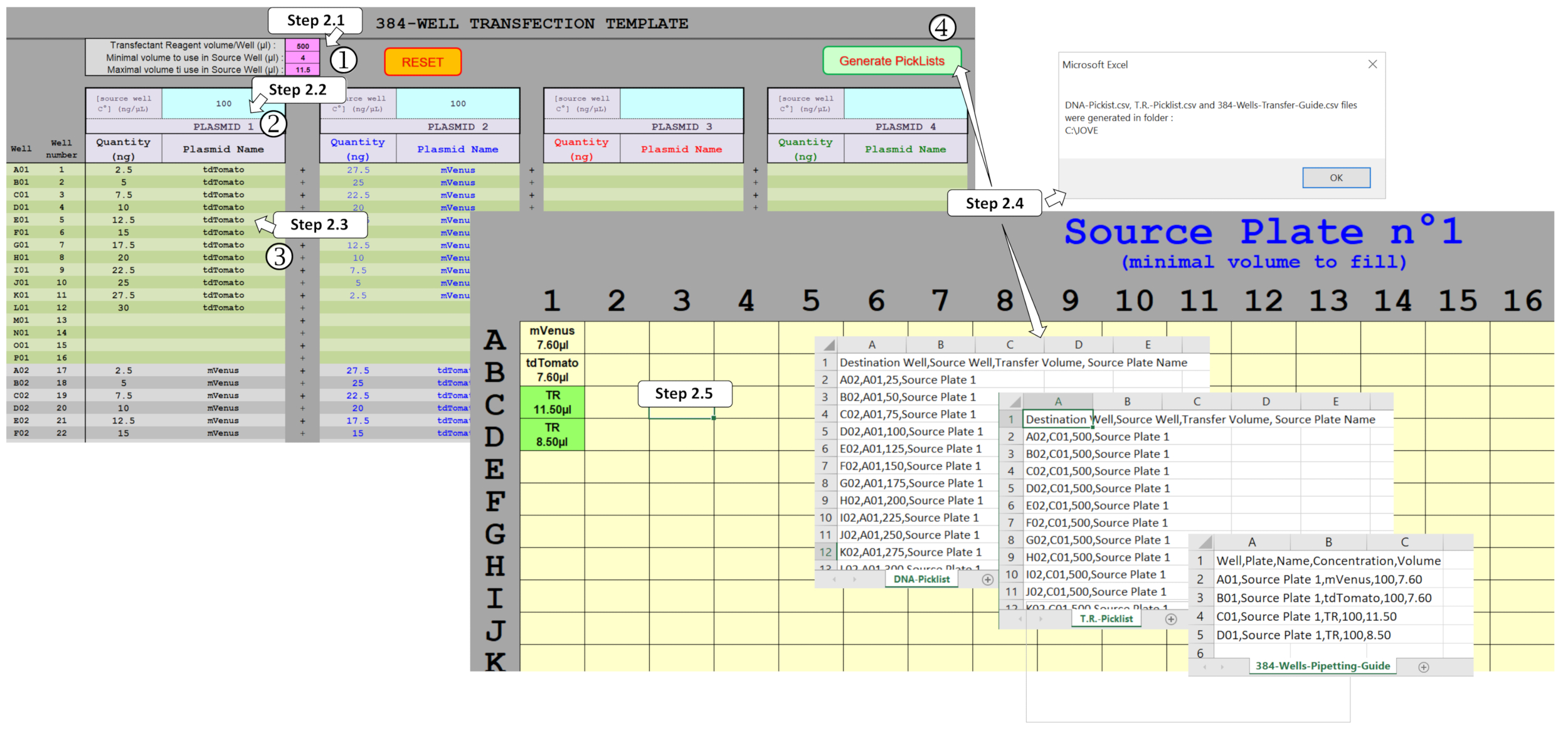{kind=link}
- Введите параметры протокола нанодчении в розовых полях. Установите значение смеси трансфективного реагента (TR) до 500 нл. Установите минимальное значение объема в исходной пластине до 4 qL. Установите максимальный объем в исходной пластине скважин до 11,25 л.
ПРИМЕЧАНИЕ: Используемый здесь нанододознар может передавать не более 500 нл за один запуск ADE. Эти розовые поля заполнены рекомендуемыми значениями, но могут быть изменены в соответствии с потребностями пользователя. - Введите 100 нг / Л концентрации ДНК, начиная концентрации в синих полях, соответствующих основной ДНК.
ПРИМЕЧАНИЕ: Это значение является оптимальной концентрацией, ранее определяемой, но может, однако, быть изменено для различных потребностей пользователя. - Введите желаемое количество ДНК в серых/зеленых полях. Введите количества и плазмидные названия для 384 скважин, обеспечивая то же правописания, если тот же плазмид используется в нескольких скважинах.
- Создайте дизайн исходной пластины, файлы picklists и файл руководства по пипетке. Нажмите на Generate Picklists, чтобы позволить макросу генерировать файлы DNA-Picklist.csv, T.R.-Picklist.csv и 384-Wells-Pipetting-Guide.csv из данных, собранных на соответствующем листе. При запросе исправьте значения ячеек, заполненных оранжевым цветом, поскольку они указывают на ошибки или объемы, которые не могут быть обработаны нанодайзером.
- Печать шаблона (ы) из листа Исходной плиты. Указаны плазмидные названия и минимальный объем для заполнения скважин. Аналогичным образом, объемы смеси трансфекционного реагента, которые затем должны быть заполнены в следующих скважинах, указаны как TR и выделены зеленым цветом.
3. Подготовка пластины источника ДНА используя применение направляющего выступа 384-ну наилучшим образом
- Разбавить хранящуюся плазмид ДНК со ступени 1.2.14 до 100 нг/Л с помощью дистиллированной воды.
- Калибровать 384-колодец сетки на пластины размеров: открыть 384-колодец пипеттинг направляющий приложение на планшете (Рисунок 3). Поместите исходную пластину на сетку на нижнем экране, а в верхнем левом меню калибровки, нажмите кнопку или - (или использовать красный курсор) для повышения или уменьшения размера сетки и колодцев для того, чтобы настроить зеленые колодцы на четыре угловых колодца пластины .

Рисунок 3 : Использование приложения направляющих направляющих 384 колодца. (1) Калибровка 384-колодца сетки до размера пластины; (2) ) Гора универсального 3D-печатного адаптера пластины к планшету с помощью двусторонней ленты; (3) Размещение пластины на адаптере; (4) Перемещение сетки, чтобы центр его на установленную пластину. (5) Блокировка шага калибровки. (6) Открытие 384 скважин pipetting guide.csv файл. (7) Учитывая список файлов, приложение будет указывать ожидаемое название исходной пластины, реагент (ДНК или трансфекционный реагент), концентрацию и объем для раздачи в целевые скважины, которые будут освещены один за 1. (8) Левая и правая кнопки стрелки позволяют пользователю следовать пипетки руководство легко распределять реагенты в соответствии с электронной таблицей макро шаблон амплитуды (ы). Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}
- Используя двусторонню ленту, установите 3D-печатный адаптер пластины на экране, чтобы избежать движения исходной пластины при распределении. При необходимости переместите калиброванную сетку, используя стрелки вращения и кнопки Вверх/Вниз/Левая, чтобы настроить сетку на экране в положение пластины. После того, как сетка и размеры скважин правильно откалиброваны и расположены, отметьте калибровку lock.
- Нажмите на FILE и откройте 384 скважины pipetting guide.csv файл. Следуйте инструкциям экрана вручную распределять указанный объем указанной плазмиды при указанной концентрации в белом выделенном хорошо соответствующем надлежащему целевому назначению ожидаемой пластины. Используйте - или стрелки, чтобы вернуться или дальше в процессе дозирования ДНК. Прекратите дозирование при достижении первого раствора реагента Трансфекции для загрузки.
- Как только диспенсации ДНК закончены, удалите исходную пластину из адаптера. Если несколько исходных пластин должны быть заполнены, то поместите новую пластину источника на адаптер и следуйте инструкциям дозирования. После того, как диспенсация ДНК закончена, центрифуга ДНК заполненные источник пластины (ы) (на 1500 х г в течение 2 мин), чтобы обеспечить надлежащее выравнивание жидкости и удалить пузырьки, ведущие к неточности в ADE основе передач.
4. Перистальтический жидкий обработчик на основе 1 л разбавитель диспенсации в пункт назначения пластины
ПРИМЕЧАНИЕ: Выполните шаги 4.1-4.5 в шкафе биологической безопасности.
- Дезинфицировать 1 л кассеты голову, распыляя его с распылителем дезинфицирующее средство(Таблица материалов), и позволяют этому решению, чтобы войти в наконечник держателя. Поглотить остатки дезинфицирующего средства на поглощающей бумаге. Установите кассету 1 зл на перистальтическое устройство обработчика жидкости. Включите устройство и убедитесь, что параметр типа кассеты является правильным (1 зл), а также формат пластины (384 скважины).
- Дезинфекция всего просвета труб: вставьте трубку организатора (держа восемь труб вместе) в стерильный сосуд и заполнить его с 5 мл 70% алкоголя. Используя первоздавую функцию перистальтической жидкости обработчик, сначала промыть спирт в трубке, а затем промыть его, пройдя 5 мл дистиллированной воды и 5 мл сыворотки свободной среды (Dulbecco в модифицированных Eagle в среднем "DMEM" дополняется 100 U /mL пенициллин-стрептомицин; ТаблицаМатериала), последовательно заполняя в одном и том же сосуде. Убедитесь, что ни один из кончик засоряется путем визуального осмотра потока жидкости из всех из них.
- Прайм трубки с сыворотки свободной среде, заполнив новый стерильный сосуд с 10 мл прегретой сыворотки свободной среды и дайвинг трубки организатора в нем. Нажмите на кнопку перистальтики жидкости обработчик около 10 с. Еще раз, убедитесь, что не кончик засоряется путем визуального осмотра потока жидкости из всех из них.
- Заполните тарелку 1 л разбавитель. Поместите стерильную 384-хорошую культурную пластину (направление) на перистальтический жидкий обработчик пластины перевозчика и удалить его крышку.
- Запустите предварительно калиброванные программы, чтобы обойтись 1 Л л в каждой скважине из 384-хорошо пластины. Время дозирования составляет около 8 с. Затем замените крышку 384-хорошо пластины.
ПРИМЕЧАНИЕ: Кроме того, этот шаг может быть обработан вручную, в шкафу безопасности, используя многоканальный микропипетли.
5. Проведение обследования для управления объемами, выданными вручную
ПРИМЕЧАНИЕ: Для получения подробной информации, см. Рисунок 4.
- Запустите программу нанодайзера, перейдите к диагностической вкладке, отметьте исходную пластину Out Box, загрузите исходную пластину на держатель пластины и зайдите в тарелку, чтобы войти в тарелку. При запросе выберите 384LDV-A-B2, чтобы настроить нанододознат в режим раздавливания буфера, и нажмите Ok.
- Выберите Опрос в меню «Разное» и нажмите на Launch. Выберите заполненные скважины для анализа и нажмите на кнопку Go. Убедитесь, что измеренные объемы соответствуют ожидаемым, и убедитесь, что скважины не были загружены с объемами более 12 Зл, так как это позволит избежать передачи.

Рисунок 4 : Определение параметров программного обеспечения для обследования. (1) Запуск программы наноддазатора. (2) Откройте вкладку Диагностика. (3) Вставьте исходную пластину, тикая для исходной пластины, а затем, В. (4) Определите тип исходной тарелки в меню, когда это вызвано. (5) В разное поле выберите Обзор в выпадающем меню. (6) Запуск программы обследования, нажав на запуск. (7) Выберите заполненные скважины для измерения. (8) Начните анализ, нажав на Go. (9) После проведения обследования измеренные объемы записываются в соответствующие выбранные скважины. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
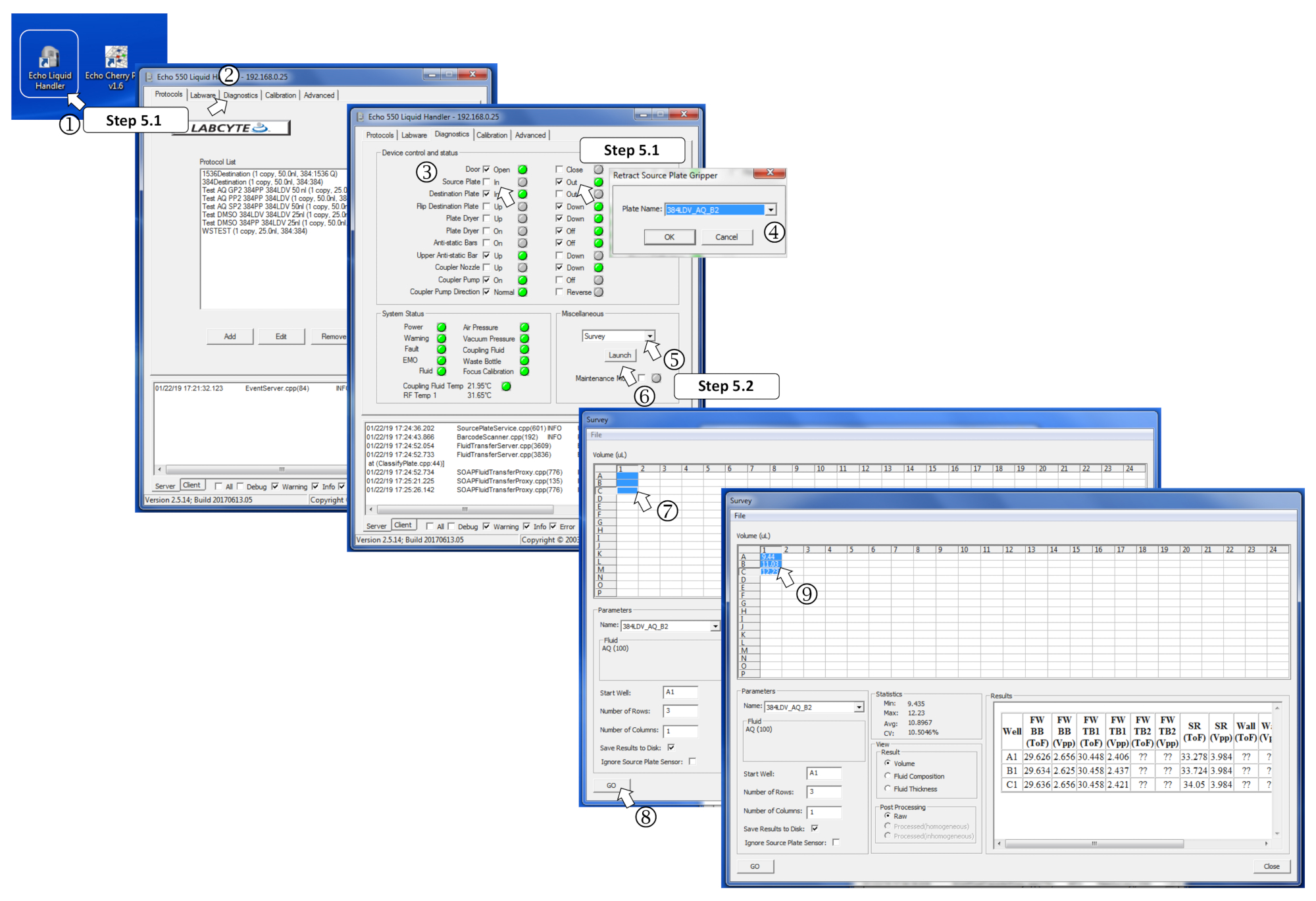{kind=link}
6. Диспенсация ДНК на основе АДЕ в пункт назначения
- Запустите программное обеспечение для выбора, установите 384-ну хорошо источник и назначения типа пластины 384-LDV и Greiner 384PS-781096, соответственно(Рисунок 5). Установите устройство в режим раздавливания буфера, выбрав 384LDV-A-B2и отключите "оптимизацию пропускной перевалки".

Рисунок 5 : Производительность диспенсации на основе выбора. (1) Запуск программного обеспечения нанодчении. Во вкладке Протокола выберите (2) формат пластины образца, (3)тип пластины назначения и (4)отсетики "оптимизировать пропускную стоимость передачи". (5) Выберите вкладку Список выбора. (6) Нажмите на импорт и выберите правильный файл q.csv (DNA-PickList или T.R.-Picklist). (7) После выбора, нажмите на импорт. (8) Нажмите на воспроизведение и сохранить протокол. (9) Выполните моделирование диспенсации, нажав на Имитацию, или ( 10 ) Начать запрограммированное диспенсацию, нажав на Run. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
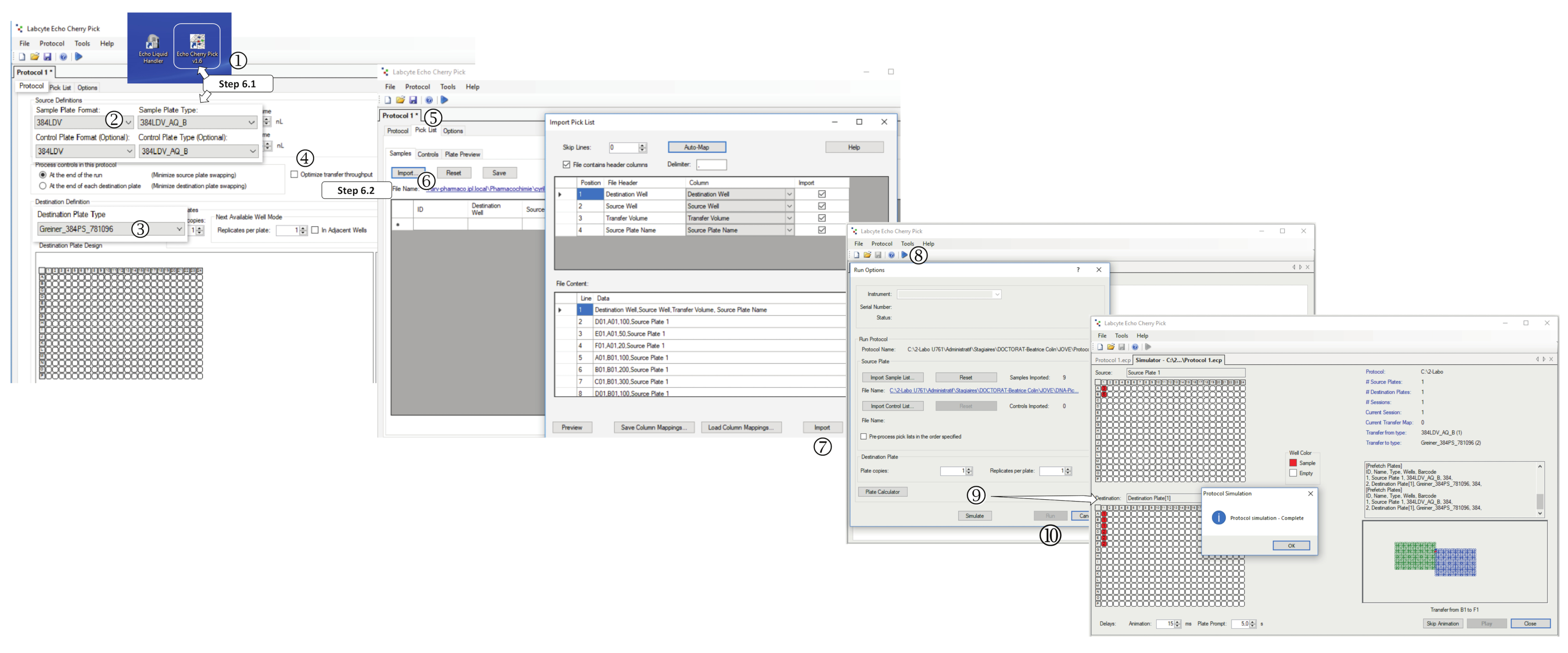{kind=link}
- Выберите вкладку "Список пик", нажмите на импорт, выберите файл DNA-Picklist.csv. Нажмите на воспроизведение и сохраните протокол. Нажмите на Simulate, чтобы выполнить моделирование запрограммированных диспенсаций, чтобы убедиться, что список соответствует ожидаемому экспериментальному дизайну. После завершения, нажмите на Закрыть.
- Нажмите на Play, а затем запустить, чтобы начать программу дозирования: когда его спросили, вставьте запрошенную пластину источника (ДНК решения вручную заполнены) и назначения пластины (разбавленной заполнены) в нанодспенсер.
ПРИМЕЧАНИЕ: Время дозирования составляет примерно 5-20 минут для полной 384-хорошо пластины, в зависимости от выбранных объемов и общее количество диспенсаций в экспериментальном дизайне. - Кроме того, приостановить протокол здесь, как разбавитель- и ДНК заполненные пластины могут обрабатывать сухой или замороженный хранения на срок до 7 дней. Для сухого хранения, пусть пластины высохнуть на скамейке при комнатной температуре, а затем хранить их таким же образом. Оттепель и центрифуга (при 1500 х г в течение 2 мин) замороженные хранимые пластины перед использованием в шаге трансфекции (раздел 7).
7. Диспенсация трансфективных реагентов, управляемых ADE
- В шкафу биобезопасности, extemporaneously разбавляют реагент трансфекционного липополиплакса в сывороточной среде до 1x окончательной концентрации. Vortex и немедленно обойтись этой трансфекционной реагента смесь в соответствии с предопределенным источником пластины (ы), разработанный макро с использованием предварительно калиброванных 384-колодец пипеттинг руководство приложение, как описано в шаге 3.4.
ПРИМЕЧАНИЕ: Не центрифуги исходной пластины, как только он загружается с реагентом, как не трансфекция замечена после центрифугирования. - Запустите программу нанодчебирования для выполнения «обследования», как описано в разделе 5, для того, чтобы контролировать объемы всех заполненных вручную ТРУБ TR исходной пластины (ы), чтобы избежать ошибок дозирования из-за объемов, превышающих 12 Зл.
- Нажмите на reset, чтобы очистить список образцов списка ДНК в программном обеспечении picklist, и убедитесь, что параметры устройства по-прежнему настроены на водные буферы и типы исходных и назначения, используемые, как в шаге 6.1.
- Нажмите на импорт и выберите файл TR-Picklist.csv. Нажмите на Play и сохранить протокол, если предложено, и (это необязательно, но настоятельно рекомендуется) выполнить моделирование запрограммированных трансфекционных реагента распределения смеси для обеспечения надлежащего проектирования диспенсаций, нажав на Кнопка имитации. После завершения, нажмите на Закрыть.
- Нажмите на кнопку Play , а затем запустите кнопку, чтобы начать программу дозирования: по запросу, поместите исходную пластину (s) (TR-смесь заполнены) и назначения пластины (разбавитель и ДНК-заполненные) в нанодчене.
ПРИМЕЧАНИЕ: Время дозирования составляет менее 20 минут для полной 384-хорошо пластины при распределении 500 нл смеси TR. - Инкубировать 15-30 мин при комнатной температуре после добавления TR в ДНК, как указано в протоколе производителя.
8. Перистальтический обработчик жидкости на основе клеточного диспенсации
- Подготовьте перистальтический жидкий обработчик для раздавливания клеток. Дезинфицировать 10 л кассеты голову, распыляя его с Aniospray Surf 29 дезинфицирующее средство и поглощая остатки на бумаге. Установите кассету на перистальтический жидкий обработчик устройства, изменить настройки типа кассеты до 10 qL, и убедитесь, что формат пластины установлен на 384 скважин.
- Дезинфекция 10 л кассетных труб, как это было описано ранее в шаге 4.2. Погружение трубки организатора в стерильный сосуд и промыть трубки с 5 мл 70% алкоголя, затем с 5 мл дистиллированной воды, и, наконец, с 5 мл сыворотки свободной среде, последовательно заполнены в том же сосуде и до тех пор, пока каждая трубка пуста.
- Подготовьте суспензию клетки для того чтобы dispense. Из сопливого HeLa ячейки B10-культуры блюдо, мыть клетки 1x с 1x фосфат-буфера солей (PBS) раствор, а затем разъединить клетки с трипсином / EDTA в течение 5 мин при 37 градусов по Цельсию.
- Проверьте диссоциацию клеток под микроскопом и остановите действие трипсина/ЭДТА, добавив 10 мл полной среды (DMEM, дополненной 10% сывороткой крупного рогатого скота плода и 100 U/mL пенициллина-стрептомицина; см. Таблицу Материала) в блюде культуры. Урожай клетки в 50 мл трубки и рассчитывать клетки под микроскопом, используя клетку Malassez или автоматический счетчик клеток.
- Подготовьте не менее 25 мл клеточной подвески HeLa при концентрации 37 500 клеток/мл в полной среде (т.е. 1500 ячеек/40 л) для полной 384-хорошей пластины, чтобы обеспечить грунтовку трубки и 40 л/ну диспансеризации.
- Чтобы распределить клетки, заполните новый стерильный сосуд с подготовленной клеточной подвеской и перемешайте его, чтобы избежать осадки, ведущие к неточности в плотности клеток диспенсации. Вставьте организатора трубки в этом растворе и нажмите кнопку Prime до тех пор, пока подвеска ячейки не начнет промывать от дозирующей головы. Убедитесь, что ни один из кончик засоряется путем визуального осмотра потока жидкости из всех из них, и убедитесь, что каждая трубка загружается с клеточной подвеской.
- Загрузите ДНК и TR-заполненные 384-ну хорошо назначения пластины на перистальтический жидкости обработчик пластины перевозчика и удалить его крышку. Запустите предварительно калиброванные программы, чтобы обойтись 40 л клеточной подвески на полной 384-хорошо пластины (т.е., 1500 клеток / хорошо). Время дозирования составляет около 8 с. Заменить крышку 384-колодца пластины.
ПРИМЕЧАНИЕ: Кроме того, 40 л клеточной подвески можно вручную обойтись с помощью многоканального микропипетля.
9. Пользовательский биологический контроль (мониторинг эффективности трансфекции клеток)
ПРИМЕЧАНИЕ: Следуя экспериментальным настройкам и намерениям эксперимента, используйте необходимые методы для люминесценции, флуоресценции, скрининга высокого содержания и обратной транскрипции количественной полимеразной цепной реакции (RT-qPCR). В этом разделе протокола эффективность трансфекции клеток оценивается с помощью автоматизированной флуоресценционной микроскопии и анализа изображений.
- Инкубировать пластину при 37 градусах Цельсия с 5% CO2 в насыщенной водой атмосфере и до правильного выражения белка.
ПРИМЕЧАНИЕ: Здесь, 48 ч инкубации время используется для клеток HeLa для мониторинга эффективности трансфекции, используя tdTomato- и mVenus-выражения плазмиды. - Удалите среду культуры 48 ч после трансфекции путем инвертирования пластины, добавьте 30 л/также 10% формалина с помощью перистальтической жидкой обработчика (10 qL кассеты), и инкубировать в течение 15 минут при комнатной температуре.
- Удалите формалин, инвертируя пластину; затем, инкубировать клетки в течение 15 минут при комнатной температуре с 0,1 нг /мл Hoechst разбавленной в 1x PBS растворе.
- Вымойте клетки 3x в течение 15 минут с 80 qL 1x PBS скорректированы до рН No 8 для того, чтобы восстановить сигнал высокой флуоресценции потеряли 6,9 рН формелин раствор инкубации шаг.
- Используя автоматизированный флуоресцентный микроскоп, приобретайте изображения двух или трех флуоресцентных каналов (Hoechst, tdTomato, и mVenus) последовательно с 10x целями и надлежащим набором фильтров выбросов (4',6-diamidino-2-penylindole ,DAPI), dsRed и флуоресценсея изотиоцианат (FITC) соответственно).
- Для оценки эффективности трансфекции используйте программное обеспечение для анализа изображений для определения эффективности трансфекции с помощью анализа скриптов на основе окрашивания ядер.
Результаты
f Для того, чтобы определить, может ли технология ADE быть использована для автоматизированного протокола обратного трансфекции, мы отслеживали эффективность трансфекции клеток с помощью флуоресцентной микроскопии, используя красный флуоресцентный tdTomato, выражающий п...
Обсуждение
Создание и оптимизация точного метода трансфекции высокой пропускной связи для данной клеточной линии требует от ученых следовать некоторым ключевым параметрам, описанным в этом разделе. Мы настоятельно рекомендуем начать с рекомендуемых значений во всем протоколе, так как эти парам...
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Авторы раскрыли получение следующей финансовой поддержки для исследования, авторства и/или публикации этой статьи: Inserm, Лилльский университет, Институт Лилля Пастера, Conseil Regional du Nord, и PRIM-HCV1 и 2 (Пале де Рехерш Междисциплинарное сюр-ле-Медикамент), Национальная эгенство Речерче (ANR-10-E-PX-04-01), Федер (12001407 (D-AL) Equipex Imaginex BioMed) и Европейское сообщество (ERC-STG INTRACETB. Авторы хотели бы поблагодарить д-ра С. Моуре, д-ра Б. Виллеман, д-ра Р. Ферру-Клемента и д-ра Х. Гроульта за их критический обзор и исправление рукописи.
Материалы
| Name | Company | Catalog Number | Comments |
| 384LDV Microplate | Labcyte | LP-0200 | |
| 384-well Microplate μClear Black | Greiner | 781906 | |
| Ampicilin | Sigma | A9393-5G | Selection antibiotic for bacteria transformed with ampicilin expressing vector |
| Android Tablet | Samsung | Galaxy Note 8 | used to guide the user while the source plate manual dispense |
| Aniospray Surf 29 | Anios | 2421073 | disinfectant to clean the MicroFlo head |
| Columbus software | Perkin Elmer | image analysis software | |
| Dulbecco's Modified Eagle Medium (DMEM), high glucose, GlutaMAX Supplement, pyruvate | Thermo Fisher Scientific | 10566032 | cell culture medium |
| Echo Cherry Pick 1.5.3 software | Labcyte | Software enabling ADE-based dispenses by the Echo550 device from a *.csv file; nanodispenser software | |
| Echo550 | Labcyte | ADE-based dispenser | |
| Fetal Bovine Serum | Thermo Fisher Scientific | 16000044 | to add in cell culture medium |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | HT501128-4L | to fix cell |
| HeLa cells | ATCC | HeLa (ATCC® CCL-2™) | |
| Hoechst 33342, Trihydrochloride, Trihydrate | Thermo Fisher Scientific | H3570 | 10 mg/mL Solution in Water |
| INCell Analyzer 6000 | GE Healthcare | 29043323 | automated laser-based confocal imaging platform |
| LB medium | Thermoischer Scientific LB Broth Base (Lennox L Broth Base)®, powder | 12780052 | culture medium for bacteria growth |
| Lysis Buffer (A2) | Macherey-Nagel | 740912.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| MicroFlo 10µL cassette | Biotek Instruments Inc | 7170013 | to use with the Microflo Dispenser |
| MicroFlo 1μL cassette | Biotek Instruments Inc | 7170012 | to use with the Microflo Dispenser |
| MicroFlo Dispenser | Biotek Instruments Inc | 7171000 | peristaltic pump-based liquid handler device |
| Microvolume spectrophotometer | Denovix | DS-11 Spectrophotometer | Measure the DNA concentration of samples |
| mVenus plasmid | mVenus cDNA was cloned by enzymatic restriction digestion and ligation in Age1/BsrG1 sites of the tdTomato-N1 plasmid | Vector type: Mammalian Expression, Fusion Protein: mVenus | |
| Neutralization Buffer (A3) | Macherey-Nagel | 740913.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| NucleoSpin Plasmid kit | Macherey-Nagel | 740588.50 | used to prepare plasmid from bacterial culture |
| Optimal-Modified Eagle Medium (Opti-MEM) Medium | Thermo Fisher Scientific | 31985070 | |
| optional Wash bufferWash Buffer (A4) | Macherey-Nagel | 740914.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| orbital shaker | incubated large capacity shaker | 444-7084 | Used to grow bacteria under gentle agitation and 37°C |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140122 | 10,000 U/mL |
| Phosphate-Buffered Saline | Thermo Fisher Scientific | 10010001 | |
| Plasmid mini-columns | Macherey-Nagel | 740499.250 | Silica membrane mini-column to prepare plasmid from bacterial culture |
| Resuspension Buffer (A1) | Macherey-Nagel | 740911.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| RNAse A | Macherey-Nagel | 740505 | Enzyme from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| tdTomato-N1 plasmid | Addgene | Plasmid #54642 | Vector type: Mammalian Expression, Fusion Protein: tdTomato |
| TransIT-X2 Dynamic Delivery System | Mirus Bio | MIR 6000 | |
| Wash Buffer (AW) | Macherey-Nagel | 740916.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| 3D printer | Creality | CR10S | used to print the plate adapter |
| Blender Software | https://www.blender.org/ Free software under GNU General Public License (GPL). | version 2.79b | used to design the plate adapter |
Ссылки
- Colin, B., Deprez, B., Couturier, C. High-Throughput DNA Plasmid Transfection Using Acoustic Droplet Ejection Technology. SLAS Discovery: Advancing Life Sciences R & D. , 2472555218803064 (2018).
- Mirus Bio. . Optimising Transfection Performance. , (2019).
- Thermo Fisher Scientific. . Factors Influencing Transfection Efficiency | Thermo Fisher Scientific - FR. , (2019).
- Boussif, O., et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 92 (16), 7297-7301 (1995).
- Figueroa, E., et al. A mechanistic investigation exploring the differential transfection efficiencies between the easy-to-transfect SK-BR3 and difficult-to-transfect CT26 cell lines. Journal of Nanobiotechnology. 15 (1), 36 (2017).
- Kirchenbuechler, I., Kirchenbuechler, D., Elbaum, M. Correlation between cationic lipid-based transfection and cell division. Experimental Cell Research. 345 (1), 1-5 (2016).
- Zhang, Z., Qiu, S., Zhang, X., Chen, W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnology. 18 (1), 4 (2018).
- Cao, D., et al. Transfection activity and the mechanism of pDNA-complexes based on the hybrid of low-generation PAMAM and branched PEI-1.8k. Molecular bioSystems. 9 (12), 3175-3186 (2013).
- Bos, A. B., et al. Development of a semi-automated high throughput transient transfection system. Journal of Biotechnology. 180, 10-16 (2014).
- Colosimo, A., et al. Transfer and expression of foreign genes in mammalian cells. BioTechniques. 29 (2), 314-318 (2000).
- Villa-Diaz, L. G., Garcia-Perez, J. L., Krebsbach, P. H. Enhanced transfection efficiency of human embryonic stem cells by the incorporation of DNA liposomes in extracellular matrix. Stem Cells and Development. 19 (12), 1949-1957 (2010).
- Sabatini, D. M. . Reverse transfection method. , WO2001020015A1 (2001).
- Raymond, C., et al. A simplified polyethylenimine-mediated transfection process for large-scale and high-throughput applications. Methods (San Diego, CA). 55 (1), 44-51 (2011).
- Junquera, E., Aicart, E. Recent progress in gene therapy to deliver nucleic acids with multivalent cationic vectors. Advances in Colloid and Interface Science. 233, 161-175 (2016).
- Woodruff, K., Maerkl, S. J. A High-Throughput Microfluidic Platform for Mammalian Cell Transfection and Culturing. Scientific Reports. 6, 23937 (2016).
- Vasileiou, T., Foresti, D., Bayram, A., Poulikakos, D., Ferrari, A. Toward Contactless Biology: Acoustophoretic DNA Transfection. Scientific Reports. 6, 20023 (2016).
- Hadimioglu, B., Stearns, R., Ellson, R. Moving Liquids with Sound: The Physics of Acoustic Droplet Ejection for Robust Laboratory Automation in Life Sciences. Journal of Laboratory Automation. 21 (1), 4-18 (2016).
- Grant, R. J., et al. Achieving accurate compound concentration in cell-based screening: validation of acoustic droplet ejection technology. Journal of Biomolecular Screening. 14 (5), 452-459 (2009).
- Sackmann, E. K., et al. Technologies That Enable Accurate and Precise Nano- to Milliliter-Scale Liquid Dispensing of Aqueous Reagents Using Acoustic Droplet Ejection. Journal of Laboratory Automation. 21 (1), 166-177 (2016).
- Day, R. N., Davidson, M. W. The fluorescent protein palette: tools for cellular imaging. Chemical Society Reviews. 38 (10), 2887-2921 (2009).
- Zielinski, D., Gordon, A., Zaks, B. L., Erlich, Y. iPipet: sample handling using a tablet. Nature Methods. 11 (8), 784-785 (2014).
- Brunner, S., et al. Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Therapy. 7 (5), 401-407 (2000).
- Nii, T., et al. Single-Cell-State Culture of Human Pluripotent Stem Cells Increases Transfection Efficiency. BioResearch Open Access. 5 (1), 127-136 (2016).
- Noonan, D. J., Henry, K., Twaroski, M. L. A High-Throughput Mammalian Cell-Based Transient Transfection Assay. Signal Transduction Protocols. 284, 051-066 (2004).
- . . Transfection | TransIT Transfection Reagents | Mirus Bio. , (2015).
- American Type Culture Collection. . General protocol for transfection of stem cells, primary cells, and continuous cell lines with ATCC TransfeX Transfection Reagent. , (2017).
- American Type Culture Collection. . Transfection Reagents for Nucleic Acid Transfer into ATCC Cells. , (2017).
- de Los Milagros Bassani Molinas, M., Beer, C., Hesse, F., Wirth, M., Wagner, R. Optimizing the transient transfection process of HEK-293 suspension cells for protein production by nucleotide ratio monitoring. Cytotechnology. 66 (3), 493-514 (2014).
- Promega. . FuGENE® 6 Transfection Reagent. , (2019).
- Olden, B. R., Cheng, Y., Yu, J. L., Pun, S. H. Cationic polymers for non-viral gene delivery to human T cells. Journal of Controlled Release: Official Journal of the Controlled Release Society. 282, 140-147 (2018).
- Park, E., Cho, H. B., Takimoto, K. Effective gene delivery into adipose-derived stem cells: transfection of cells in suspension with the use of a nuclear localization signal peptide-conjugated polyethylenimine. Cytotherapy. 17 (5), 536-542 (2015).
- Wood, R. W., Loomis, A. L. The physical and biological effects of high-frequency sound-waves of great intensity. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science. 4 (22), 417-436 (1927).
- Mamat, U., et al. Eliminating Endotoxin at the Source - A Novel Competent Cell Line with Modified Lipopolysaccharide for Low-Endotoxin Plasmid Production. , (2014).
- Ivanova, N. V., Kuzmina, M. L. Protocols for dry DNA storage and shipment at room temperature. Molecular Ecology Resources. 13 (5), 890-898 (2013).
- Lesnick, J., Lejeune-Dodge, A., Ruppert, N., Jarman, C. . High-Precision Cell Dispensing with the Labcyte Echo® Liquid Handler. , (2017).
- Yang, X., et al. A public genome-scale lentiviral expression library of human ORFs. Nature Methods. 8 (8), 659-661 (2011).
- Peng, J., Zhou, Y., Zhu, S., Wei, W. High-throughput screens in mammalian cells using the CRISPR-Cas9 system. The FEBS journal. 282 (11), 2089-2096 (2015).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены