
Method Article
Моделирование активного сайта фермента с помощью бесплатного программного обеспечения для молекулярной визуализации
В этой статье
Резюме
Ключевым навыком в биомолекулярном моделировании является отображение и аннотирование активных сайтов в белках. Эта техника демонстрируется с помощью четырех популярных бесплатных программ для макромолекулярной визуализации: iCn3D, Jmol, PyMOL и UCSF ChimeraX.
Аннотация
Навыки биомолекулярной визуализации имеют первостепенное значение для понимания ключевых концепций в биологических науках, таких как структурно-функциональные отношения и молекулярные взаимодействия. Различные программы позволяют учащемуся манипулировать 3D-структурами, а биомолекулярное моделирование способствует активному обучению, развивает вычислительные навыки и преодолевает разрыв между двумерными изображениями учебника и тремя измерениями жизни. Критическим навыком в этой области является моделирование активного сайта белка, отображающего части макромолекулы, которые могут взаимодействовать с небольшой молекулой или лигандом таким образом, чтобы показать связывающие взаимодействия. В этом протоколе мы описываем этот процесс с помощью четырех свободно доступных программ макромолекулярного моделирования: iCn3D, Jmol/JSmol, PyMOL и UCSF ChimeraX. Это руководство предназначено для студентов, стремящихся изучить основы конкретной программы, а также инструкторов, включающих биомолекулярное моделирование в свою учебную программу. Протокол позволяет пользователю моделировать активный сайт с помощью определенной программы визуализации или выбрать несколько доступных бесплатных программ. Моделью, выбранной для этого протокола, является глюкокиназа человека, изоформа фермента гексокиназы, которая катализирует первую стадию гликолиза. Фермент связан с одним из своих субстратов, а также с нереакционноспособным аналогом субстрата, что позволяет пользователю анализировать взаимодействия в каталитическом комплексе.
Введение
Понимание представлений молекулярного мира имеет решающее значение для того, чтобы стать экспертом в биомолекулярных науках1, потомучто интерпретация таких изображений является ключом к пониманию биологической функции2. Введение учащегося в макромолекулы обычно происходит в виде двумерных хрестоматийных изображений клеточных мембран, органелл, макромолекул и т. Д., Но биологическая реальность заключается в том, что это трехмерные структуры, и понимание их свойств требует способов визуализации и извлечения смысла из 3D-моделей.
Соответственно, развитие биомолекулярной визуальной грамотности на курсах молекулярных наук о жизни высшего дивизиона привлекло внимание, с рядом статей, сообщающих о важности и трудностях обучения и оценки навыков визуализации1,3,4,5,6,7,8,9 . Ответом на эти статьи стало увеличение числа вмешательств в классе, как правило, в течение семестра в одном учреждении, где программы и модели молекулярной визуализации используются для нацеливания на сложные концепции2,10,11,12,13,14,15 . Кроме того, исследователи стремились охарактеризовать, как студенты используют программы и / или модели биомолекулярной визуализации для подхода к конкретной теме16,17,18,19. Наша собственная группа, BioMolViz, описала структуру, которая подразделяет всеобъемлющие темы визуальной грамотности на цели и задачи обучения, чтобы направлять такие вмешательства20,21,и мы проводим семинары, которые обучают преподавателей использовать Структуру в обратном дизайне оценок для измерения навыков визуальной грамотности22.
В центре всей этой работы находится критический навык: умение манипулировать структурами макромолекул с помощью программ для биомолекулярной визуализации. Эти инструменты разрабатывались независимо с использованием различных платформ; поэтому они могут быть довольно уникальными в своей работе и использовании. Это требует инструкций для конкретной программы, и идентификация программы, которая удобна пользователю, важна для облегчения дальнейшей реализации.
Помимо самых основ манипулирования структурами в 3D (вращение, выбор и изменение модели), основной целью является моделирование активного сайта белка. Этот процесс позволяет учащемуся развивать свое понимание в трех всеобъемлющих темах, описанных в структуре BioMolViz: молекулярные взаимодействия, лиганды / модификации и структурно-функциональные отношения20,21.
Четыре популярных варианта программ для биомолекулярной визуализации включают: Jmol / JSmol23,iCn3D24,PyMOL25и UCSF Chimera26,27. Мы призываем новичков в Chimera использовать UCSF ChimeraX, следующее поколение программы молекулярной визуализации Chimera, которая в настоящее время поддерживается версией программы.
В этом протоколе мы демонстрируем, как использовать каждую из этих четырех программ для моделирования активного сайта глюкокиназы человека с ассоциированным аналоговым комплексом субстрата (PDB ID: 3FGU) и для отображения измерений для иллюстрации конкретных связывающих взаимодействий28. Модель представляет собой каталитический комплекс фермента. Для захвата активного центра в состоянии прекатализа негидролизуемый аналог АТФ связывали с активным центром глюкокиназы. Этот эфир фосфоаминофосфоновой кислоты-аденилата (ANP) содержит фосфорно-азотную связь вместо обычной фосфорно-кислородной связи в этом положении. Активный сайт также содержит глюкозу (обозначенную БЦЖ в модели) и магний (обозначаемую МГ). Кроме того, в структуре присутствует ион калия (K), полученный из хлорида калия, используемого в кристаллизационном растворителе. Этот ион не критичен для биологической функции и расположен вне активного центра.

Рисунок 1:Структуры СПС/АНП. Структура аденозинтрифосфата (АТФ) по сравнению со фосфоаминофосфоновой кислотой-аденилатным эфиром (ANP). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
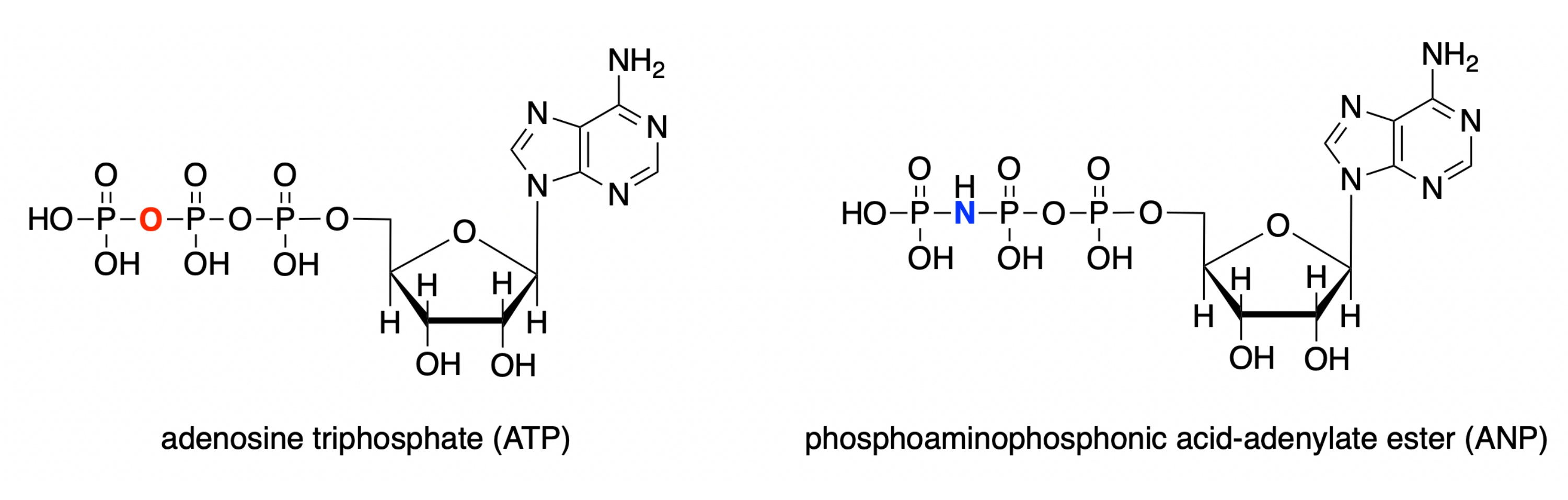{kind=link}
Протокол демонстрирует выбор связанных лигандов аналогового комплекса субстрата и идентификацию остатков активного сайта в пределах 5 Å связанного комплекса, который захватывает аминокислоты и молекулы воды, способные создавать соответствующие молекулярные взаимодействия, включая гидрофобные и ван-дер-Ваальсовые взаимодействия.
Дисплей первоначально манипулируется, чтобы показать большую часть белка в мультяшном представлении, с активными аминокислотными остатками сайта в представлении палочки, чтобы показать соответствующие атомы белка и выделить молекулярные взаимодействия. После шага 3 протокола для каждой программы эти представления были применены, и вид белка аналогичен в разных программах(рисунок 2). В конце протокола белковый мультфильм скрыт, чтобы упростить просмотр и сосредоточиться на активном сайте.

Рисунок 2:Сравнение структуры между программами. Сравнение структуры 3FGU в каждой программе после шага Adjust the Representation (шаг 2 или 3 каждого протокола). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
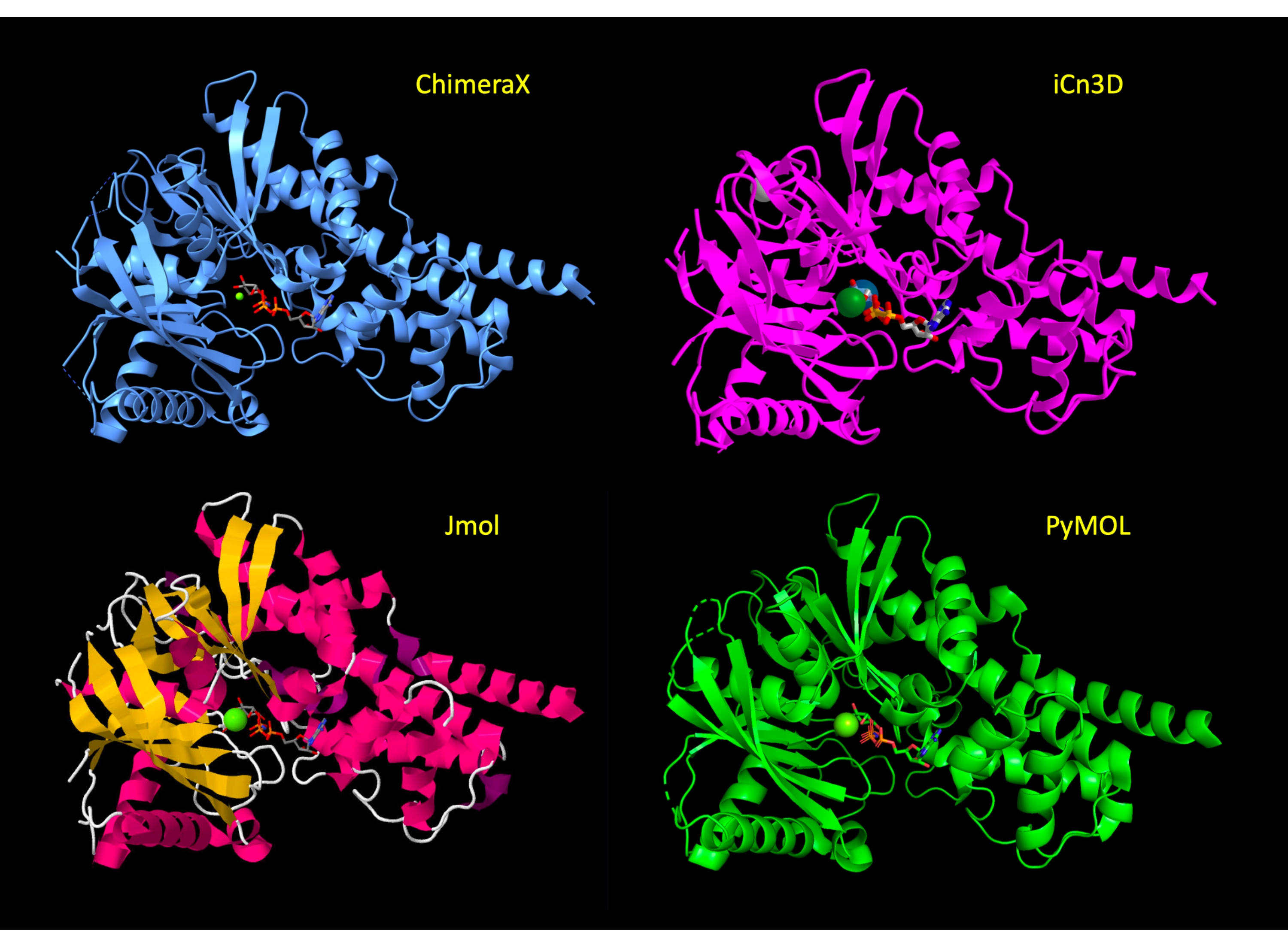{kind=link}
Краситель КФК наносится на активный центр аминокислот и связанных лигандов29,30. Эта цветовая схема различает атомы различных химических элементов в молекулярных моделях, показанных в линейных, палочных, шаровых и стиковых представлениях, а также в представлениях заполнения пространства. Водород белый, азот синий, кислород красный, сера желтая, а фосфор оранжевый в цветовой схеме CPK. Традиционно черный используется для углерода, хотя в современном использовании углеродная окраска может варьироваться.
Атомы водорода не видны в кристаллических структурах, хотя каждая из этих программ способна предсказать их местоположение. Добавление атомов водорода к большой высокомолекулярной структуре может скрыть вид, поэтому они не отображаются в этом протоколе. Соответственно, водородные связи будут показаны путем измерения от центра двух гетероатомов (например, кислород к кислороду, кислород к азоту) в этих структурах.
Обзоры программ
Загружаемые графические интерфейсы пользователя (GUI): PyMOL (версия 2.4.1), ChimeraX (версия 1.2.5) и Jmol (версия 1.8.0_301) являются инструментами молекулярного моделирования на основе графического интерфейса. Эти три интерфейса имеют командные строки для ввода типизированного кода; многие из тех же возможностей доступны через меню и кнопки в графическом интерфейсе. Общей особенностью командной строки этих программ является то, что пользователь может загружать и повторно выполнять предыдущие команды с помощью клавиш со стрелками вверх и вниз на клавиатуре.
Веб-графические интерфейсы: iCn3D (I-see-in-3D) - это средство просмотра на основе WebGL для интерактивного просмотра трехмерных макромолекулярных структур и химических веществ в Интернете без необходимости установки отдельного приложения. Он не использует командную строку, хотя полная веб-версия имеет редактируемый журнал команд. JSmol - это JavaScript или HTML5 версия Jmol для использования на веб-сайте или в окне веб-браузера, и очень похожа по работе на Jmol. JSmol можно использовать для создания онлайн-учебников, включая анимацию.
Proteopedia31,32,FirstGlance в Jmol33и веб-интерфейс JSmol (JUDE) в Центре биомолекулярного моделирования Школы инженерии Милуоки являются примерами таких онлайн-сред проектирования на основе Jmol34. Proteopedia wiki - это обучающий инструмент, который позволяет пользователю моделировать структуру макромолекул и создавать страницы с этими моделями на веб-сайте35. Инструмент создания сцен Proteopedia, созданный с использованием JSmol, интегрирует графический интерфейс с дополнительными функциями, недоступными в графическом интерфейсе Jmol.
Jmol и iCn3D основаны на языке программирования Java; JSmol использует Java или HTML5, а PyMOL и ChimeraX основаны на языке программирования Python. Каждая из этих программ загружает файлы банка белковых данных, которые можно загрузить из rcSB Protein Data Bank под 4-значным буквенно-цифровым PDB ID36,37. Наиболее распространенными типами файлов являются файлы Банка данных белка (PDB), содержащие расширение .pdb, и Файл кристаллографической информации (CIF или mmCIF), содержащий расширение .cif. CIF заменил PDB в качестве типа файла по умолчанию для банка данных белка, но оба формата файлов функционируют в этих программах. Могут быть небольшие различия в способе отображения последовательности/структуры при использовании CIF в отличие от PDB-файлов; тем не менее, файлы функционируют аналогично, и различия не будут подробно рассмотрены здесь. База данных молекулярного моделирования (MMDB), продукт Национального центра биотехнологической информации (NCBI), представляет собой подмножество структур PDB, с которыми связана категориальная информация (например, биологические особенности, сохраненные белковые домены)38. iCn3D, продукт NCBI, способен загружать PDB-файлы, содержащие данные MMDB.
Для просмотра модели пользователь может загрузить нужный файл с выделенной страницы Банка данных белка для структуры (например, https://www.rcsb.org/structure/3FGU),а затем использовать выпадающее меню Файл программы, чтобы открыть структуру. Все программы также способны загружать файл структуры непосредственно через интерфейс, и этот метод подробно описан в протоколах.
Графические интерфейсы ChimeraX, Jmol и PyMOL содержат одно или несколько окон консоли, размер которых можно изменить путем перетаскивания угла. iCn3D и JSmol полностью содержатся в веб-браузере. При использовании iCn3D пользователю может потребоваться прокрутка во всплывающих окнах, чтобы отобразить все пункты меню, в зависимости от размера экрана и разрешения.
Протоколы, подробно описанные здесь, предоставляют простой метод отображения активного сайта фермента с помощью каждой программы. Следует отметить, что существует несколько способов выполнения шагов в каждой программе. Например, в ChimeraX та же задача может быть выполнена с помощью раскрывающихся меню, панели инструментов вверху или командной строки. Пользователям, заинтересованным в детальном изучении конкретной программы, рекомендуется изучить онлайн-учебники, руководства и вики, доступные для этих программ39,40,41,42,43,44,45,46.
Существующие руководства и учебные пособия для этих программ представляют элементы этого протокола как дискретные задачи. Чтобы отобразить активный сайт, пользователь должен синтезировать необходимые операции из различных руководств и учебников. Эта рукопись дополняет существующие учебные пособия, представляя линейный протокол для моделирования помеченного активного сайта с молекулярными взаимодействиями, предоставляя пользователю логику для активного моделирования сайта, которая может быть применена к другим моделям и программам.

Рисунок 3:Графический интерфейс ChimeraX. Графический интерфейс ChimeraX с выпадающими меню, панелью инструментов, средством просмотра структуры и командной строкой. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 4:iCn3D GUI. Интерфейс iCn3D GUI с выпадающими меню, панелью инструментов, средством просмотра структуры, журналом команд, всплывающим окном выбора наборов, а также всплывающими меню последовательностей и аннотаций. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 5:Графический интерфейс Jmol. Интерфейс Jmol GUI с выпадающими меню, панелью инструментов, средством просмотра структуры, всплывающим меню и консолью / командной строкой с метками. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

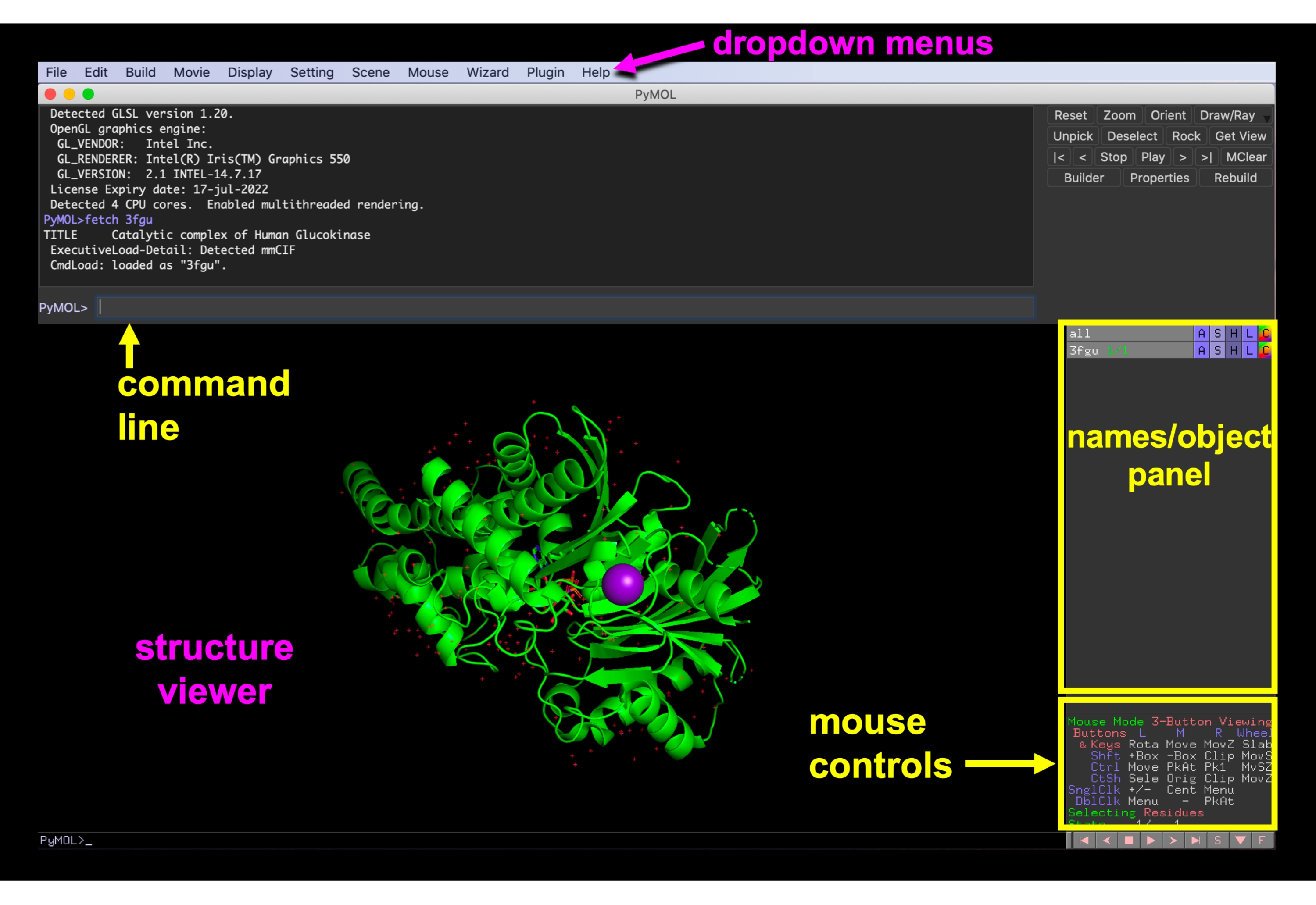
Рисунок 6:Графический интерфейс PyMOL. Графический интерфейс PyMOL с выпадающими меню, просмотрщиком структуры, панелью имен/объектов, меню управления мышью и командной строкой. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
протокол
ПРИМЕЧАНИЕ: Протокол для каждой программы состоит из десяти всеобъемлющих этапов: (1) Загрузка структуры в программу, (2) Идентификация лигандов в активном центре, (3) Корректировка представления, (4) Выбор остатков в пределах 5 Å для определения активного сайта, (5) Отображение взаимодействия фермента с лигандами активного сайта, (6) Отображение боковых цепей в виде палочек и отображение/корректировка молекул воды активного сайта, (7) Упрощение структуры, (8) Маркировка лигандов и водородно-связанных боковых цепей, (9) Сохранение рендеринга в любой момент, чтобы вернуться к работе над ним или поделиться с другими, (10) Сохранение изображения для встраивания или печати. Шаги 1, 4 и 7-10 идентичны для каждого протокола; однако, из-за уникальной работы каждой программы, некоторые протоколы более эффективно выполняются при обмене шагами 2/3 и 5/6.
1. Протокол UCSF ChimeraX
ПРИМЕЧАНИЕ: Управление трекпадом и мышью. Чтобы повернуть, щелкните и перетащите или используйте перетаскивание двумя пальцами (мышь: щелчок левой кнопкой мыши и перетаскивание). Масштабирование, сжатие и распространение (Mac) или управление + движение двумя пальцами (ПК) (мышь: колесо прокрутки). Чтобы перевести (т.е. переместить всю структуру), нажмите опцию + щелчок и перетащите (Mac) или shift + щелчок и перетащите (PC) (мышь: щелчок правой кнопкой мыши и перетаскивание). Чтобы перецентрировать, используйте раскрывающиеся меню в верхней части интерфейса, чтобы щелкнуть Действия > Вид.
- Загрузка структуры в ChimeraX: в командной строке, расположенной в нижней части графического интерфейса, которой предшествует "Command:", введите:
открыть 3fgu
ПРИМЕЧАНИЕ: После ввода любой введенной команды строки нажмите клавишу return на клавиатуре, чтобы выполнить ее. - Идентификация лигандов в активном сайте: убедитесь, что есть два представления, мультяшная лента и палочки. Используя мышь, поверните / масштабируйте белок, чтобы наилучшим образом визуализировать лиганды, отображаемые вблизи центра белка, которые показаны в виде палочек. Наведите курсор на лиганд, чтобы показать его название.
- Корректировка представления: используйте команды в подшагах ниже, чтобы перекрасить белок и лиганды, применить окраску CPK к атомам, не относящимся к углероду, а затем отменить выделение. Выбранные части молекулы подсвечиваются зеленым цветом.
- Используйте раскрывающиеся меню в верхней части интерфейса, чтобы изменить раскраску: Нажмите «Действия» > «Цвет» > «Васильково-синий». Затем нажмите «Выбрать > структуру > лиганда». Чтобы выбрать цвет, нажмите на Действия > Цвет > Серый. Чтобы применить раскраску CPK, нажмите «Выбрать > все»,а затем нажмите «Действия > цветом > by Heteroatom». Наконец, очистите выделенный фрагмент, щелкнув Выбрать > Очистить.
ПРИМЕЧАНИЕ: Выделение также может быть очищено нажатием клавиши CONTROL и щелчком на черном фоне средства просмотра структуры или в командной строке, введя: ~select. По умолчанию для большинства структур, содержащих 1-4 цепи, ChimeraX автоматически показывает молекулы воды и аминокислотные остатки в пределах 3,6 Å лигандов и ионов. - Используйте раскрывающееся меню, чтобы скрыть отображаемые в данный момент атомы, щелкнув Действия > Атомы / Связи > Скрыть.
- Используйте раскрывающееся меню, чтобы показать лиганды и Mg-ион в активном сайте, щелкнув Выбрать > структуру > лиганда. Затем нажмите на Действия > Атомы / Связи > Показать. Затем щелкните Выбрать остатки > > MG,а затем действия > атомов / связей > показать. Чтобы очистить выделенную область, нажмите выбрать > очистить.
ПРИМЕЧАНИЕ: После выбора с помощью выпадающего меню шаг 1.3.3 можно выполнить, нажав на кнопки Скрыть и Показать на панели инструментов Atoms.
- Используйте раскрывающиеся меню в верхней части интерфейса, чтобы изменить раскраску: Нажмите «Действия» > «Цвет» > «Васильково-синий». Затем нажмите «Выбрать > структуру > лиганда». Чтобы выбрать цвет, нажмите на Действия > Цвет > Серый. Чтобы применить раскраску CPK, нажмите «Выбрать > все»,а затем нажмите «Действия > цветом > by Heteroatom». Наконец, очистите выделенный фрагмент, щелкнув Выбрать > Очистить.
- Выбор остатков в пределах 5 Å для определения активного сайта: В средстве просмотра структуры для выбора лигандов нажмите control + shift и выполните щелчок мыши по любому отдельному атому или связи в каждом из трех лигандов, т.е. BCG, ANPи Mg.
- Затем нажимайте клавишу со стрелкой вверх на клавиатуре, пока все атомы трех лигандов не будут выделены зеленым свечением. Определите этот выбор для использования в будущем, щелкнув в раскрывающемся меню Выбрать > Определить селектор. Во всплывающем меню введите:
Лиганды, чтобы назвать текущий выбор, а затем нажмите OK.
ПРИМЕЧАНИЕ: Щелкнув стрелку вверх слишком много раз на шаге 1.4.1, вы выберете весь белок. В этом случае нажимайте кнопку со стрелкой вниз, пока не будут выбраны только атомы трех лигандов. - Используйте раскрывающееся меню, чтобы выбрать остатки в пределах 5 Å от лигандов: Нажмите Выбрать > зону. Во всплывающем окне переключите раскрывающееся меню Выбрать на значение Остаткии убедитесь, что установлен верхний флажок (установите флажок расстояние меньше (<) и установите значение 5,0 Å). Затем нажмите OK. Будут выделены только остатки, которые находятся на расстоянии менее 5 Å.
ПРИМЕЧАНИЕ: Шаги 1.4-1.4.2 можно значительно упростить с помощью командной строки, введя:
название замороженных лигандов :BGC:MG:ANP
Выбрать зону лигандов 5 расширить истинные остатки true
- Затем нажимайте клавишу со стрелкой вверх на клавиатуре, пока все атомы трех лигандов не будут выделены зеленым свечением. Определите этот выбор для использования в будущем, щелкнув в раскрывающемся меню Выбрать > Определить селектор. Во всплывающем меню введите:
- Отображение боковых цепочек в виде палочек и отображение / настройка активных молекул воды сайта: Используйте раскрывающееся меню для отображения выделения и центра и масштабирования выделения, щелкнув Действия > Атомы / Связи > Показать, чтобы показать их. Чтобы центрировать выделение, нажмите «Действия» > «Вид». Затем, чтобы очистить выделенный фрагмент, нажмите «Выбрать > «Очистить» или щелкните в любом месте пустого пространства.
- Отображение взаимодействия фермента с активными лигандами сайта: Используйте раскрывающиеся меню и нажмите «Выбрать > пользовательских селекторов > лигандов. Затем нажмите на Инструменты > Структурный анализ > H-bonds. Во всплывающем окне убедитесь, что установлен флажок Ограничить по выделению, в раскрывающемся меню установлено значение С выбранным хотя бы одним концом, установлен флажок Выбрать атомы, а затем нажмите кнопку ОК. Чтобы очистить выделенную область, нажмите выбрать > очистить.
ПРИМЕЧАНИЕ: Установите флажок Метка расстояния, чтобы увидеть длину связи в Å; однако это делает представление очень занятым. Наконец, вы можете изменить цвет H-bonds, нажав на поле «Цвет» и выбрав новый цвет во всплывающем окне. - Упрощение структуры: используйте верхнюю панель инструментов мультфильма, чтобы скрыть мультфильм или нажмите на выпадающее меню: Действия > Мультфильм > Скрыть.
- Маркировка лигандов и водородно-связанных боковых цепей: Используйте мышь для выбора остатков, которые являются водородом, связанными с лигандами (соединенными пунктирными линиями), как на этапе 1.4. Затем в раскрывающихся меню нажмите «Действия», > «Метка > остатки > комбо «Имя». Затем нажмите «Выбрать > пользовательских селекторов > лигандов». Затем нажмите «Действия», > «Метить остатки >, > выкл. Наконец, очистите выделенный фрагмент, нажав на Выбрать > Очистить.
- Сохранение рендеринга в любой момент, чтобы вернуться к работе над ним или поделиться с другими: в выпадающем меню выберите Файл > Сохранить. Выберите расположение, введите имя файла и нажмите кнопку Сохранить.
ПРИМЕЧАНИЕ: Убедитесь, что формат установлен на: Сеанс ChimeraX *.cxs. - Сохранение изображения для встраивания или печати: сначала используйте мышь, чтобы сориентировать молекулу по желанию. Измените цвет фона на белый, введя в командной строке:
набор bgЦвет белый
Наконец, нажмите на значок снимка на панели инструментов. Изображение будет сохранено на рабочем столе.
ПРИМЕЧАНИЕ: Цвет фона также доступен в раскрывающемся меню; на Mac нажмите на UCSF ChimeraX > настройки; на ПК нажмите «Избранное» > «Настройки» > «Фон».
2. Протокол iCn3D
ПРИМЕЧАНИЕ: Элементы управления трекпадом и мышью:Чтобы повернуть, щелкните и перетащите (мышь: левый щелчок и перетаскивание). Масштабирование, сжатие и распределение (мышь: поворот колеса прокрутки). Для перевода (т.е. перемещения всей структуры) щелкните и перетащите двумя пальцами (мышь: щелкните правой кнопкой мыши и перетащите). Чтобы перецентрировать, наведите указатель мыши на Вид в верхних раскрывающихся меню, а затем щелкните Выбор центра.
- Загрузка структуры в iCn3D: перейдите в iCn3D Web-средство просмотра 3D-структур и введите 3FGU в поле Input MMDB или PDB ID для загрузки файла.
- Идентификация лигандов в активном сайте: наведите указатель мыши на Анализ в раскрывающемся меню, а затем щелкните Seq. и Аннотации. Последовательности, в данном случае Белки и Химические/Ионы/Вода, показаны в сложенной таблице. Прокрутите вниз, чтобы увидеть активные лиганды сайта ANP, BGC и Mg в списке. В средстве просмотра структуры наведите курсор на лиганды в активном месте (показаны в виде палочек в центре белкового мультфильма), чтобы просмотреть их названия.
- Корректировка представления: Для этого протокола не требуется никаких первоначальных корректировок.
- Выбор остатков в пределах 5 Å для определения активного сайта: Чтобы выбрать лиганды, используйте раскрывающееся меню Выбрать и нажмите Выбрать на 3D. Убедитесь, что установлен флажок Остаток .
- Чтобы выбрать лиганды, удерживайте кнопку ALT на ПК или кнопку Option на Mac и нажмите на первый лиганд (например, BCG)с помощью мыши или трекпада. Затем нажмите control и нажмите на лиганды ANP и MG, чтобы добавить их в выбор.
ПРИМЕЧАНИЕ: Лиганды будут подсвечиваться желтым цветом по мере их выбора. - Сохраните этот выбор с помощью раскрывающегося меню: нажмите «Выбрать > сохранить выбор» и с помощью клавиатуры введите имя во всплывающем окне (например, 3 Лиганды),а затем нажмите «Сохранить». Появится всплывающее окно «Выбор наборов».
ПРИМЕЧАНИЕ: Если выбор неправильный, нажмите «Выбрать > «Очистить выделение». - Выберите остатки в пределах 5 Å от лигандов: В раскрывающемся меню нажмите Выбрать > по расстоянию. Во всплывающем меню измените второй пункт (Сфера с радиусом) на 5 Å, введя блок. Нажмите на слово Displayв поле, а затем закройте окно, нажав на крестовый знак в правом верхнем углу.
ПРИМЕЧАНИЕ: Во всплывающем меню, которое появляется на шаге 2.4.3, оставьте первый набор с вводом «выбрано», а второй набор как «не выбран». Обратите внимание, что атомы/структуры в пределах 5 Å подсвечиваются желтым свечением при нажатии кнопки Display. - Сохраните сайт 5 Å active с помощью выпадающего меню: наведите курсор на Select и нажмите Save Selection, введите имя во всплывающем окне с помощью клавиатуры (например, 5Ang)и нажмите Save.
- Затем создайте новый выбор, объединяющий два набора (5Ang и 3Ligands): во всплывающем меню «Выбрать наборы» щелкните, удерживая нажатой клавишу CTRL (PC) или удерживая нажатой клавишу Command (Mac) 5Ang и 3Ligands. Нажмите Выбрать > Сохранить выбор,используйте клавиатуру, чтобы ввести имя (например, 5AFull),а затем нажмите Сохранить.
- Чтобы выбрать лиганды, удерживайте кнопку ALT на ПК или кнопку Option на Mac и нажмите на первый лиганд (например, BCG)с помощью мыши или трекпада. Затем нажмите control и нажмите на лиганды ANP и MG, чтобы добавить их в выбор.
- Показывая взаимодействие фермента с активными лигандами сайта, такими как водородные связи: наведите курсор на Анализ в раскрывающемся меню и нажмите «Взаимодействия». Появится полное всплывающее меню всех нековалентных взаимодействий.
- Снимите все флажки, кроме флажков «Водородные связи» и «Соляной мост/Ионик». Нажмите на 3 Лиганды, чтобы выбрать первый набор и 5Ang для второго набора. Нажмите на текст в коробке, который читает взаимодействия с 3D-дисплеем. Закройте окно, нажав на крестный знак в правом верхнем углу.
ПРИМЕЧАНИЕ: Контакт/Взаимодействия, по-видимому, представляют собой индуцированное диполь-индуцированное дипольное взаимодействие, которое часто делает дисплей занятым. При желании измените расстояние для любого типа взаимодействия. - Чтобы отобразить только водородные связи, нажмите на 5Afull во всплывающем окне выбора наборов. Затем наведите указатель мыши на Анализ в раскрывающемся меню, а затем щелкните Chem. Привязка > Показать.
- Снимите все флажки, кроме флажков «Водородные связи» и «Соляной мост/Ионик». Нажмите на 3 Лиганды, чтобы выбрать первый набор и 5Ang для второго набора. Нажмите на текст в коробке, который читает взаимодействия с 3D-дисплеем. Закройте окно, нажав на крестный знак в правом верхнем углу.
- Отображение боковых цепочек в виде палочек и отображение / настройка активных молекул воды сайта: Используйте всплывающее меню выбора наборов и нажмите на 5AFull. Затем в раскрывающихся меню нажмите Style > Side Chains > Stick. Чтобы применить раскраску CPK, нажмите на Color > Atom. Наконец, нажмите на Style > Water > Spheres (если вы предпочитаете более крупные молекулы воды).
- Упрощение структуры: во всплывающем меню выбора наборов нажмите на 5AFull. Затем в раскрывающихся меню нажмите View > View Selection (чтобы просто увидеть сайт привязки 5AFull). Затем нажмите style > Proteins > Stick (чтобы показать белковую цепь как палочку вместо ленты).
- Чтобы окрасить атомы углерода лигандов в контрастный цвет, нажмите «Химические вещества» во всплывающем окне «Выбор наборов». Затем в раскрывающемся меню нажмите Просмотреть > Просмотр выбора. Затем нажмите выбрать > Выбрать на 3D (убедитесь, что установлен флажок «атом»). Используя элементы управления, описанные в шаге 2.4.1, используйте мышь и клавиатуру для выбора всех атомов углерода в BGC и ANP. Затем в раскрывающемся меню нажмите на Color > Unicolor > Cyan > Cyan.
- Чтобы повторно отобразить весь активный сайт, используйте всплывающее окно «Выбор наборов», чтобы щелкнуть 5AFull. Затем в раскрывающемся меню нажмите «Просмотреть > «Просмотреть выбор».
- Маркировка лигандов и водородно-связанных боковых цепей: используйте всплывающее окно выбора наборов, чтобы выбрать Interface_all,а затем в раскрывающемся меню нажмите «Анализ > метка > на остаток и число».
ПРИМЕЧАНИЕ: Вам придется повторно выбирать Per Residue & Number каждый раз, когда вы хотите добавить метку, даже если пункт меню уже будет отмечен из предыдущей метки. - Сохранение рендеринга в любой момент, чтобы вернуться к работе над ним или поделиться с другими: В раскрывающемся меню нажмите Файл > Поделиться Ссылкой. Скопируйте короткий URL-адрес (например, https://structure.ncbi.nlm.nih.gov/icn3d/share.html?r83NqCz41bu7cmcs8) и вставьте его в браузер.
- Сохранение изображения для встраивания или печати: в раскрывающемся меню нажмите Выбрать > Переключить выделение. Затем нажмите на Стиль > Фон > Белый. Наконец, нажмите «Файл > «Сохранить файлы» > iCn3D PNG Image и выберите нужный размер.
3. Протокол Jmol
ПРИМЕЧАНИЕ: Элементы управления трекпадом и мышью: Чтобы повернуть, щелкните и перетащите (мышь: левый щелчок и перетаскивание). Для масштабирования прокрутите по вертикали двумя пальцами (мышь: shift + щелчок левой кнопкой мыши + перетаскивание по вертикали). Для перевода (т.е. перемещения всей структуры) элемента управления + ALT + щелчок и перетаскивание (ПК), управление + опция + щелчок и перетаскивание (Mac). Для повторного центрирования: shift + двойной щелчок в пустом пространстве окна просмотра структуры.
- Загрузка структуры в Jmol: используйте раскрывающееся меню в верхней части графического интерфейса, чтобы настроить рабочее пространство со структурой, щелкнув Файл > Консоли. Затем нажмите на Файл > Получить PDB. Во всплывающем окне введите: 3fgu
Затем нажмите OK.
ПРИМЕЧАНИЕ: В качестве альтернативы, используйте консоль Jmol для загрузки структуры, набрав: load = 3fgu
ПРИМЕЧАНИЕ: После ввода любой введенной команды строки нажмите клавишу return на клавиатуре, чтобы выполнить ее. - Настройка представления: откройте всплывающее меню, щелкнув правой кнопкой мыши (или элемент управления + щелчок) в любом месте окна средства просмотра структур.
- Чтобы изменить белок на мультяшное представление, во всплывающем меню нажмите выбрать > Выбор ореолов. Затем нажмите «Выбрать > белок > «Все». Наконец, нажмите на Стиль > Схема > Мультфильм.
ПРИМЕЧАНИЕ: Выделение гало помещает желтый контур (свечение) вокруг всех выбранных атомов. - Используйте верхнее раскрывающееся меню, чтобы скрыть воды, нажав на Display > Select > Water. Затем щелкните Display > Atom > Noneи, наконец, нажмите Display > Select > None.
- Чтобы изменить белок на мультяшное представление, во всплывающем меню нажмите выбрать > Выбор ореолов. Затем нажмите «Выбрать > белок > «Все». Наконец, нажмите на Стиль > Схема > Мультфильм.
- Идентификация лигандов в активном сайте: используйте мышь, чтобы увеличить масштаб активного сайта, а затем используйте команды на подшагах, чтобы отобразить лиганды в виде палочек.
ПРИМЕЧАНИЕ: Имена лигандов отображаются в консоли Jmol при загрузке файла. Вы также можете просмотреть связанные названия лигандов с помощью всплывающего меню, нажав на Выбрать > Hetero > HETATM.- Наведите указатель мыши на лиганды, чтобы просмотреть их имена. Активный участок находится вблизи центра сооружения; лиганды MG, BGC и ANP расположены в активном месте.
- Выберите лиганды BCG и ANP: Используя консоль Jmol, введите:
выберите БГК, АНП - Чтобы отобразить лиганды BCG и ANP в виде палочек, используйте всплывающее меню и нажмите «Стиль > схемы» > Sticks.
- Выбор остатков в пределах 5 Å для определения активного сайта: В консоли Jmol введите следующую команду, чтобы выбрать атомы в пределах 5 Å из трех лигандов:
выбрать в пределах (5, (bgc,anp,mg))- Чтобы выбрать полные аминокислотные остатки, введите в консоли следующую команду и нажмите клавишу ВВОД
выбрать внутри(группа, выбрано)
ПРИМЕЧАНИЕ: Консоль Jmol является лучшим способом выбора остатков в пределах 5 Å.
- Чтобы выбрать полные аминокислотные остатки, введите в консоли следующую команду и нажмите клавишу ВВОД
- Отображение боковых цепочек в виде палочек и отображение / настройка активных молекул воды сайта: щелкните правой кнопкой мыши, чтобы открыть всплывающее меню, и наведите курсор на Style > Scheme > Sticks.
ПРИМЕЧАНИЕ: Шаг 3.5 показывает активные боковые цепи сайта в представлении палочки. В структуре все еще будут некоторые пустые ореолы, которые представляют молекулы воды в активном центре.- В консоли Jmol повторно выполните следующую команду:
выбрать в пределах (5, (bgc,anp,mg))
ПРИМЕЧАНИЕ: Чтобы повторно выполнить команду, щелкните в консоли, а затем используйте клавиши со стрелками на клавиатуре, пока эта команда не появится, и нажмите enter, чтобы повторно выполнить ее. - Чтобы отобразить атомы молекулы воды, удалите лиганды и белок из выделения, набрав следующие две команды:
выбрать удалить группу белка
выберите удалить группу гетеро, а не воду - Чтобы отобразить молекулы воды, нажмите на выпадающее меню Дисплей. Наведите курсор на Atom и нажмите на 20% van der Waals. Зеленые ионы магния по-прежнему будут показаны в виде палочек. Отображение иона магния в более распространенном представлении сферы путем ввода следующих команд в консоли Jmol:
выберите Mg
заполнение пространства 50% - Перекрасьте лиганды, чтобы отличить их от белка: в консоли Jmol введите следующую команду, чтобы выполнить команду, которая перекрашивает лиганды в более светлой цветовой схеме:
выбрать (bgc,anp) и углерод; цвет [211,211,211]
выделение (bgc, anp) и кислорода; цвет [255,185,185]
отбор (bgc,anp) и азот; цвет [150,210,255]
выделение (bgc,anp) и фосфора; цвет [255,165,75]
выберите Mg; цвет бледно-зеленый
- В консоли Jmol повторно выполните следующую команду:
- Показывая взаимодействие фермента с активными лигандами сайта: С помощью консоли Jmol выполните каждую строку следующей команды:
определить лигбинд (ANP, BGC, MG)
выбрать в пределах (5, (bgc,anp,mg))
выберите удалить группу гетеро, а не воду- Чтобы отобразить строки для иллюстрации водородных связей, введите следующую команду в консоли Jmol:
соединить 3.3 (лигбинд и (кислород или азот)) (выбрано и (кислород или азот)) стойка желтая
Затем измените толщину строк, введя в консоли следующую команду:
выбрать все; стойка 0,1; выберите нет
- Чтобы отобразить строки для иллюстрации водородных связей, введите следующую команду в консоли Jmol:
- Упрощение структуры: Чтобы скрыть мультфильм о белке и очистить выделение, в консоли Jmol введите:
выбрать все; мультфильм выключен; выберите нет - Маркировка лигандов и водородно-связанных боковых цепей: во всплывающем окне нажмите «Установить выбор > «Выбрать атом». Нажмите на атом в одном из водородных связанных остатков. Аминокислоты и остатки отображаются в консоли. Затем с помощью консоли введите метку, например:
этикетка Glu-256 - Сохранение рендеринга в любой момент, чтобы вернуться к работе над ним или поделиться с другими: В верхнем меню нажмите на значок камеры. Введите имя файла и выберите папку для сохранения.
ПРИМЕЧАНИЕ: Экспортированный файл JPEG (.jpg) содержит информацию как для изображения, отображаемого в окне отображения во время экспорта, так и для текущего состояния модели. Чтобы перезагрузить модель, откройте Jmol и перетащите сохраненный файл JPEG в окно отображения Jmol. - Сохранение изображения для внедрения или печати: в консоли Jmol перекрасьте фон в белый цвет, введя:
фон белый
Как и в шаге 3.9, нажмите на значок камеры и сохраните файл.
4. Протокол PyMOL
ПРИМЕЧАНИЕ: Элементы управления трекпадом и мышью: Чтобы повернуть, щелкните и перетащите (мышь: левый щелчок и перетаскивание). Масштабирование, сжатие и распределение (мышь: щелкните правой кнопкой мыши и перетащите). Чтобы перевести (т.е. переместить всю структуру), управление + щелчок и перетаскивание (мышь: команда + левый щелчок и перетаскивание). Для повторного центрирования перейдите на правую панель объектов и нажмите на A > Orient или Center.
- Загрузка структуры в PyMOL: в командной строке в верхней части графического интерфейса (перед "PyMOL>") введите:
получить 3FGU
ПРИМЕЧАНИЕ: После ввода любой введенной команды строки нажмите клавишу return на клавиатуре, чтобы выполнить ее. - Настройка представления: В панели имен /объектов в правой части окна PyMOL, справа от «3FGU», нажмите на H > Waters.
- Идентификация лигандов в активном сайте: сначала включите средство просмотра последовательностей, щелкнув в верхнем раскрывающемся меню: Отображение > последовательности.
- Прокрутите серую полосу вправо, пока не найдете названия лигандов (BCG, ANP, MG, K).
ПРИМЕЧАНИЕ: Есть два изображения, мультяшная лента и палочки; лиганды показаны в виде палочек. Убедитесь, что режим выбора в элементах управления мышью на нижней правой панели установлен в режим Остаточный и 3-кнопочный режим просмотра, щелкнув по этим именам, чтобы переключиться между параметрами. - С помощью мыши поверните и масштабируйте, чтобы сделать лиганды видимыми.
- Прокрутите серую полосу вправо, пока не найдете названия лигандов (BCG, ANP, MG, K).
- Выбор остатков в пределах 5Å для определения активного сайта: Чтобы выбрать лиганды в активном сайте, нажмите на каждый из них(BCG, ANP, MG)в средстве просмотра структуры. На панели имен/объектов появится новая выделенная область; справа от этого нового объекта с именем «sele» нажмите на кнопку A, а затем нажмите «Переименовать» во всплывающем меню.
ПРИМЕЧАНИЕ: Чтобы очистить нежелательный выбор, щелкните пустое место в средстве просмотра структуры, чтобы отменить выбор.- С помощью клавиатуры удалите буквы «sele», которые появляются в верхней левой части окна просмотра структуры, и вместо них введите:
Лигандов
ПРИМЕЧАНИЕ: Шаги 4.4-4.4.1 можно выполнить с помощью командной строки; тип:
селе лиганды, резн BGC+ANP+MG - Используйте этот выбор, чтобы определить область вокруг лигандов, сначала дублируя ее, нажмите на лиганды > A > Duplicate. Затем нажмите на sel01 > A > Переименовать
С помощью клавиатуры удалите буквы "se101" и введите:
активный - Измените эту выделенную область, чтобы отобразить остатки в пределах 5 Å: На панели имен/объектов щелкните активные > A > Изменить > Развернуть > на 5 A, Остатки. Затем, чтобы показать эти остатки в виде палочек, нажмите на активные палочки > S > солодки >. Наконец, щелкните пустое место в средстве просмотра структур, чтобы очистить выделенную область.
ПРИМЕЧАНИЕ: Шаг 4.4.3 можно выполнить с помощью командной строки, введите:
селе активный, byres все в пределах 5 лигандов
показать палочки, активные
- С помощью клавиатуры удалите буквы «sele», которые появляются в верхней левой части окна просмотра структуры, и вместо них введите:
- Отображение боковых цепей в виде палочек и отображение / корректировка активных молекул воды сайта: На панели имен / объектов нажмите на лиганды > A > Duplicate. Чтобы переименовать выделенную область, нажмите на Sel02 > A > Переименовать выбор. Удалите буквы в меню переименования, которое отображается в правом верхнем углу средства просмотра структуры, и введите:
active_water- Чтобы настроить новый выбор для содержания активных молекул воды, нажмите на active_water > A > Изменить > вокруг > атомов в пределах 4 ангстрем. Чтобы изменить это дальше и ограничить молекулы воды, нажмите на active_water > A > Изменить > ограничить > растворителем. Наконец, нажмите на active_water > A > Предустановленный > ball and Stick.
ПРИМЕЧАНИЕ: Графический интерфейс позволяет выбирать в пределах 4 Å; команды line позволяют выбрать более подходящее расстояние 3,3 Å для молекул воды, связанных водородом. Радиусы ван-дер-Ваальса сфер не могут быть установлены в графическом интерфейсе, но выбор «мяч и палка» близок к 0,5 Å.
ПРИМЕЧАНИЕ: Шаги 4.5-4.5.1 могут быть выполнены с помощью командной строки, путем ввода каждой строки следующего кода:
выберите active_water, ((лиганды) около 3.3) и (resn HOH)
показать сферы, active_water
alter active_water, vdw=0.5
перестраивать
- Чтобы настроить новый выбор для содержания активных молекул воды, нажмите на active_water > A > Изменить > вокруг > атомов в пределах 4 ангстрем. Чтобы изменить это дальше и ограничить молекулы воды, нажмите на active_water > A > Изменить > ограничить > растворителем. Наконец, нажмите на active_water > A > Предустановленный > ball and Stick.
- Показаны взаимодействия фермента с активными лигандами сайта. Увеличьте масштаб активного сайта, нажав на активную > A > Zoom. Чтобы найти полярные контакты между лигандами и активным сайтом, нажмите на лиганды > A > Найти > полярные контакты > к любым атомам. Отображение расстояний в виде меток, нажав на ligands_polar_contacts > S > Метки.
- Упрощение структуры: Скрыть мультфильм белка, который скрывает часть белка, которая не находится в активном сайте, нажав на 3FGU > H > Cartoon в панели имен /объектов. Затем скройте метки длины водородной связи, нажав на ligands_polar_contacts > H > Метки на панели имен / объектов.
- Чтобы окрасить лиганды, чтобы дифференцировать их от белка, нажмите на лиганды > C > по элементам > CHNOS и выберите вариант, где «C» голубой (светло-голубой).
ПРИМЕЧАНИЕ: Шаг 4.7.1 может быть выполнен с помощью командной строки. Тип:
цвет голубой, лиганды
цвет атомарный, лиганды и !elem C
- Чтобы окрасить лиганды, чтобы дифференцировать их от белка, нажмите на лиганды > C > по элементам > CHNOS и выберите вариант, где «C» голубой (светло-голубой).
- Маркировка лигандов и водородно-связанных боковых цепей: На панели имен/объектов на кнопках справа от названия любого объекта нажмите на активную > L > Остатки.
- Сохранение рендеринга в любой момент, чтобы вернуться к работе над ним или поделиться с другими: В раскрывающемся меню выберите Файл > Сохранить сеанс как. Затем выберите местоположение во всплывающем окне, введите имя файла и нажмите «Сохранить».
- Сохранение изображения для встраивания или печати: во-первых, измените фон на белый в раскрывающемся меню, щелкнув Display > Background > White. Экспортируйте изображение как новый файл, нажав на Файл > Экспорт изображения как > PNG.
Результаты
Успешно выполненный протокол для каждой из программ приведет к молекулярной модели, увеличенной на активном сайте, с остатками активного сайта и лигандами, показанными в виде палочек, скрытым белковым мультфильмом и лигандами, отображаемыми с контрастной цветовой схемой. Взаимодействующие аминокислотные остатки должны быть помечены их идентификаторами, а водородные связи и ионные взаимодействия показаны линиями. Наличие этих особенностей можно определить при визуальном осмотре модели.
Чтобы облегчить эту проверку и позволить пользователю определить, правильно ли он выполнил шаги протокола, мы предоставили анимированные рисунки, которые представляют изображение структуры после каждого шага. Для ChimeraX, iCn3D, Jmol и PyMOL это проиллюстрировано на рисунках 7-10соответственно.
Рисунок 7:Вывод протокола ChimeraX. Анимированный рисунок, иллюстрирующий шаги 1.1-1.8 протокола ChimeraX. Пожалуйста, нажмите здесь, чтобы загрузить этот рисунок.
Рисунок 8:Вывод протокола iCn3D. Анимированный рисунок, иллюстрирующий шаги 2.1-2.8 протокола iCn3D. Пожалуйста, нажмите здесь, чтобы загрузить этот рисунок.
Рисунок 9:Вывод протокола Jmol. Анимированный рисунок, иллюстрирующий шаги 3.1-3.8 протокола Jmol. Пожалуйста, нажмите здесь, чтобы загрузить этот рисунок.
Рисунок 10:Вывод протокола PyMOL. Анимированный рисунок, иллюстрирующий шаги 4.1-4.8 протокола PyMOL. Пожалуйста, нажмите здесь, чтобы загрузить этот рисунок.
Наиболее распространенной ошибкой, которая может повлиять на результат этих протоколов, является ошибочный выбор, в результате чего часть структуры отображается в нежелательном рендеринге. Обычно это результат неправильного щелчка либо на самой структуре, либо на одной из кнопок меню отображения. Примером неоптимального результата может быть модель, содержащая остатки за пределами активного сайта, отображаемые в виде палочек. Пользователь может начать анализировать, произошла ли эта ошибка, визуально осмотрев остатки, отображаемые в виде палочек, и убедившись, что они находятся в непосредственной близости от активных лигандов сайта. Усовершенствованный метод оценки того, находятся ли отображаемые остатки в пределах 5Å от лигандов активного сайта, заключается в использовании инструментов измерения, встроенных в каждую программу, для измерения расстояния между соседним лигандом и остатком активного сайта. Измерительные инструменты выходят за рамки данной рукописи; тем не менее, мы рекомендуем заинтересованным пользователям изучить множество онлайн-учебников, подробно описывающих этот тип анализа.
Приведем конкретный пример неоптимального выполнения этого протокола, возникшего в результате неправильного щелчка по панели имен/объектов в PyMOL. Эта ошибка отображает весь белок в виде палочек, а не показывает только активный сайт с помощью этого представления, как показано на рисунке 11.

Рисунок 11:Отрицательный результат. Пример отрицательного результата. Неправильный выбор полного мультфильма в PyMOL и отображение палочек. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Для устранения неполадок пользователю необходимо скрыть флешки для всей модели (помеченные 3FGU на панели имен/объектов), а затем показать представление джойстика только для выделения с именем «активный», используя кнопки скрытия и отображения / команд в PyMOL. Восстановление модели после этого типа ошибки относительно просто, как только пользователь может создать соответствующие выборки для разных частей модели, а также эффективно отображать и скрывать их. Заманчиво перезапустить протокол и проработать шаги в другой раз; Тем не менее, мы призываем пользователя не бояться выходить «за рамки сценария» и экспериментировать с моделью. По нашему опыту, работа с ошибками отображения облегчает прогресс в понимании программы моделирования.
Параллельное отображение конечных выходных данных успешно выполненного протокола для каждой программы показано на рисунке 12. Представления ориентированы одинаково, чтобы пользователь мог сравнивать внешний вид моделей, созданных в разных программах.

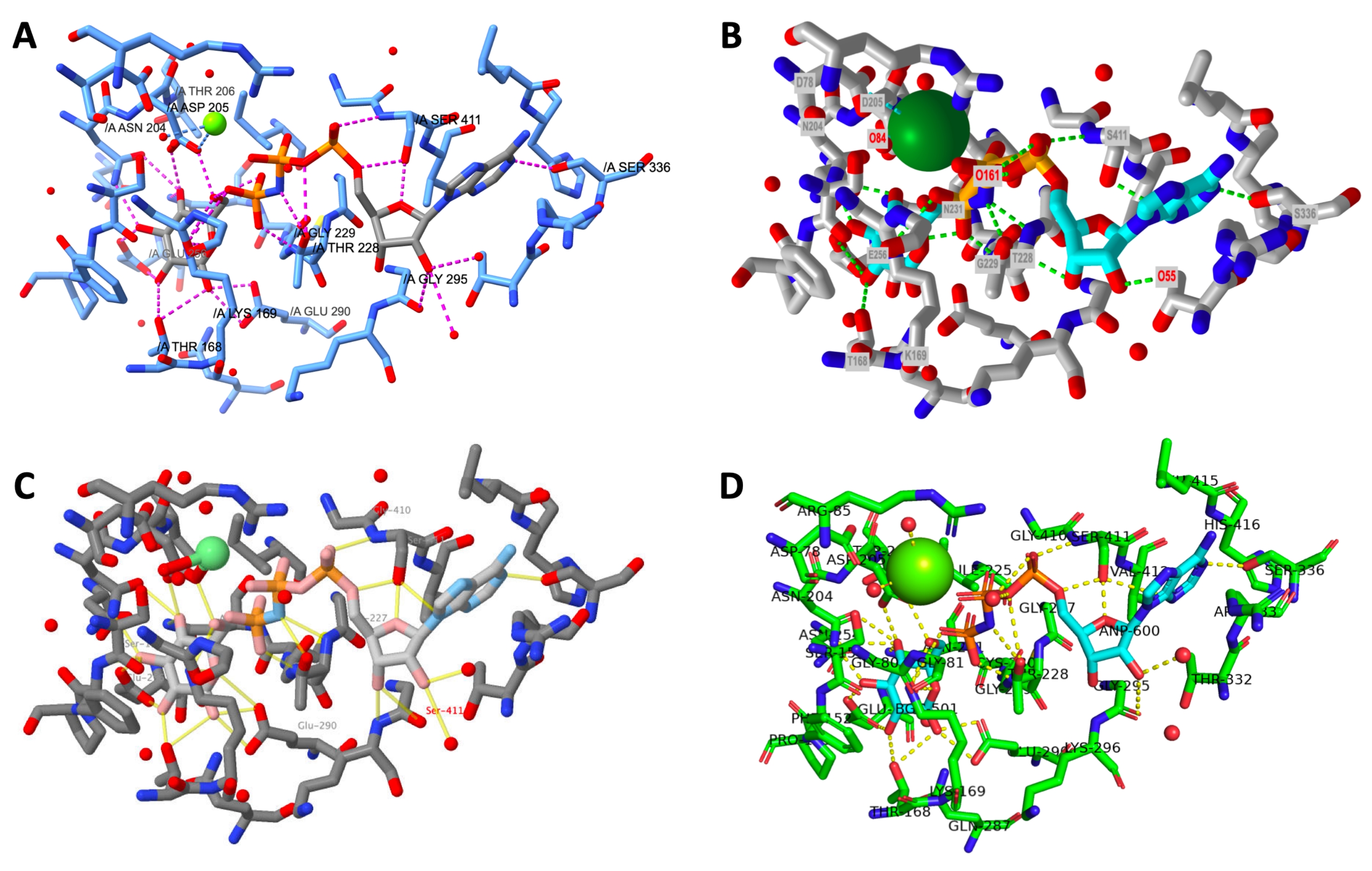
Рисунок 12:Окончательное сравнение структуры по программам. Сравнение структуры рендеринга каждого активного сайта в конце протокола. A: ChimeraX, B: iCn3D, C: Jmol, D: PyMOL. Метка активного сайта PyMOL включает все остатки активного сайта и лиганды. Другие выходы имеют только водородные связанные боковые цепи. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Обсуждение
Этот протокол описывает десятиэтапный процесс моделирования активного центра фермента, применяемый к четырем популярным программам биомолекулярного моделирования. Критическими этапами протокола являются: идентификация лигандов в активном центре, выбор остатков в пределах 5 Å для определения активного сайта и демонстрация взаимодействия фермента с лигандами активного центра. Различение лигандов, относящихся к биологической функции, имеет первостепенное значение, поскольку это позволяет пользователю определить аминокислотные остатки в пределах 5 Å, которые могут играть роль в связывании лигандов. Наконец, использование программы для отображения молекулярных взаимодействий позволяет пользователю развивать навыки, необходимые для понимания молекулярных взаимодействий, которые способствуют связыванию.
Ограничением компьютерных протоколов молекулярного моделирования является зависимость от конкретных команд и синтаксиса. В то время как биохимические протоколы могут быть терпимы к небольшим изменениям в процедуре, компьютерные исследования могут дать совершенно разные конечные продукты, если процедура не соблюдается строго. Это особенно важно при использовании интерфейсов командной строки, где для достижения определенного результата требуется синтаксис конкретной программы, а кажущееся незначительным изменение пунктуации или заглавных букв может привести к сбою команды. Существуют различные вики и руководства для каждой программы, где пользователь может найти и устранить неполадки ввода командной строки; пользователь должен обратить пристальное внимание на детали синтаксиса команды. Хотя большинство программ молекулярной визуализации включают команды отмены, из-за сложности интерфейсов команда отмены не всегда точно отменяет последний выполненный шаг. Поэтому сохранение текущего рабочего состояния часто поощряется, особенно для новых пользователей.
Дополнительные ограничения могут возникнуть из-за данных, используемых для создания самой модели. В то время как стандарты, присущие банку данных белка, обеспечивают определенный уровень согласованности, пользователи программ молекулярной визуализации часто сталкиваются с неожиданными эффектами при рендеринге белка. Во-первых, большинство структур определяются с помощью рентгеновской кристаллографии, которая обеспечивает единую модель белка; однако структуры ЯМР часто состоят из нескольких моделей, которые могут быть визуализированы по одной. Во-вторых, структуры, определенные в результате экспериментов по кристаллографии или криогенной электронной микроскопии, могут содержать атомы, положение которых не может быть выяснено, и появляться в виде пробелов в определенных представлениях белка. Белковые структуры могут иметь альтернативные конформации боковых цепей, которые при отображении в виде палочки выглядят как две группы, выступающие из одной и той же аминокислотной основы. Даже короткие участки позвоночника могут иметь такие альтернативные конформации, и иногда лиганды накладываются в активном центре в более чем одной конформации связывания.
Для кристаллической структуры осажденные 3D-координаты включают все компоненты асимметричной единицы, что дает достаточно информации для воспроизведения повторяющейся единицы кристалла белка. Иногда эта структура будет содержать дополнительные белковые цепи по сравнению с биологически активной формой белка (например, мутант фетального гемоглобина, PDB ID: 4MQK). И наоборот, некоторые программы могут не загружать автоматически все цепочки биологически активного блока. Например, основная протеаза SARS-CoV2 (PDB ID: 6Y2E) загружает половину биологически активного димера (состоящего из двух белковых цепей) при извлечении с помощью команд, описанных в этом протоколе в ChimeraX, PyMOL и Jmol. Хотя небольшая модификация команды загрузит биологически активный димер, это соображение может быть непростым для начинающего пользователя программы моделирования. Другой вопрос, который может возникнуть, заключается в идентификации активного сайта или самого субстрата. Кристаллографические эксперименты проводятся с использованием различных молекул, которые могут быть смоделированы в конечную структуру. Например, молекулы сульфата могут связывать фосфатные сайты связывания в активном центре или они могут связывать другие области, которые не имеют отношения к механизму. Эти молекулы могут скрывать правильную идентификацию самого активного сайта и могут даже предложить студенту, что они являются частью механизма.
Предположительно, пользователь захочет применить эту процедуру к другим активным/обязательным сайтам. Чтобы применить этот протокол в будущей работе, включающей анализ новых белковых активных сайтов, пользователю необходимо будет определить, какие из связанных лигандов имеют отношение к функционированию. Некоторые лиганды не связаны с функцией белка, а вместо этого являются результатом условий растворителя или кристаллизации, используемых для проведения эксперимента (например, ион калия, присутствующий в модели 3FGU). Ключевые лиганды следует определить, обратившись к оригинальной рукописи. С практикой и, где это применимо, пониманием синтаксиса команды строки, пользователь сможет применить протокол для нужной программы моделирования к любому активному сайту фермента и смоделировать другие макромолекулы по своему выбору.
Идентификация и анализ связанных субстратов и лигандов имеет центральное значение для выяснения молекулярных механизмов и структурных усилий по разработке лекарств, которые непосредственно привели к улучшению лечения заболеваний, включая синдром приобретенного иммунодефицита (СПИД) и COVID-1947,48,49,50,51,52 . В то время как отдельные программы молекулярной визуализации предлагают различные интерфейсы и пользовательский опыт, большинство из них предлагают сопоставимые функции. Для развития грамотности биомолекулярной визуализации важно, чтобы студенты высшего уровня биохимии ознакомились с визуализацией структуры и инструментами для генерации таких изображений4,20,53. Это позволяет студентам выйти за рамки интерпретации двумерных изображений в учебниках и журнальных статьях и легче разрабатывать свои собственные гипотезы из структурных данных54,что подготовит ученых-разработчиков к решению будущих проблем общественного здравоохранения и улучшению понимания биохимических процессов.
Таким образом, этот протокол подробно описывает активное моделирование сайта с использованием четырех ведущих бесплатных программ макромолекулярного моделирования. Наше сообщество, BioMolViz, использует непрограммный подход к биомолекулярному моделированию. Мы специально избегали критики или сравнения функций программы, хотя пользователь, выборка каждой программы, скорее всего, обнаружит, что он предпочитает определенные аспекты макромолекулярного моделирования в одной программе по сравнению с другой. Мы приглашаем читателей использовать структуру BioMolViz, в которой подробно описываются цели и задачи обучения на основе биомолекулярной визуализации, поставленные в этом протоколе, и изучить ресурсы для преподавания и изучения биомолекулярной визуализации через веб-сайт сообщества BioMolViz по адресу http://biomolviz.org.
Раскрытие информации
Авторы заявляют, что у них нет соответствующих или материальных финансовых интересов, которые относятся к исследованиям, описанным в этой статье.
Благодарности
Финансирование этой работы было предоставлено Национальным научным фондом:
Грант на улучшение stem-образования для студентов (Награда No 1712268)
Сети координации исследований в бакалавриате в области биологического образования (награда No 1920270)
Мы благодарны Карстену Тайсу, доктору философии Вестфилдского университета, за полезные дискуссии о Jmol.
Материалы
| Name | Company | Catalog Number | Comments |
| ChimeraX (Version 1.2.5) https://www.rbvi.ucsf.edu/chimerax/ | |||
| Computer | Any | ||
| iCn3D (web-based only: https://www.ncbi.nlm.nih.gov/Structure/icn3d/full.html) | |||
| Java (for Jmol) https://java.com/en/download/ | |||
| Jmol (Version 1.8.0_301) http://jmol.sourceforge.net/ | |||
| Mouse (optional) | Any | ||
| PyMOL (Version 2.4.1 - educational): https://pymol.org/2 educational use only version: https://pymol.org/edu/?q=educational |
Ссылки
- Loertscher, J., Green, D., Lewis, J. E., Lin, S., Minderhout, V. Identification of threshold concepts for biochemistry. CBE Life Sciences Education. 13 (3), 516-528 (2014).
- Jaswal, S. S., O’Hara, P. B., Williamson, P. L., Springer, A. L. Teaching structure: Student use of software tools for understanding macromolecular structure in an undergraduate biochemistry course: Teaching structure in undergraduate biochemistry. Biochemistry and Molecular Biology Education. 41 (5), 351-359 (2013).
- Tibell, L. A. E., Rundgren, C. -. J. Educational challenges of molecular life science: Characteristics and implications for education and research. CBE Life Sciences Education. 9 (1), 25-33 (2010).
- Schönborn, K. J., Anderson, T. R. The importance of visual literacy in the education of biochemists. Biochemistry and Molecular Biology Education. 34 (2), 94-102 (2006).
- Anderson, T. R. Bridging the educational research-teaching practice gap: The importance of bridging the gap between science education research and its application in biochemistry teaching and learning: Barriers and strategies. Biochemistry and Molecular Biology Education. 35 (6), 465-470 (2007).
- Schönborn, K. J., Anderson, T. R. Bridging the educational research-teaching practice gap: Foundations for assessing and developing biochemistry students’ visual literacy. Biochemistry and Molecular Biology Education. 38 (5), 347-354 (2010).
- Bateman, R. C., Craig, P. A. Education corner: A proficiency rubric for biomacromolecular 3D literacy. PDB Newsletter. 45, 5-7 (2010).
- Mnguni, L., Schönborn, K., Anderson, T. Assessment of visualization skills in biochemistry students. South African Journal of Science. 112, 1-8 (2016).
- Craig, P. A., Michel, L. V., Bateman, R. C. A survey of educational uses of molecular visualization freeware. Biochemistry and Molecular Biology Education. 41 (3), 193-205 (2013).
- Loertscher, J., Villafañe, S. M., Lewis, J. E., Minderhout, V. Probing and improving student’s understanding of protein α-Helix structure using targeted assessment and classroom interventions in collaboration with a faculty community of practice. Biochemistry and Molecular Biology Education. 42 (3), 213-223 (2014).
- Abualia, M., et al. Connecting protein structure to intermolecular interactions: A computer modeling laboratory. Journal of Chemical Education. 93 (8), 1353-1363 (2016).
- Carvalho, I., Borges, A. D. L., Bernardes, L. S. C. Medicinal chemistry and molecular modeling: An integration to teach drug structure–activity relationship and the molecular basis of drug action. Journal of Chemical Education. 82 (4), 588 (2005).
- Forbes-Lorman, R. M., et al. Physical models have gender-specific effects on student understanding of protein structure-function relationships. Biochemistry and Molecular Biology Education. 44 (4), 326-335 (2016).
- Terrell, C. R., Listenberger, L. L. Using molecular visualization to explore protein structure and function and enhance student facility with computational tools. Biochemistry and Molecular Biology Education. 45 (4), 318-328 (2017).
- Zhang, S., et al. Structure-based drug design of an inhibitor of the SARS-CoV-2 (COVID-19) main protease using free software: A tutorial for students and scientists. European Journal of Medicinal Chemistry. 113390, (2021).
- Roberts, J. R., Hagedorn, E., Dillenburg, P., Patrick, M., Herman, T. Physical models enhance molecular three-dimensional literacy in an introductory biochemistry course. Biochemistry and Molecular Biology Education. 33 (2), 105-110 (2005).
- Jenkinson, J., McGill, G. Visualizing protein interactions and dynamics: Evolving a visual language for molecular animation. CBE Life Sciences Education. 11 (1), 103-110 (2012).
- Bussey, T. J., Orgill, M. What do biochemistry students pay attention to in external representations of protein translation? The case of the Shine–Dalgarno sequence. Chemistry Education Research and Practice. 16 (4), 714-730 (2015).
- Harle, M., Towns, M. H. Students’ understanding of primary and secondary protein structure: Drawing secondary protein structure reveals student understanding better than simple recognition of structures. Biochemistry and Molecular Biology Education. 41 (6), 369-376 (2013).
- Dries, D. R., et al. An expanded framework for biomolecular visualization in the classroom: Learning goals and competencies. Biochemistry and Molecular Biology Education. 45 (1), 69-75 (2017).
- Procko, K., et al. Meeting report: BioMolViz workshops for developing assessments of biomolecular visual literacy. Biochemistry and Molecular Biology Education. 49 (2), 278-286 (2021).
- Wang, J., et al. iCn3D, a web-based 3D viewer for sharing 1D/2D/3D representations of biomolecular structures. Bioinformatics. 36 (1), 131-135 (2020).
- PyMOL . . The PyMOL Molecular Graphics System. Version 2.0. , (2021).
- Goddard, T. D., et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Science. 27 (1), 14-25 (2018).
- Pettersen, E. F., et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science. 30 (1), 70-82 (2021).
- Petit, P., et al. The active conformation of human glucokinase is not altered by allosteric activators. Acta Crystallographica. Section D. 67 (11), 929-935 (2011).
- Corey, R. B., Pauling, L. Molecular models of amino acids, peptides and proteins. Review of Scientific Instruments. 24, 621-627 (1953).
- Koltun, W. L. Precision space-filling atomic models. Biopolymers. 3 (6), 665-679 (1965).
- Hodis, E., et al. Proteopedia - a scientific 'wiki' bridging the rift between three-dimensional structure and function of biomacromolecules. Genome Biology. 9 (8), 1-10 (2008).
- Prilusky, J., et al. Proteopedia: A status report on the collaborative, 3D web-encyclopedia of proteins and other biomolecules. Journal of Structural Biology. 175 (2), 244-252 (2011).
- . FirstGlance in Jmol Available from: https://www.bioinformatics.org/firstglance/fgij/ (2021)
- Jmol User Design Environment (JUDE). MSOE Centerfor BioMolecular Modeling Available from: https://cbm.msoe.edu/modelingResources/jmolUserDesignEnvironment/#forward (2021)
- Castro, C. R., et al. A practical guide to teaching with Proteopedia. Biochemistry and Molecular Biology Education. 49 (5), 707-719 (2021).
- Berman, H. M., et al. The protein data bank. Nucleic Acids Research. 28, 235-242 (2000).
- . The Protein Data Bank Available from: https://www.rcsb.org/ (2021)
- Wang, Y., et al. MMDB: 3D structure data in Entrez. Nucleic Acids Research. 28 (1), 243-245 (2000).
- . iCn3D Help Page Available from: https://www.ncbi.nlm.nih.gov/Structure/icn3d/docs/icn3d_help.html (2021)
- . MSOE Center for BioMolecular Modeling Jmol Training Guide Available from: https://cbm.msoe.edu/modelingResources/jmolTrainingGuide/started.html (2021)
- . Jmol/JSmol Interactive Scripting Documentation Available from: https://chemapps.stolaf.edu/jmol/docs/ (2021)
- . PyMOL Wiki Available from: https://pymolwiki.org/index.php/Main_Page (2021)
- . PyMOL Advanced Scripting Workshop by Schrödinger Available from: https://pymol.org/tutorials/scripting/index.html (2021)
- . UCSF ChimeraX User Guide Available from: https://www.cgl.ucsf.edu/chimerax/docs/user/index.html (2021)
- . UCSF ChimeraX Tutorials Available from: https://www.rbvi.ucsf.edu/chimerax/tutorials.html (2021)
- Kuntz, I. D. Structure-based strategies for drug design and discovery. Science. 257 (5073), 1078-1082 (1992).
- Hubbard, R. E. . Structure-based drug discovery: an overview. , (2006).
- Patrick, G. L. . An introduction to medicinal chemistry, 6th ed. , (2017).
- Van Montfort, R. L., Workman, P. Structure-based drug design: aiming for a perfect fit. Essays in Biochemistry. 61 (5), 431-437 (2017).
- Holdgate, G. A., Meek, T. D., Grimley, R. L. Mechanistic enzymology in drug discovery: a fresh perspective. Nature Reviews. Drug Discovery. 17 (2), 115-132 (2018).
- Wang, M. Y., et al. SARS-CoV-2: structure, biology, and structure-based therapeutics development. Frontiers in Cellular and Infection Microbiology. 10, (2020).
- White, B., Kim, S., Sherman, K., Weber, N. Evaluation of molecular visualization software for teaching protein structure differing outcomes from lecture and lab: Differing outcomes from lecture and lab. Biochemistry and Molecular Biology Education. 30 (2), 130-136 (2002).
- Canning, D. R., Cox, J. R. Teaching the structural nature of biological molecules: Molecular visualization in the classroom and in the hands of students. Chemistry Education Research and Practice. 2 (2), 109-122 (2001).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены