
Method Article
Редактирование генов первичных В-клеток макак-резусов
В этой статье
Резюме
Мы представляем метод культивирования и редактирования генов первичных В-клеток макак-резусов с использованием CRISPR/Cas9 и рекомбинантного аденоассоциированного вируса серотипа 6 для изучения терапии В-клетками.
Аннотация
В-клетки и их потомство являются источниками высокоэкспрессируемых антител. Их высокая способность экспрессировать белок вместе с их изобилием, легким доступом через периферическую кровь и способностью к простым приемным переносам сделали их привлекательной мишенью для подходов к редактированию генов для экспрессии рекомбинантных антител или других терапевтических белков. Редактирование генов первичных В-клеток мыши и человека является эффективным, и мышиные модели для исследований in vivo показали многообещающие результаты, но осуществимость и масштабируемость для более крупных моделей животных до сих пор не были продемонстрированы. Поэтому мы разработали протокол редактирования первичных В-клеток макак-резусов in vitro , чтобы сделать возможными такие исследования. Мы сообщаем об условиях культивирования in vitro и редактирования генов первичных В-клеток макак-резуса из мононуклеарных клеток периферической крови или спленоцитов с использованием CRISPR/Cas9. Для достижения адресной интеграции больших (<4,5 кб) кассет был включен быстрый и эффективный протокол для подготовки рекомбинантного аденоассоциированного вируса серотипа 6 в качестве гомологически направленного шаблона репарации с использованием самоподавляющего аденовирусного вектора-помощника с поддержкой тетрациклина. Эти протоколы позволяют изучать перспективную терапию В-клетками у макак-резусов.
Введение
В-клетки являются основой гуморального иммунитета. При активации родственным антигеном и вторичными сигналами наивные В-клетки дают начало В-клеткам зародышевого центра, В-клеткам памяти и плазматическим клеткам1. Последний является источником секретируемых антител, которые опосредуют защитные функции большинства доступных в настоящее время вакцин2. Плазматические клетки были описаны как фабрики антител, поскольку они выделяют огромное количество антител в сыворотку - около 2 нг / день / клетка3, что составляет 7-16 г / л сыворотки, что делает антитела одним из трех наиболее распространенных белков в сыворотке4. В-клетки в изобилии содержатся в крови и, таким образом, могут быть легко получены и введены обратно в человека.
Эти черты сделали В-клетки мишенью клеточной терапии для редактирования генов рецептора В-клеток (BCR) и экспрессии широко нейтрализующих антител (bNAbs) к вирусу иммунодефицита человека (ВИЧ)5,6,7,8,9,10,11,12,13,14,15 и другим белкам 16, 17,18,19,20,21. Такие подходы показали потенциал в многочисленных исследованиях на мышах in vivo 7,8,10,11,16,22. Тем не менее, для клинического перевода 9,15,23 все еще необходимо преодолеть несколько препятствий, среди которых безопасность, продолжительность и величина терапевтической эффективности, а также масштабирование на более крупных животных, таких как нечеловекообразные приматы (NHP). Действительно, NHP и, в частности, макаки-резусы, которые имеют долгую историю исследований антител и ВИЧ24,25, являются наиболее подходящей моделью для проверки этих параметров.
Здесь мы разработали протоколы, которые позволяют решать эти проблемы. На сегодняшний день в нескольких исследованиях предпринимались попытки культивировать В-клетки макак-резуса ex vivo, и сообщалось только о положительном отборе с использованием CD20 для очистки В-клеток макак-резусов26,27,28. Мы разработали протокол выделения нетронутых В-клеток макак-резусов путем отрицательного истощения других типов клеток. Кроме того, определены условия культивирования для целенаправленного редактирования генов В-клеток макак-резусов. В этом протоколе описывается использование рибонуклеопротеинов (РНП) CRISPR/Cas9 и рекомбинантного аденоассоциированного вируса серотипа 6 (rAAV6) в качестве гомологически направленного шаблона репарации (HDRT) для редактирования генов культивируемых В-клеток макаки-резуса. С помощью этого протокола была достигнута эффективность редактирования до 40% при больших (~1,5 кб) вставках. Мы также представляем быстрый и экономичный способ получения rAAV6 с использованием самоподавляющего аденовирусного хелпера29 с поддержкой тетрациклина, чтобы обеспечить быстрое тестирование HDRT в этом формате. В совокупности эти протоколы описывают эффективный рабочий процесс редактирования генов В-клеток макак-резусов (рис. 1), что позволяет оценивать терапию В-клетками в модели NHP.
Для начала экспериментов донорский материал может быть заказан из коммерческих источников или получен с помощью флеботомии или спленэктомии. В этом исследовании флеботомия и забор крови были выполнены, как описано ранее30 , с использованием антикоагулянта ЭДТА. Для получения В-клеток селезенки, первичных макак-резусов, частичная (25-50%) или полная спленэктомия проводилась с использованием методов, о которых сообщалось ранее31. Животные голодали в течение ночи перед операцией. Вкратце, во время операции брюшная полость была обрезана и подготовлена чередующимися скрабами из хлоргексидина и 70% изопропилового спирта три раза. В брюшной полости был сделан разрез (5-10 см) для идентификации и изоляции селезенки. Сосудистую сеть селезенки перевязывали либо швами, либо сосудистыми зажимами. Разрез был закрыт в два слоя полидиоксаноновыми швами 4-0 PDS. Спленэктомия была выполнена один раз для отдельного животного. Одноклеточные суспензии получали из селезенки макак путем мацерации через клеточные ситечки. Мононуклеарные клетки из суспензий клеток крови и селезенки готовили с использованием центрифугирования градиента плотности и хранили в жидком азоте.
протокол
Все процедуры и эксперименты на животных проводились в соответствии с протоколами, утвержденными Комитетом по уходу за животными и их использованию Национального института аллергии и инфекционных заболеваний, Национальных институтов здравоохранения. Краткое изложение следующих протоколов представлено на рисунке 1. Самцы и самки макак-резусов (Macaca mulatta) индийского генетического происхождения в возрасте от 2 до 8 лет содержались и ухаживали за ними в соответствии с руководящими принципами Комитета по уходу за лабораторными животными и их использованию на объекте 2-го уровня биобезопасности.
ВНИМАНИЕ: Все эксперименты проводились с соблюдением универсальных мер предосторожности в отношении патогенов, передающихся через кровь, с использованием стерильных/асептических методов и надлежащего оборудования уровня биобезопасности 2 в вытяжных шкафах с ламинарным потоком.
1. Производство rAAV6
- Подготовьте реагенты для производства rAAV6.
- Спроектируйте и клонируйте шаблон репарации, направленный на гомологию, между инвертированными концевыми повторами (ITR) AAV2 в векторе pAAV с использованием стандартных методов. Убедитесь, что плечи гомологии имеют не менее ~ 250.н. с каждой стороны, но может быть достаточно всего 60.н., хотя более длинные плечи гомологии предпочтительнее, если конструкция конструкции позволяет это. Если в HDRT присутствуют целевые последовательности какой-либо из используемых сгРНК, удалите их с помощью тихих мутаций, которые наиболее эффективны в соседнем мотиве протоспейсера или затравочной области целевого сайта.
ПРИМЕЧАНИЕ: Синтез генов в сочетании со сборкой Гибсона для эффективного клонирования32 может быть выполнен. Подготовьте Maxiprep правильного клона для трансфекции. Для дизайна sgRNA рекомендуется CHOPCHOP33, а список других инструментов можно найти на https://zlab.bio/guide-design-resources. Максимальная емкость упаковки для AAV, включая РМЭ, составляет ~ 4,7 КБ. AAV6 является наиболее часто используемым серотипом для редактирования гемопоэтических клеток, особенно В-клеток9. Другие серотипы AAV для редактирования генов В-клеток макак-резусов не тестировались, но AAV28 и AAV-DJ10,11 использовались в исследованиях на мышах. - Подготовьте питательную среду 293AAV и продукционную среду в соответствии с таблицей 1 и таблицей 2. Стерильный фильтр через мембранный фильтрующий блок из полиэфирсульфона (PES) 0,2 мкм. Хранить при температуре 4 °C.
- Приготовьте 1x раствор полиэтиленимина (PEI) (1 мг / мл, 100 мл).
- В стеклянном стакане объемом 250 мл нагрейте ~ 70 мл H2O в микроволновой печи в течение ~ 30 с, а затем добавьте 100 мг PEI. Добавьте магнитную мешалку и перемешивайте, пока PEI не растворится.
- Отрегулируйте pH до 7 с помощью 1 M HCl, затем долейте до 100 мл H2O, подождите 10 минут, снова проверьте pH и при необходимости отрегулируйте.
- Стерильно отфильтруйте раствор PEI через мембранный фильтрующий блок PES 0,2 мкм, аликвоту и храните при температуре -20 °C. После размораживания раствор можно хранить при температуре 4 °C до 2 месяцев.
- Приготовьте 5-кратный раствор полиэтиленгликоля (ПЭГ)/NaCl.
- Взвесьте 400 г ПЭГ, 8 000 и 24 г NaCl.
- Добавьте магнитную мешалку в стеклянный стакан объемом 2 л, добавьте взвешенный PEG 8,000 и NaCl и промойте ~ 550 мл деионизированной воды.
- Перемешайте при нагревании и доведите до кипения или 80-90 °C до полного растворения.
- Отрегулируйте pH до ~ 7,4 с помощью 1 М NaOH, затем отрегулируйте объем до 1 л с помощью мерного цилиндра и перелейте его в стеклянную бутылку объемом 2 л с помощью магнитной мешалки.
- Автоклавную бутылку, магнитную мешалку и раствор на водяной бане в течение 30 мин при 121 °С.
- После автоклавирования охладите раствор в холодном помещении, помешивая, используя магнитную мешалку, чтобы предотвратить разделение на разные фазы. При необходимости аликвотировать и хранить при температуре 4 °C.
- Подготовьте буфер для рецептуры.
- Смешайте 500 мл DPBS с 50 мкл 10% Pluronic F-68. Стерильный фильтр через мембранный фильтрующий блок PES 0,2 мкм и хранение при комнатной температуре (RT).
- Спроектируйте и клонируйте шаблон репарации, направленный на гомологию, между инвертированными концевыми повторами (ITR) AAV2 в векторе pAAV с использованием стандартных методов. Убедитесь, что плечи гомологии имеют не менее ~ 250.н. с каждой стороны, но может быть достаточно всего 60.н., хотя более длинные плечи гомологии предпочтительнее, если конструкция конструкции позволяет это. Если в HDRT присутствуют целевые последовательности какой-либо из используемых сгРНК, удалите их с помощью тихих мутаций, которые наиболее эффективны в соседнем мотиве протоспейсера или затравочной области целевого сайта.
- Клеточная культура, трансфекция и трансдукция для производства rAAV6
- Размораживают, культивируют и замораживают клетки 293AAV, как описано производителем, используя вышеуказанную питательную среду 293AAV и трипсин-ЭДТА для расщепления. Рекомендуется заморозить некоторые ранние проходы и использовать клетки для производства AAV до того, как они достигнут прохода 40.
- Для производства rAAV6 посейте четыре чашки для клеточных культур диаметром 15 см по 5 x 106 клеток по 30 мл каждая. Клетки готовы к трансфекции обычно через 1-2 дня после посева, когда они достигают 80%-90% слияния.
- Разморозьте Maxiprep плазмиды pAAV, содержащей HDRT, для упаковки в AAV6. Ресуспендируют 85,6 мкг плазмиды pAAV в 3 мл чистой среды DMEM.
- Растворите 342 мкл 1 мг/мл раствора PEI в 3 мл чистой среды DMEM. Инкубируйте оба раствора в течение 10 мин при ЛТ.
- Смешайте обе пробирки по 3 мл в одну пробирку с ~ 6,4 мл смеси для трансфекции и инкубируйте в течение 20 мин при ЛТ.
- Тем временем разморозьте самомолчающий вспомогательный вектор RepCap6 с поддержкой тетрациклина из морозильной камеры с температурой -80 °C на водяной бане с температурой 37 °C. Чтобы трансдуцировать клетки 293AAV, добавьте вспомогательный вектор при кратности инфекции (MOI) 25, используя среднюю инфекционную дозу культуры тканей (TCID50) и предполагая, что 1,15 x 107 клеток / чашка; обычно используется 2-10 мкл на 15-сантиметровую тарелку. Аккуратно покачайте и взболтайте посуду, чтобы распределить.
- После инкубации смеси для трансфекции добавьте по каплям 1,6 мл в каждую из четырех 15-сантиметровых посуд. Инкубировать при 37 °C и 5% CO2 в течение ночи.
ПРИМЕЧАНИЕ: В качестве альтернативы, если интересующие векторы rAAV6 уже доступны, эти векторы могут быть использованы для обеспечения вирусного генома, подлежащего упаковке, что сводит на нет необходимость в каких-либо плазмидах с этой системой и дает сопоставимые титры rAAV6. Для этого подхода клетки 293AAV трансдуцируются совместно с желаемым rAAV6 при MOI 50 (на основе копий генома rAAV6 [GC] / мл) вместе со вспомогательным вектором. - На следующий день тщательно аспирируйте и выбросьте питательную среду и замените 30 мл предварительно подогретой производственной среды. Инкубируйте еще 96 ч до сбора урожая. Для получения максимальной урожайности не рекомендуется дальнейшая смена среды.
- Сбор и очистка рекомбинантного AAV6 из среды
- Не вытесняя клетки из чашки, соберите всю надосадочную жидкость клеток в фильтрующий блок с мембраной PES размером 0,2 мкм, по крайней мере, на 50% больше, чем объем фильтруемой среды. Затем отфильтруйте надосадочную жидкость.
ПРИМЕЧАНИЕ: Если желателен более высокий выход rAAV6, клетки могут быть собраны и rAAV извлечен из клеточного гранулирования с использованием коммерческих наборов или установленных протоколов34,35. Поскольку AAV6 в основном секретируется в среду36, использовался только надосадочная жидкость, что сокращало трудозатраты, затраты и время. - Добавьте 5 растворов ПЭГ/NaCl в отфильтрованную надосадочную жидкость на 25% от собранного объема; обычно это 30 мл, если используются четыре 15-сантиметровые чашки по 30 мл.
- Хорошо перемешайте, перевернув, а затем инкубируйте в течение ночи при температуре 4 ° C, чтобы вирусные частицы выпали в осадок.
ПРИМЕЧАНИЕ: Частицы AAV стабильны до 2 дней в этом растворе. - Предварительно охладите центрифугу с поворотным ведром с 250 мл пробирочных вкладышей до 4 °C. Подготовьте центробежный фильтрующий блок объемом 4 мл с отсечкой 100 кДа и гидрофильный шприцевой фильтр PES 0,22 мкм путем предварительной обработки каждой мембраны 2 мл 10% плюроника F-68 в течение не менее 1 ч при ЛТ.
- Переложите смесь AAV-PEG/NaCl в пробирку объемом 250 мл, центрифугу при 2 500 x g в течение 1 ч при 4 °C, а затем осторожно удалите всю надосадочную жидкость путем аспирации.
- Ресуспендируйте вирусную гранулу от бежевого до белого путем вихривания в 4 мл 1 M HEPES до полной ресуспендирования. При необходимости дайте ему постоять 5 минут и снова встряхните. Ресуспендируют с помощью серологической пипетки объемом 5 мл и переносят общий объем в пробирку объемом 15 мл.
- В вытяжном шкафу добавьте равный объем хлороформа к вирусной суспензии - обычно 4 мл.
- Энергично вихревите в течение 2 мин, а затем центрифугу при 1000 x g в течение 5 мин при RT.
- Соберите верхний слой (надосадочную жидкость, содержащую AAV) в новую пробирку объемом 50 мл и выбросьте нижний слой (хлороформ).
ВНИМАНИЕ: Растворы, содержащие хлороформ, являются опасными отходами. Следуйте институциональным рекомендациям по его утилизации. - Поместите надосадочную жидкость, содержащую AAV, под вытяжной шкаф и дайте оставшимся хлороформу испариться в течение 30 минут.
- Тем временем промойте предварительно обработанный центробежный фильтрующий блок и шприцевой фильтр.
- Добавьте 1,5 мл буфера для рецептуры в предварительно обработанный центробежный фильтрующий блок. Центрифуга при 3 500 x g в течение 10 мин при 15 °C в роторе качающегося ковша. Повторите этот шаг с 4 мл буфера состава, чтобы промыть мембрану.
- Дважды промойте фильтр шприца 5 мл буфера для рецептуры с помощью шприца объемом 5 мл.
- Загрузите ~ 4 мл надосадочной жидкости, содержащей AAV, полученной при экстракции хлороформа, в шприц объемом 5 мл, прикрепите промытый шприцевой фильтр и отфильтруйте непосредственно в центробежный фильтрующий блок.
- Центрифугу при 3,500 x g в течение 25 мин при 15 °C, а затем убедитесь, что раствор AAV в фильтре составляет около 50-100 мкл. Если объем раствора составляет >100 мкл, продолжайте центрифугу.
- После удаления фильтрата добавьте 4 мл буфера для рецептуры в чашку центробежного фильтрующего блока и равномерно перемешайте раствор пипеткой. Центрифугу при 3,500 x g в течение 25 мин при 15 °C, а затем убедитесь, что раствор AAV в фильтре составляет 50-100 мкл. Если объем раствора составляет >100 мкл, продолжайте центрифугу. Повторите этот шаг для еще одной стирки.
- После окончательного центрифугирования убедитесь, что объем раствора составляет 50-70 мкл; Если нет, продолжайте центрифугу. Переведите препарат в пробирку объемом 1,5 мл. При желании аликвотировать и хранить при температуре −80 °C.
- Не вытесняя клетки из чашки, соберите всю надосадочную жидкость клеток в фильтрующий блок с мембраной PES размером 0,2 мкм, по крайней мере, на 50% больше, чем объем фильтруемой среды. Затем отфильтруйте надосадочную жидкость.
- Определение титра рекомбинантного AAV6 методом кПЦР
ПРИМЕЧАНИЕ: Праймеры для кПЦР отжигаются в области ИТР и, таким образом, должны быть пригодны для всех конструкций, клонированных в pAAV.- Разморозьте аликвоту rAAV6, подлежащего титру, и аликвоту эталонного материала AAV6. Эталонный материал AAV6 должен быть близок к 4 x 1011 ГЦ/мл; В противном случае отрегулируйте разбавление соответствующим образом.
- Проведите дижест ДНКазы I, чтобы удалить оставшуюся свободную плазмидную ДНК в препарате rAAV6, объединив 2,0 мкл образца или эталонного материала AAV6 с 15,6 мкл безнуклеазного H 2 O,2,0мкл 10-кратного буфера ДНКазы I и 0,4 мкл ДНКазы I.
- Аккуратно перемешайте и выдержите в течение 30 минут при 37 °C, а затем переложите на лед. Это разбавление 1 (см. таблицу 3).
- Приготовьте пятикратное серийное разбавление всех образцов и эталонного материала AAV6, как показано в таблице 3 ниже, водой.
- Приготовьте мастер-микс для кПЦР SYBR Green. На лунку смешайте 4,7 мкл безнуклеазной воды с 10 мкл основной смеси SYBR Green, 0,15 мкл праймера ITR вперед при 100 мкМ и 0,15 мкл праймера ITR в обратном направлении при 100 мкМ.
ПРИМЕЧАНИЕ: Каждая проба измеряется в двух экземплярах, с 16 скважинами для эталонного стандарта, 8 скважинами на образец и 2 скважинами для контроля без шаблона. Приготовьте на 10% больше мастер-микса, чтобы учесть ошибку пипетирования. - В оптическую 96-луночную или 384-луночную реакционную пластину загрузите 15 мкл на лунку мастер-смеси SYBR Green qPCR.
- Затем загрузите 5 мкл образцов и эталонный материал AAV6 или воду без нуклеазы для контроля без шаблона. Для эталонного стандарта AAV6 разбавление нагрузки 2 до разбавления 9. Для образцов загружают разбавление 5 до разбавления 8. Измеряйте каждое разведение в двух экземплярах. Избегайте пузырьков.
- Запечатайте загруженную пластину оптической прозрачной пленкой, центрифугу при 800 x g в течение 1 минуты при RT и загрузите пластину в прибор qPCR с соответствующей установкой на 96 или 384 лунки.
- Настройте и запустите прибор qPCR с помощью детектирования SYBR со следующими условиями циклирования: 98 °C в течение 3 мин, затем 40 циклов 98 °C в течение 15 с и 58 °C в течение 30 с, после чего следует кривая плавления.
- Проанализируйте данные с помощью программного обеспечения прибора, используя концентрацию эталонного материала AAV6 в копиях генома на миллиметр (ГХ/мл) в качестве стандартной кривой (см. Таблицу 3). Рассчитайте конечную концентрацию образца путем умножения на коэффициент разбавления.
- Убедитесь, что стандартная кривая R2 близка к 1,0, эффективность ПЦР составляет 90% -110%, базовый уровень удален, кривая плавления показывает один пик, значения C t изменяются в соответствии с разведениями, а дубликаты находятся в пределах 0,5 Ct; В противном случае исключите выбросы. Ожидаем урожайность, как показано на рисунке 2.
2. Подготовка В-клеточных сред и стимулов
- Подготовьте размораживающую среду: смешайте RPMI-1640 с 20% FCS. Стерильный фильтр через мембранный фильтрующий блок PES 0,2 мкм. Хранить при температуре 4 °C.
- Подготовьте среду для культивирования В-клеток: смешайте реагенты, указанные в таблице 4, а затем проведите стерильную фильтрацию через мембранный фильтрующий блок PES 0,2 мкм. Хранить при температуре 4 °C.
- Ресуспендировать каждый из стимуляторов В-клеток, указанных в таблице 5 , в исходных концентрациях в среде для культивирования В-клеток, за исключением CpG ODN, который следует ресуспендировать в воде, не содержащей нуклеаз. Хранить при температуре −80 °C.
- При отрицательном истощении неВ-клеток (необязательный шаг 4) приготовьте DPBS (без кальция, без магния) с 2% FCS (DPBS 2% FCS). Стерильный фильтр через мембранный фильтрующий блок PES 0,2 мкм. Хранить при температуре 4 °C.
3. Получение и культивирование В-клеток макак-резусов
ПРИМЕЧАНИЕ: Криоконсервированные макак-резусы PBMC или спленоциты используются для создания клеточной культуры30,31.
- Предварительно подогрейте размораживающую среду и питательные среды В-клеток на водяной бане с температурой 37 °C. Разморозьте стимуляторы В-клеток из таблицы 5 на льду.
- Подготовьте трубку подходящего размера, содержащую предварительно разогретую талую среду. В идеале это должно быть более чем в 10 раз больше объема размороженных клеток.
- Разморозьте один-два криовиала PBMC или спленоцитов за раз на водяной бане с температурой 37 ° C и сцедите в подготовленную пробирку с предварительно подогретой средой. Промойте криотрубки, чтобы собрать все клетки.
- Центрифугируйте клетки при 200 x g в течение 10 мин при RT.
ПРИМЕЧАНИЕ: Эти настройки центрифугирования снижают загрязнение тромбоцитов, сохраняя при этом выход PBMC. Можно использовать более высокие скорости, такие как 350 x g в течение 5 минут. - Ресуспендируйте клетки в 10 мл размораживающей среды для промывки.
- Повторите шаги 3.4 и 3.5, в общей сложности три центрифугирования для удаления замораживающей среды. После последнего центрифугирования ресуспендируют клетки в расчетной дозе ~ 5 x 106 клеток/мл в среде для культивирования В-клеток.
ПРИМЕЧАНИЕ: В приведенном выше протоколе культивируют целые препараты PBMC или спленоцитов с контаминацией другими клетками. Если требуются более чистые культуры В-клеток, хотя и при значительно сниженном общем выходе В-клеток, переходите к шагу 4. Никаких различий в эффективности редактирования между этими двумя методами не наблюдалось. - При необходимости разбавьте аликвоту 10 мкл клеток питательной средой В-клеток для подсчета. Подсчитывают с помощью гемоцитометра и окрашивают трипановым синим, сочетая равные объемы ресуспендированных клеток и 0,4% раствора трипанового синего.
- Отрегулируйте концентрацию клеток до 3 x 106 клеток / мл со средой для культивирования В-клеток в соответствии с количеством клеток. Затем добавьте стимуляторы В-клеток к их конечным концентрациям в соответствии с таблицей 5 и перемешайте.
- Перенесите клетки в соответствующую чашку для клеточных культур. В целом, рекомендуется 0,6 х 10 6-0,7 х 106 клеток/см2. Инкубируют клетки при 37 °C с 5%CO2 в течение 48 ч ± 2 ч.
4. Необязательное отрицательное истощение не-В-клеток
ПРИМЕЧАНИЕ: Выход и чистота зависят от процентного содержания В-клеток на входе среди PBMC, который может сильно различаться у отдельных макак-резусов27. Ожидайте чистоту 80%-95%, эффективность 60% и 1 x 10 6-1,5 x 106 ячеек от 1 x 107 PBMC.
- После последней промывки (этап 3.6) ресуспендируйте клетки в концентрации 1 x 108 клеток/мл в DPBS 2% FCS и человеческом Fc-блоке, разбавленном в соотношении 1:200. Количество ячеек основано на количестве размороженных ячеек.
- Инкубировать в течение 15 мин на льду, чтобы блокировать Fc-рецепторы, а затем добавить биотинилированные антитела, указанные в таблице 6. Инкубировать еще 20 мин на льду.
- Долейте в тюбик DPBS 2% FCS и отжимайте при 200 x g в течение 10 мин при 4 °C.
- Ресуспендируют клетки в DPBS 2% FCS на 80% объема из шага 4.1 (т.е. 80 мкл на 1 x 107 клеток).
- Добавьте магнитные шарики стрептавидина в суспензию клетки в количестве 20% от объема из шага 4.1 (т.е. 20 мкл шариков на 1 x 107 ячеек).
- Инкубируйте клетки в течение 15 минут на льду и периодически перемешивайте.
- Между тем, на 1 х 108 ячеек подготовьте магнитный сепаратор с большой магнитной обеднительной колонной и фильтром предварительного разделения. Промойте фильтр и колонку предварительного разделения 2 мл DPBS 2% FCS под действием силы тяжести и откажитесь от проточного потока. Установите пробирку для сбора 15 мл.
ПРИМЕЧАНИЕ: Использование других колонн, таких как колонны с положительным отбором или другие системы очистки магнитных шариков, может резко снизить чистоту. - После инкубации дополните клетки до 0,5 мл DPBS 2% FCS, если объем составляет <0,5 мл. Если объем составляет ≥0,5 мл, просто продолжайте.
- Загрузите клеточную суспензию в фильтр предварительного разделения на подготовленной колонке и соберите проточный поток в пробирку объемом 15 мл.
- Дважды упраздните несвязанные обогащенные В-клетки, добавив 1 мл DPBS 2% FCS в фильтр предварительного разделения. Соберите несвязанные клетки в одну и ту же трубку под действием силы тяжести.
ПРИМЕЧАНИЕ: Дополнительное элюирование может незначительно увеличить урожайность. Чистота и эффективность могут быть оценены с помощью проточной цитометрии входных клеток, обогащенных клеток и клеток, удерживаемых на колонке. Чтобы получить ячейки, удерживаемые на колонке, снимите колонку с магнита и промойте 3 мл DPBS 2% FCS с помощью прилагаемого поршня. При желании оцените чистоту с помощью проточной цитометрии, как показано на рисунке 3 , используя реагенты в таблице 7. - Центрифугируйте обогащенные В-клетки в дозе 200 x g в течение 10 мин при 4 °C.
- Ресуспендируют клетки при расчетной дозе ~ 5 x 106 клеток / мл в среде для культивирования В-клеток и продолжайте на шаге 3.7.
5. Первичное редактирование генов В-клеток макак-резусов
- После активации В-клеток макак-резусов в течение 48 ч ± 2 ч готовят реагенты для электропорации и трансдукции.
- Предварительно разогрейте ДМСО, безнуклеазный дуплексный буфер, буфер T и буфер E (набор для электропорации 10 мкл) или E2 (набор для электропорации 100 мкл) из набора для электропорации в RT.
- Разморозьте rAAV6 HDRT и В-клеточные стимуляторы из таблицы 5 на льду.
- Ресуспендировать сгРНК CRISPR-Cas9 при 100 мкМ в дуплексном буфере. Восстановите в течение 10 минут при RT и смешайте вихревом и щелчком. Храните восстановленные сгРНК на льду до использования. Хранить при температуре −80 °C.
ПРИМЕЧАНИЕ: СгРНК CRISPR-Cas9 могут быть разработаны с помощью различных онлайн-инструментов (см. 1.1.1) и могут сильно различаться по эффективности резки. Эмпирическое тестирование эффективности резки рекомендуется проводить с использованием таких анализов, как TIDE37 или ICE38. - На 10 мкл электропорации готовят 550 мкл среды для культивирования В-клеток со всеми стимуляторами из таблицы 5 и добавляют 1% ДМСО. Увеличьте объемы в 10 раз для электропорации 100 мкл. Опционально 10% этой среды может быть приготовлено без антибиотика-антимикотика, что несколько повышает жизнеспособность клеток после трансфекции.
- На 10 мкл электропорации готовят лунку из 48-луночной клеточной культуральной пластины с 50 мкл питательной среды В-клеток со стимуляторами и без антибиотика-антимикотика, если он используется. Для электропорации 100 мкл пипеткой 500 мкл вводят в лунки 6-луночной пластины.
- Добавьте rAAV6 HDRT в среду в скважинах, до 20% объема в скважине. Стремитесь к MOI в диапазоне от 1 x 10 5-1 x 10 6 в зависимости от количества клеток на трансфекцию (электропорация 10 мкл: 5 x 10 5 клеток; электропорация 100 мкл:5 x 106 клеток) и GC в препарате rAAV6. Высокие концентрации запасов rAAV6 от 5 x 1013 ГХ/мл до 5 x 1014 ГХ/мл рекомендуются для достижения высоких MOI при небольших объемах.
ПРИМЕЧАНИЕ: Более низкие MOI могут привести к снижению эффективности редактирования, а MOI 5 x 105 , как правило, близки к максимальной эффективности редактирования, которую мы видели. Влияние различных MOI на жизнеспособность В-клеток не наблюдалось. Рекомендуется включать контрольную группу без rAAV6 HDRT, без трансфекции RNP и без обоих. - Предварительно разогрейте подготовленную посуду и оставшуюся среду, переложив их в инкубатор при 37 °C с 5% CO2.
- На 10 мкл электропорации приготовьте 1,15 мкл рибонуклеопротеина (РНП): смешайте 0,4 мкл 61 мкМ Cas9 с 0,75 мкл 100 мкМ сгРНК в дуплексном буфере. Подготовьте дополнительно (рекомендуется на 30% больше для однократной электропорации) из-за ошибки пипетирования и во избежание образования пузырьков при загрузке наконечников для электропорации. Уменьшите масштаб в 10 раз для наконечников объемом 100 мкл.
- Инкубируйте RNP в течение не менее 15 минут при RT перед смешиванием с клетками. После инкубации несколько RNP могут быть объединены, если необходимо одновременно нацеливаться на более чем один локус. Никаких существенных различий в эффективности не наблюдалось одновременно с тремя локусами.
- Тем временем подготовьте клетки к электропорации. Всегда держите клетки в режиме RT, чтобы избежать температурных скачков. Заготавливают клетки через 48 ч ± 2 ч культуры в соответствующий сосуд. Промойте посуду DPBS, чтобы собрать максимальное количество ячеек.
- Центрифугируйте клетки при 200 x g в течение 10 мин при RT. Откажитесь от надосадочной жидкости и ресуспендируйте клетки в DPBS при ~ 2 x 106 клеток / мл.
- Соедините 10 мкл 0,4% раствора трипанового синего с 10 мкл клеточной суспензии и посчитайте с помощью гемоцитометра.
ПРИМЕЧАНИЕ: На этом этапе, из-за потери во время сбора урожая и промывки, ожидайте около 60% клеток, которые были помещены в культуру через 48 часов ± 2 часа раньше. - Тем временем центрифугируйте клетки при 200 x g в течение 10 минут при RT. Выбросьте надосадочную жидкость, убедившись, что все оставшиеся DPBS сведены к минимуму. Ресуспендируют клетки в предварительно нагретом (RT) буфере T при 5,55 x 107 клеток / мл в зависимости от приведенного выше количества клеток.
- Настройте систему трансфекции, включив аппарат и установив его на 1,350 В, 15 мс и 1 импульс. Поместите дозаторную станцию внутрь вытяжного шкафа с ламинарным потоком.
- Для каждого набора из 10 электропораций готовят трансфекционную трубку с 3 мл буфера E (для трансфекций 10 мкл) или E2 (для трансфекций 100 мкл). Вставьте трубку в дозатор.
- На 10 мкл электропорации объедините 1,15 мкл РНП с 9 мкл клеток. Убедитесь, что у вас достаточный объем (+ 30%), чтобы избежать аспирации воздуха в наконечник электропорации. Инкубировать при РТ в течение 1-2 мин перед электропорацией.
- Аспирируйте 10 мкл или 100 мкл РНП и клеточной смеси в электропорационный наконечник соответствующего размера на электропорационной пипетке, вставьте загруженную пипетку в дозаторную станцию и начните электропорацию. Убедитесь, что на наконечниках нет пузырьков воздуха, чтобы предотвратить искрение. Следите за электропорацией, чтобы убедиться, что дуга не возникает.
- Немедленно выталкивайте электропорированные ячейки в подготовленную, предварительно нагретую, небольшой объем среды с rAAV6 или без него внутри 48-луночной (10 мкл трансфекций) или 6-луночной пластины (100 мкл трансфекций). Повторите шаги 5.15-5.17 с оставшимися образцами. Добавьте контрольные образцы без трансфекции в культуральные лунки.
- Инкубируют клетки при 37 ° C с 5% CO 2 в течение 4 ч ±2 ч, а затем добавляют подготовленную, предварительно подогретую среду для культивирования В-клеток, содержащую стимуляторы, ДМСО и антибиотик / антимикотическое средство: 450 мкл для 10 мкл трансфекции или 4,5 мл для 100 мкл трансфекций.
- Продолжайте инкубацию при 37 °C с 5% CO2 в течение12-24 часов. Затем измените среду на среду для культивирования В-клеток, содержащую стимуляторы и антибиотик / антимикотик без ДМСО, если требуется расширенное культивирование. Анализ геномной ДНК можно сделать через 24 часа. Цифровая капельная ПЦР с использованием праймера вне плеча гомологии и праймера внутри вкладыша может быть использована для количественной оценки эффективности редактирования39. Выполните ПЦР для амплификации места вставки и секвенирование по Сэнгеру, чтобы проверить правильность редактирования.
- Для анализа уровня белка культивируют клетки в течение 40-48 ч после электропорации, чтобы обеспечить изменения экспрессии белка, и проводят анализ с помощью проточной цитометрии с использованием реагентов, указанных в таблице 7.
Результаты
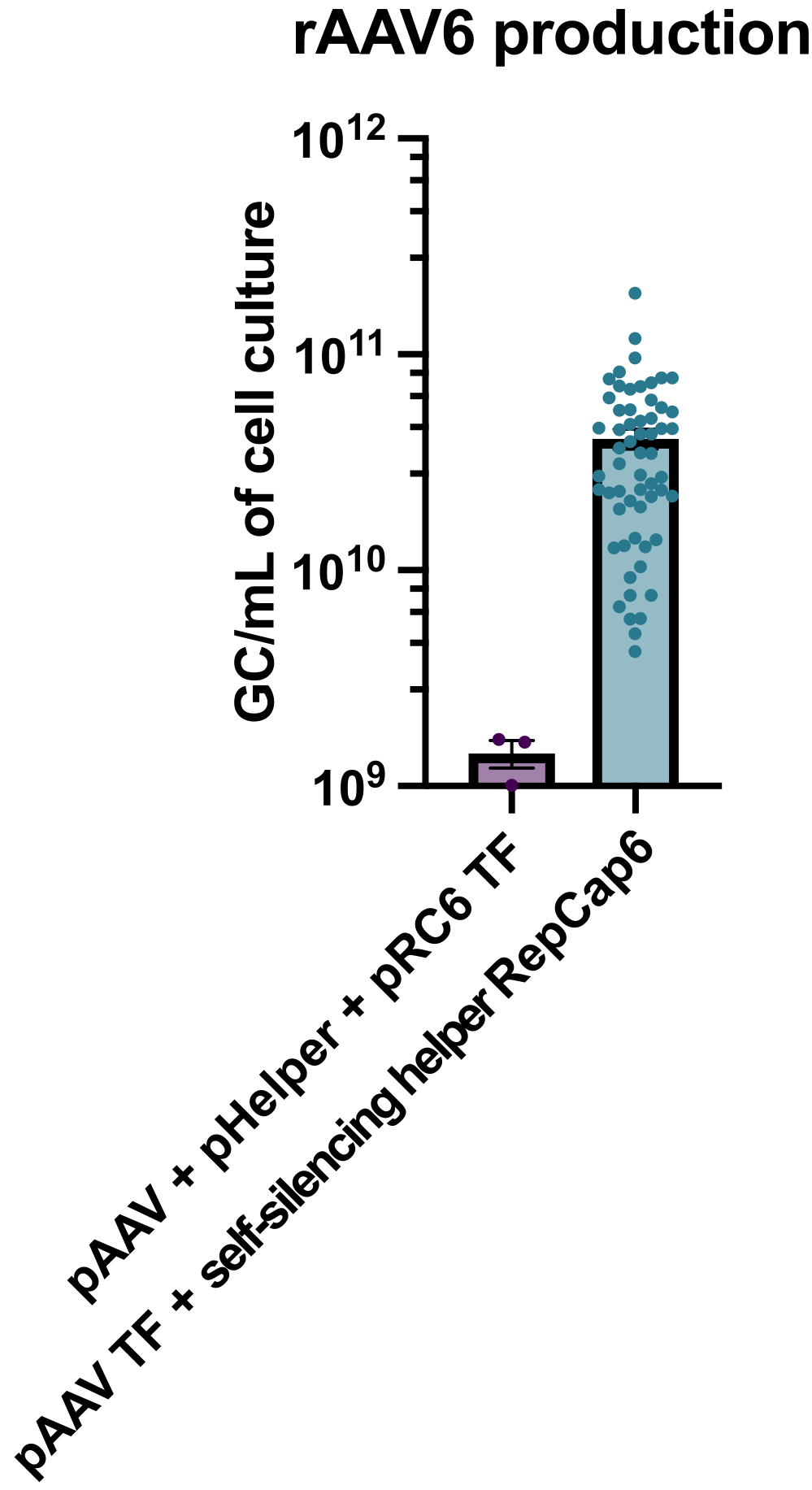
Продуцирование rAAV6 с использованием самоподавляющего аденовирусного хелпера с поддержкой тетрациклина приводило к получению в среднем 4 х 10ГХ /мл среды для культивирования клеток, что в 30-40 раз превосходило производство при использовании стандартной, безхелперной тройной трансфекции (рис. 2).
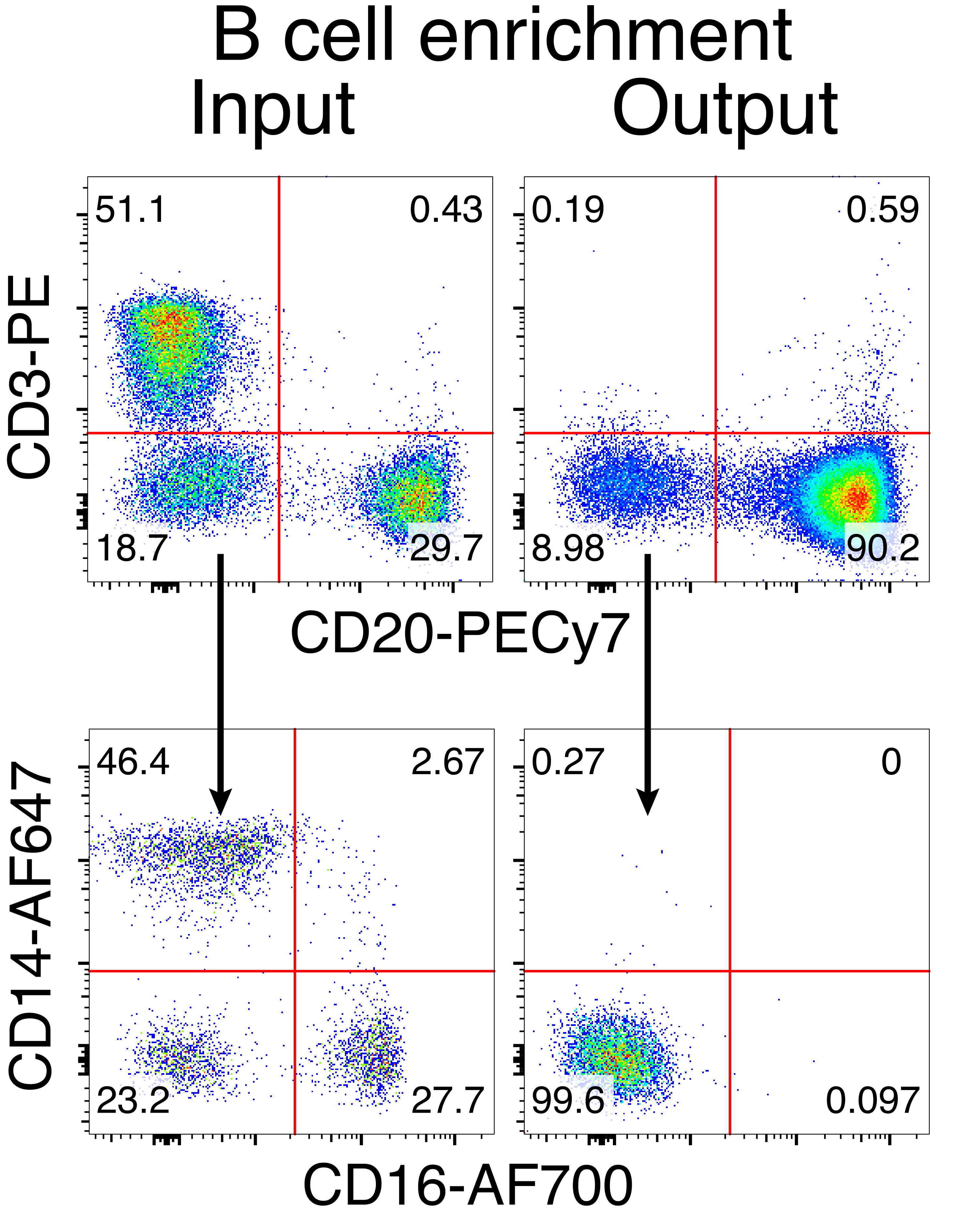
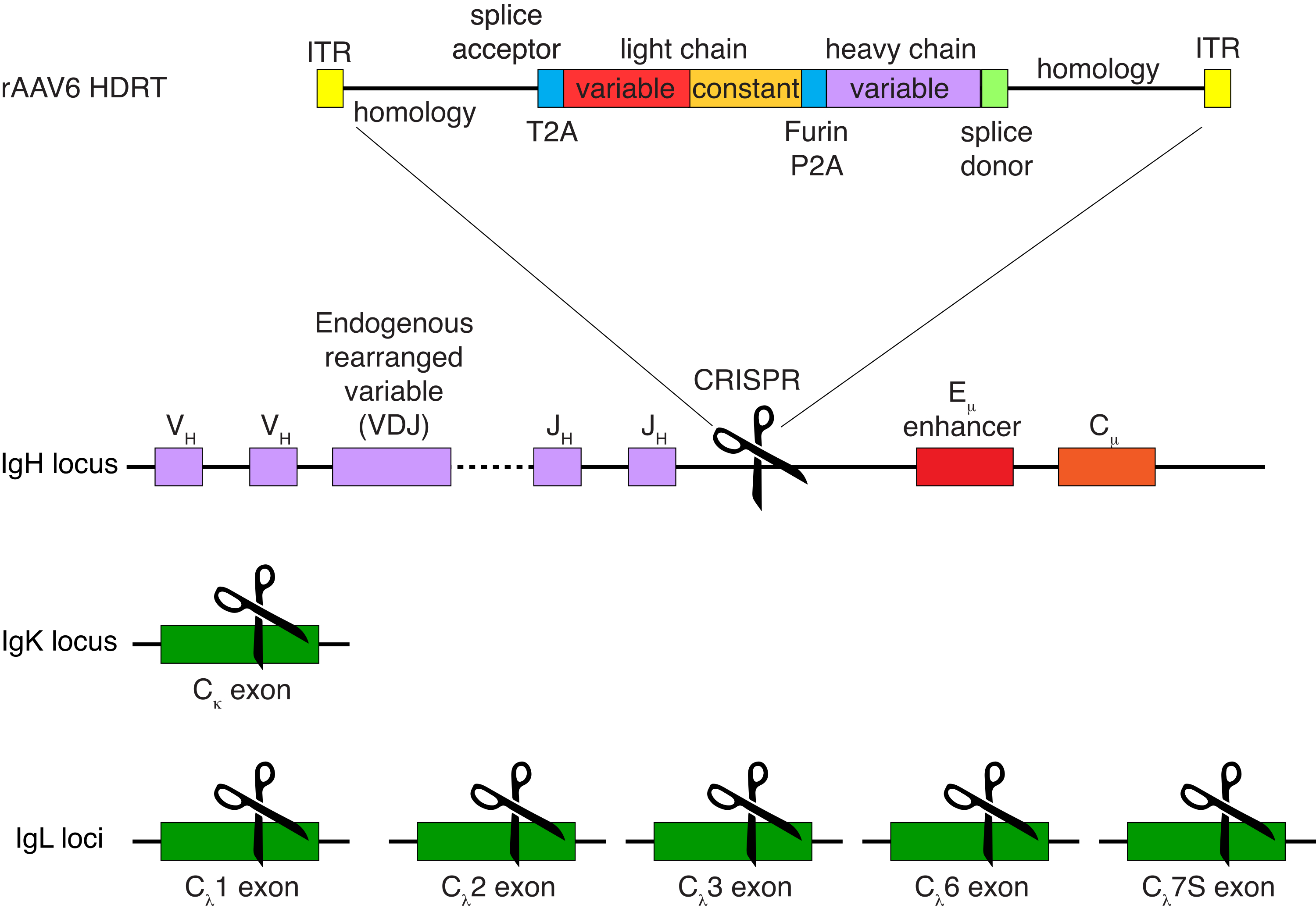
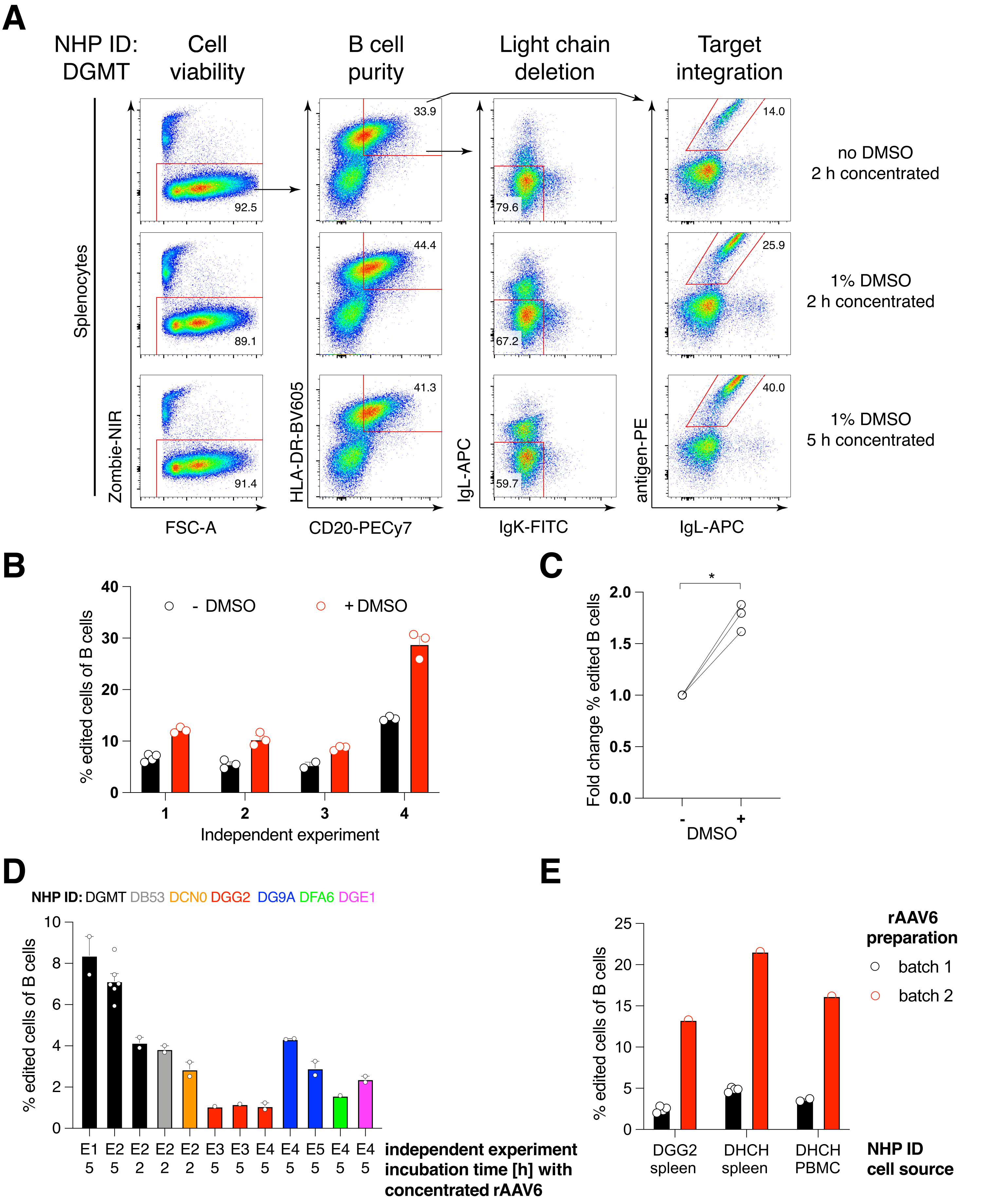
Необязательная очистка В-клеток макаки-резуса привела к элиминации подавляющего большинства CD3+ Т-клеток и CD14+ и/или CD16+ миелоидных клеток, при этом обычно получают 80-95% CD20+ В-клеток (рис. 3). Основываясь на наших предыдущих разработках в мышиных В-клетках7, мы разработали метод редактирования специфичности В-клеточного рецептора В-клеток макак-резуса при одновременном поддержании аллельного исключения в подавляющем большинстве В-клеток путем удаления легких цепей эндогенных антител путем нарушения их постоянной области. Мы сконструировали HDRT без промотора, который будет вставлен в локус IGH между последним геном IGHJ и энхансером Eμ В-клеток макак-резуса (рис. 4). Эта конструкция использует эндогенный промотор VH естественным образом перестроенным выше по течению области VDJ в зрелых В-клетках и, таким образом, не экспрессируется эписомальными геномами AAV. Кроме того, эта конструкция требует сплайсинга с нисходящими константами тяжелой цепи антитела, которые должны экспрессироваться на поверхности клетки. Таким образом, специфическое связывание антигена на поверхности клетки, показанное проточной цитометрией, указывает на правильную интеграцию целевого локуса и на то, что вставленная последовательность функциональна.
Мы упаковали такую конструкцию, кодирующую антитело Ab1485, полученное из макаки-резуса против ВИЧ bNAb40, в rAAV6 и использовали его для редактирования активированных первичных культур спленоцитов макак-резуса или PBMC, как описано выше (рис. 5A). Протокол поддерживал высокую жизнеспособность клеток (~90%) при одновременном удалении экспрессии легкой цепи в ~80% В-клеток. Большинство В-клеток по-прежнему экспрессировали изотип IgM (рис. 5B). Добавление rAAV6, кодирующего Ab1485 HDRT, привело к редактированию генов и поверхностной экспрессии Ab1485 в 16-21% В-клеток (рис. 5A), хотя и с более низкой интенсивностью флуоресценции цепей антител, чем на неотредактированных В-клетках (рис. 5A справа, рис. 5C). Это может быть результатом эпитопной конкуренции между антигенным пятном и моноклоналами, используемыми для обнаружения поверхностного BCR в проточной цитометрии, а также фактического снижения экспрессии белка из-за полицистронной природы HDRT и менее эффективного сплайсинга. Добавление 1% ДМСО и расширенная концентрированная инкубация с rAAV6 HDRT обычно повышали эффективность редактирования (рис. 6A-C). Используя этот специфический метод, обычно 5-20% и до 40%, эффективность редактирования достигается в зависимости от отдельной макаки резуса (рис. 5А, рис. 6А-Е) и качества партии rAAV6 HDRT (рис. 6Е). В целом, мы представляем протоколы для эффективного производства rAAV6, а также культивирования, очистки и редактирования генов В-клеток макак-резусов.
| Реагентов | Том | Запас | Конечная концентрация |
| DMEM, высокий уровень глюкозы | 500 мл | 1 х | ~ 88,5% |
| FCS, с отключением тепла | 50 мл | 1 х | ~ 8.85% |
| Антибиотик/Антимикотик | 5 мл | 100 х | 1 х |
| Глутамин | 5 мл | 200 мМ | 2 мМ |
| Пируват натрия | 5 мл | 100 мМ | 1 мМ |
Таблица 1: Среда для культивирования клеток 293AAV.
| Реагентов | Том | Запас | Конечная концентрация |
| DMEM, высокий уровень глюкозы | 500 мл | 1 х | ~ 95,2% |
| FCS, с отключением тепла | 10 мл | 1 х | ~ 1,9% |
| Антибиотик/Антимикотик | 5 мл | 100 х | 1 х |
| Глутамин | 5 мл | 200 мМ | 2 мМ |
| Пируват натрия | 5 мл | 100 мМ | 1 мМ |
Таблица 2: Среда для производства ячеек 293AAV.
| Серия разбавления | Объем образца (мкл) | Разбавитель и объем | Коэффициент разбавления | Полное разбавление | Артикул AAV6 |
| ГХ/мл | |||||
| Разбавление 1 | Образец 2 мкл или эталонный стандарт AAV при 4,1 x 1011 ГХ/мл | 18 мкл ДНКазы буфера и фермента | 10 х | 10 х | 4,1 х 1010 |
| Разбавление 2 | 15 мкл Dil.1 | 60 мкл H2O | 5 х | 50 х | 8,2 х 109 |
| Разбавление 3 | 20 мкл Dil.2 | 80 мкл H2O | 5 х | 250 х | 1,6 х 109 |
| Разбавление 4 | 20 мкл Dil. 3 | 80 мкл H2O | 5 х | 1250 х | 3,3 х 108 |
| Разбавление 5 | 20 мкл Dil.4 | 80 мкл H2O | 5 х | 6250х | 6,6 х 107 |
| Разбавление 6 | 20 мкл Dil. 5 | 80 мкл H2O | 5 х | 31250 х | 1,3 х 107 |
| Разбавление 7 | 20 мкл Dil. 6 | 80 мкл H2O | 5 х | 156250 х | 2,6 х 106 |
| Разбавление 8 | 20 мкл Dil. 6 | 80 мкл H2O | 5 х | 781250 х | 5,24 х 105 |
| Разбавление 9 | 20 мкл Dil. 7 | 80 мкл H2O | 5 х | 3906250 х | 1,05 х 105 |
Таблица 3: таблица разведения кПЦР.
| Реагент | Том | Запас | Конечная концентрация |
| РПМИ-1640 | 420 мл | 1 х | 84% |
| FCS, с отключением тепла | 50 мл | 1 х | 10% |
| Антибиотик/Антимикотик | 5 мл | 100 х | 1 х |
| Глутамин | 5 мл | 200 мМ | 2 мМ |
| Пируват натрия | 5 мл | 100 мМ | 1 мМ |
| ХЕПЕС | 5 мл | 1 м | 10 мМ |
| 2-B-меркапто-этанол | 550 мкл | 55 мМ | 55 мкМ |
| Заменимые аминокислоты | 5 мл | 100 х | 1 х |
| Инсулин-трансферин-селен | 5 мл | 100 х | 1 х |
Таблица 4: Среда для культивирования В-клеток.
| Реагент | Разбавление | Запас | Конечная концентрация |
| MegaCD40L | 1:1000 | 100 мкг/мл | 100 нг/мл |
| CpG ODN | 1:300 | 1 мг/мл | 3,33 мкг/мл |
| Человеческий BAFF | 1:1000 | 40 мкг/мл | 40 нг/мл |
| Человеческий Ил-2 | 1:1000 | 50 мкг/мл | 50 нг/мл |
| Человеческий Ил-10 | 1:1000 | 50 мкг/мл | 50 нг/мл |
Таблица 5: В-клеточные стимуляторы.
| Антитело | Клон | Разбавление | Заключительный конс. |
| античеловеческий CD3 | ФН-18 | 1:40 | 2,5 мкг/мл |
| античеловеческий CD8a | РПА-Т8 | 1:200 | 2,5 мкг/мл |
| античеловеческий CD14 | М5Е2 | 1:200 | 2,5 мкг/мл |
| античеловеческий CD16 | 3Г8 | 1:200 | 2,5 мкг/мл |
| античеловеческий CD33 | АС104.3Э3 | 1:50 | 1 тест |
| античеловеческий CD64 | 10.1 | 1:800 | 0,625 мкг/мл |
| античеловеческий CD66 | ТЕТ2 | 1:11 | 1 тест |
| античеловеческий CD89 | А59 | 1:800 | 0,625 мкг/мл |
Таблица 6: Антитела к необязательному истощению не-В-клеток.
| Реагент | Тип/клон | Рабочее разбавление/концентрация |
| античеловеческий CD14 AlexaFluor647 | М5Е2 | 1:50 |
| античеловеческий CD16 AlexaFluor700 | 3Г8 | 1:50 |
| античеловеческий CD20 PECy7 | 2Ч7 | 1:50 |
| античеловеческий CD3 PE | СП34-2 | 1:50 |
| Зомби-НИР | - | 1:500 |
| античеловеческий HLA-DR BV605 | Л243 | 1:200 |
| античеловеческая легкая цепь Ig лямбда-БТР | МХЛ-38 | 1:50 |
| античеловеческая легкая цепь Kappa FITC | поликлональный | 1:500 |
| античеловеческий IgM BV421 | МХМ-88 | 1:50 |
| Антиген RC1, случайно биотинилированный | - | 5 мкг/мл |
| Стрептавидин-ПЭ | - | 1:500 |
Таблица 7: Проточные цитометрические реагенты для анализа.

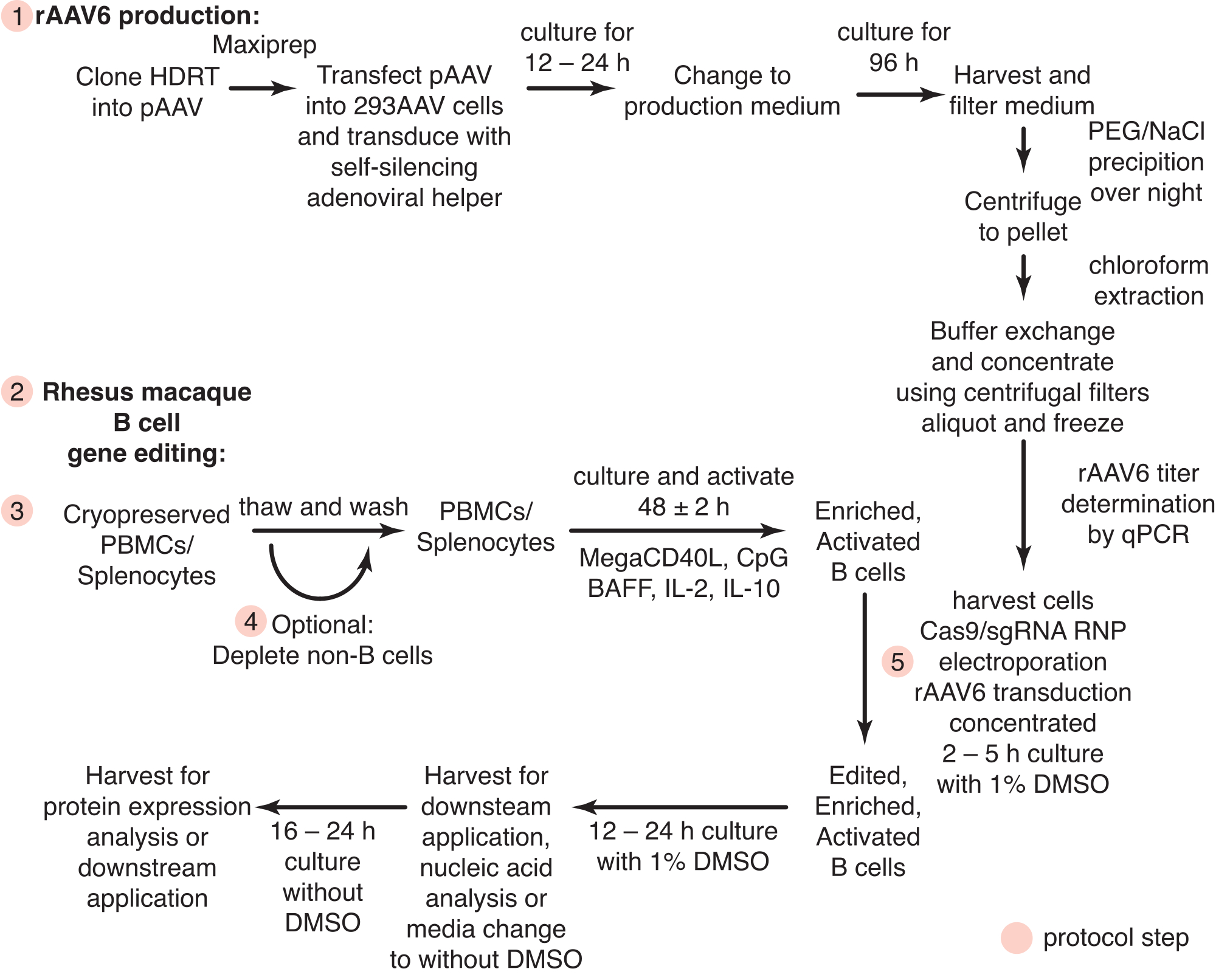
Рисунок 1: Схематический обзор продукции rAAV6 и редактирования генов первичных В-клеток макак-резусов. Протоколы разделены на продуцирование rAAV6 (этап 1) и редактирование генов В-клеток макак-резуса (этапы 2-5), включая необязательный этап истощения не-В-клеток (этап 4). Шаги в протоколах обозначены красными кружками. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 2: Высокие выходы rAAV6 с использованием самоподавляющего аденовирусного помощника. rAAV6 был получен с использованием методов, описанных здесь (трансфекция pAAV [TF] + самоподавляющий помощник RepCap6, самоподавление аденовирусного помощника) или типичная безхелперная тройная трансфекция pAAV, pHelper и pRepCap6 (pRC6). rAAV6 был очищен только из надосадочной жидкости клетки. Методы, использующие самосайленсирующие аденовирусные вспомогательные векторы, произвели в 30-40 раз больше rAAV, титрованного с помощью кПЦР, как описано выше. Каждая точка представляет собой индивидуальное производство rAAV с использованием различных конструкций pAAV от 2 до 20 независимых экспериментов. Среднее значение ± SEM построено. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 3: Обогащение В-клеток отрицательным истощением не-В-клеток. В-клетки макак-резусов были обогащены из PBMC с использованием описанного протокола и обогащены до чистоты 90%. Показаны входы и выходы предварительного обогащения после обогащения. Закрытый на живых, синглетных PBMC. Представитель пяти независимых экспериментов. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 4: Стратегия таргетинга, используемая для редактирования специфичности В-клеточного рецептора В-клеток макаки-резуса. rAAV6 был выпущен с изображением HDRT. HDRT состоит из плеча гомологии 266.н. 5', за которым следует 111.н. акцептора сращивания экзона 1 экзона 1 макаки-резуса, затем GSG-линкера с саморасщепляющейся пептидной последовательностью 2A вируса Thosea asigna (T2A ), за которой следует лидерная последовательность и полная легкая цепь антитела к макаке-резусу Ab1485 как макака-резус IGLC1. Затем следует сайт расщепления фурина, линкер GSG и последовательность пептида 2A саморасщепляющегося тешовируса свиньи (Furin-P2A), за которой следует еще одна лидерная последовательность и переменная тяжелой цепи Ab1485, за которой следуют 52.н. последовательности донора сплайсинга макаки-резуса IGHJ4, чтобы обеспечить сплайсинг в нисходящие константные области тяжелой цепи антител и плечо гомологии 514.н. Эта конструкция была нацелена на локус IGH между последним геном IGHJ и энхансером Eμ с использованием целевой последовательности сгРНК GAGATGCCAGAGCAAACCAG. Оба плеча гомологий были спроектированы так, чтобы заканчиваться на месте разреза этой сгРНК, тем самым удаляя целевую последовательность и обеспечивая оптимальную эффективность интеграции. Одновременно, чтобы поддерживать аллельное исключение и экспрессию одного В-клеточного рецептора, мы удаляли эндогенные легкие цепи с использованием sgRNA, нацеленных на IGKC макаки-резуса с целевой последовательностью GGCGGGAAGATGAAGACAGA и IGLC1, IGLC2, IGLC3, IGLC6 и IGLC7S с использованием целевой последовательности CTGATCAGTGACTTCTACCC. HDRT включал молчаливые мутации, предотвращающие расщепление последовательности IGLC1 этой сгРНК. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 5: Редактирование генов первичных В-клеток макаки-резуса . (A) Первичные спленоциты (верхняя панель) или PBMC (нижняя панель) от одной и той же макаки резусов культивировали без истощения не-В-клеток и редактировали, как описано выше. Стратегия таргетинга была такой, как показано на рисунке 4. Через два дня после электропорации клетки были собраны и окрашены на поверхность для проточного цитометрического анализа. Левый столбец был закрыт на синглетных ячейках, а другие столбцы затем были закрыты, как указано в верхней строке. Жизнеспособность клеток, чистота В-клеток, эффективность делеции легких цепей и эффективность нокаутирования Ab1485 путем окрашивания специфическим антигеном RC141 указаны в необработанных, трансфицированных RNP или трансфицированных RNP + rAAV6 трансдуцированных образцах (MOI = 5 x 105). Представитель шести независимых экспериментов с клетками разных макак-резусов. (B) экспрессия IgM на культивируемых В-клетках макак-резусов или после редактирования и (C) средняя геометрическая интенсивность флуоресценции (gMFI) IgM на В-клетках, которые не потеряли экспрессию Ig из-за нацеливания на IgLC и IgKC (неотредактированные) или В-клетки, которые связывают ожидаемый антиген (отредактированный). Красная точка указывает на gMFI культивируемых нетрансфицированных контрольных В-клеток. Указывает p < 0,0001 в парном t-критерии. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 6: Влияние ДМСО, длительной концентрированной инкубации с rAAV6 HDRT, качества партии rAAV и воспроизводимости среди различных донорских NHP на эффективность редактирования генов в первичных В-клетках макак-резусов. (A) Спленоциты культивировали и редактировали, как описано. После электропорации 5 x 10 5 клеток культивировали в среде с 1% ДМСО или без него и инкубировали в 50 мкл среды, содержащей rAAV6 HDRT, при MOI 5 x 10 5 в течение 2 ч или5 ч перед добавлением еще 450 мкл среды. Клетки анализировали через 2 дня после электропорации методом проточной цитометрии, как показано на рисунке 5. Представитель четырех независимых экспериментов. (B) Количественная оценка (A) в четырех независимых экспериментах. Точками обозначены технические реплики с настройками трансфекции 1,350 В, 10-20 мс и продолжительностью 1 импульсной электропорации и концентрациями ДМСО в диапазоне от 0.75% до 1.25%. (C) Среднее изменение эффективности редактирования по сравнению с (B). * p > 0,05 в U-критерии Манна-Уитни. (D) Эффективность редактирования по сравнению с независимыми экспериментами с различными макаками с использованием менее эффективной коммерческой партии rAAV6. (E) Эффективность редактирования с использованием двух разных коммерческих партий rAAV6, в которые одна и та же конструкция была упакована в В-клетки двух разных NHP в одном и том же эксперименте. Точками обозначены технические реплики с настройками трансфекции 1,350 В, 10-20 мс и 1 импульсной электропорацией. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Обсуждение
Представленные здесь протоколы обеспечивают быстрый и эффективный метод получения высоких урожаев и титров rAAV6 в виде HDRT и новые методы эффективного редактирования генов первичных В-клеток макак-резуса in vitro.
Производственный протокол rAAV6 сравнительно прост и быстр, что позволяет производить и тестировать множество различных конструкций одновременно без чрезмерных трудозатрат. При желании rAAV6 может быть дополнительно очищен с использованием установленных протоколов, таких как ультрацентрифугирование34 градиента йодиксанола или водное двухфазное разделение35 перед буферным обменом и концентрацией.
Несмотря на то, что это снизило общий выход, мы решили использовать только среду для культивирования клеток с восстановленной сывороткой для очистки rAAV6 вместо очистки от клеточной гранулы, поскольку большая часть rAAV6 высвобождается в среду36, а очистка из клеточной гранулы увеличивает затраты и трудозатраты. Использование самоинактивирующегося аденовирусного хелпера увеличивало урожайность в среднем в 30-40 раз, что позволяло тестировать конструкции, упакованные в AAV6 в одной 15-сантиметровой чашке. Хотя наш метод очистки является базовым, используя этот метод, мы получаем относительно небольшие вариации эффективности редактирования генов или жизнеспособности клеток от партии к партии после трансдукции с использованием различных клеточных линий или других первичных клеток (данные не показаны).
Мы разработали протокол очистки В-клеток макаки-резуса для получения нетронутых первичных В-клеток с использованием отрицательного истощения нежелательных популяций. Хотя это и не является необходимым для редактирования генов этих клеток, оно дает возможность получить относительно чистую популяцию первичных В-клеток макак-резуса для этого или других применений, если другие типы клеток мешают экспериментальным целям. Однако чистота достигается за счет снижения общего выхода В-клеток. Примечательно, что как для обогащенных, так и для необогащенных культур В-клеток доля В-клеток в исходных препаратах PBMC или спленоцитов имеет решающее значение. В частности, для PBMC мы рекомендуем проводить скрининг различных макак у людей с высоким процентом В-клеток в периферической крови, чтобы получить большое количество В-клеток для экспериментов, поскольку это значение может сильно различаться у людей27. PBMC могут быть получены путем регулярного кровотечения или лейкафереза42.
Протокол редактирования генов приводит к эффективному редактированию генов, как правило, от 60% до 80% нокаутов и 5-20% от нокаута В-клеток, хотя мы достигли до 90% нокаута BCR и 40% нокаута BCR В-клеток (рис. 5 и рис. 6).
Основными параметрами эффективного редактирования В-клеток макак-резусов являются эффективность резки сгРНК, параметры электропорации, MOI и качество препарата rAAV6. Эффективность резки сгРНК-кандидатов должна быть определена эмпирически, чтобы обеспечить оптимальное редактирование и дизайн HDRT. Представленные здесь параметры электропорации уравновешивают эффективность с жизнеспособностью для получения максимального общего количества отредактированных В-клеток, а не наибольшего процента отредактированных В-клеток. Если требуется более высокий процент отредактированных ячеек, рекомендуется повышенное напряжение (до 1,750 В) или измененная длина импульса (10-30 мс), хотя может наблюдаться большая гибель клеток. Мы также отметили несколько более высокую эффективность редактирования в В-клетках селезенки по сравнению с В-клетками из PBMC того же человека (рис. 5); Однако основная причина этого в настоящее время неизвестна.
Мы обнаружили, что добавление 1% ДМСО после электропорации значительно увеличило эффективность редактирования генов на ~ 40% в В-клетках макак-резусов, не влияя на жизнеспособность клеток (рис. 6A-C), в соответствии с отчетами в других клетках43. Однако следует избегать расширенного культивирования в 1% ДМСО, который может повлиять на жизнеспособность клеток. При желании ДМСО может быть полностью опущен.
Культивирование клеток в небольшом объеме после электропорации в течение нескольких часов вместе с rAAV6 приводит к более высокой эффективности редактирования, вероятно, из-за лучшей трансдукции HDRT rAAV6 и, таким образом, более высокой внутриклеточной концентрации HDRT в соответствующее время, когда Cas9 активен. Мы обнаружили, что культивирование клеток таким образом в течение 8 часов не влияло на жизнеспособность клеток, но эффективность редактирования не увеличивалась резко после 5 часов (рис. 6). Если требуется только нокаут, а не нокаут, этот шаг можно пропустить.
В заключение мы представляем комплексные протоколы редактирования генов В-клеток макак-резуса in vitro и производства rAAV6 HDRT, необходимого для эффективного нокаутирования желаемых конструкций. Эти протоколы позволяют быстро и экономически эффективно тестировать многие конструкции, упакованные как rAAV6, и позволяют проводить доклинические испытания осуществимости и масштабируемости терапии В-клетками в более релевантной модели приматов.
Раскрытие информации
Конкурирующие интересы не декларируются.
Благодарности
Мы хотели бы поблагодарить Гарри Б. Гристика и Памелу Бьоркман за предоставление антигена RC1, а также все лаборатории Нуссенцвейга и Мартина за критическое обсуждение. Эта работа была поддержана грантом Фонда Билла и Мелинды Гейтс INV-002777 (M.C.N.) и Программой внутренних исследований Национального института аллергии и инфекционных заболеваний Национальных институтов здравоохранения. (Р.Г. и М.А.М.). M.C.N. является следователем HHMI.
Материалы
| Name | Company | Catalog Number | Comments |
| 1.5 mL tube sterile, Dnase, Rnase and purogen free | Stellar Scientific | T17-125 | or similar |
| 10 mL serological pipette, polystyrene, sterile, nonpyrogenic, DNase-/RNase-free, and Human DNA-free | Corning | 4488 | or similar |
| 15 cm tissue culture dish | Falcon | 353025 | or similar |
| 15 mL polypropylene conical tybe | Falcon | 352097 | or similar |
| 25 mL serological pipette, polystyrene, sterile, nonpyrogenic, DNase-/RNase-free, and Human DNA-free | Corning | 4489 | or similar |
| 250 mL polypropylene conical tybe | Corning | 430776 | or similar |
| 293AAV cell line | Cell Biolabs | AAV-100 | |
| 2-B-mercapto-ethanol, 55mM (1000x) | Gibco | 21985-023 | |
| 48-well tissue culture plate | Corning | 3548 | or similar |
| 5 mL serological pipette, polystyrene, sterile, nonpyrogenic, DNase-/RNase-free, and Human DNA-free | Corning | 4487 | or similar |
| 5 mL syringes with Luer-Lok Tip | BD | 309646 | or similar |
| 50 mL polypropylene conical tybe | Falcon | 352070 | or similar |
| 50 mL serological pipette, polystyrene, sterile, nonpyrogenic, DNase-/RNase-free, and Human DNA-free | Corning | 4490 | or similar |
| 6-well tissue culture plate | Falcon | 353046 | or similar |
| AAV-6 Packaging System (plasmids) | Cell Biolabs | VPK-406 | |
| AAV6 Reference Materials (full capsids) | Charles River | RS-AAV6-FL | |
| Accu-jet S Pipette Controller | Brand | 26350 | or similar pipette controller |
| Antibiotic/Antimycotic 100x | Gibco | 15260-062 | |
| anti-human CD14 AlexaFluor647 | Biolegend | 301812 | |
| anti-human CD14 biotin | BioLegend | 301826 | |
| anti-human CD16 AlexaFluor700 | BD Biosciences | 557920 | |
| anti-human CD16 biotin | BioLegend | 302004 | |
| anti-human CD20 PECy7 | Biolegend | 302312 | |
| anti-human CD3 biotin | Thermo Fisher | APS0309 | |
| anti-human CD3 PE | BD Biosciences | 552127 | |
| anti-human CD33 biotin | Miltenyi | 130-113-347 | |
| anti-human CD64 biotin | BioLegend | 305004 | |
| anti-human CD66 biotin | Miltenyi | 130-100-143 | |
| anti-human CD89 biotin | BioLegend | 354112 | |
| anti-human CD8a biotin | BioLegend | 301004 | |
| anti-human HLA-DR BV605 | Biolegend | 307640 | |
| anti-human Ig light chain lambda APC | Biolegend | 316610 | |
| anti-human IgM BV421 | Biolegend | 314516 | |
| anti-Human Kappa Light Chain FITC | Fisher Scientific | A18854 | |
| Autoclave | Steris | Amsco Lab 250 | or similar |
| Cell culture CO2 incubator | Fisher Scientific | 51026331 | or similar |
| Centrifugal Filter Unit (Amicon Ultra - 4, 100 kDa) | Millipore | UFC810024 | |
| Centrifuge 5920 R | Eppendorf | EP022628188 | or any other, coolable swinging bucket centrifuge with inserts for 96-well plates, 15, 50 and 250 mL size tubes |
| Chloroform | Fisher Scientific | C298SK-4 | |
| Cpg ODN | Invivogen | tlrl-2395 | |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | 34869-500ML | |
| DMEM, High Glucose | Gibco | 11965092 | |
| DNaseI (RNase-free) | New England Biolabs | M0303L | |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | |
| Electroporation kit (Neon Transfection System 10 µL) | Fisher Scientific | MPK1096 | or other sizes or 100 uL transfection kit MPK 10096 |
| Electroporation system (Neon Transfection System) | Fisher Scientific | MPK5000 | |
| FCS | Hyclone | SH30910.03* | |
| Ficoll-PM400 (Ficoll-Paque PLUS) | Cytiva | 17144002 | or similar |
| Fume Hood | Fisher Scientific | FH3943810244 | or similar |
| Glutamine 200 mM | Gibco | 25030-081 | |
| Graduated Cylinder 1L | Corning | 3022-1L | or similar |
| Hemocytometer | Sigma-Aldrich | Z375357-1EA | or similar |
| HEPES 1M | Gibco | 15630-080 | |
| HEPES 1M | Gibco | 15630-080 | |
| Hot Plate Magnetic Stirrer | Fisher Scientific | SP88857200 | or similar |
| Human BAFF | Peprotech | 310-13 | |
| Human BD Fc Block | BD | 564220 | |
| Human IL-10 | Peprotech | 200-10 | |
| Human IL-2 | Peprotech | 200-02 | |
| Hydrochloric acid | Fisher Scientific | A144S-500 | |
| Hydrophilic Polyethersulfone Syringe Filters, (Supor membrane), Sterile - 0.2 µm, 25 mm | Pall | 4612 | |
| Insulin-Transferin-Selenium, 100x | Gibco | 41400-045 | |
| ITR primer forward: GGAACCCCTAGTGATGGAGTT | Integrated DNA Technologies | custom | |
| ITR primer reverse: CGGCCTCAGTGAGCGA | Integrated DNA Technologies | custom | |
| Laminar flow biosafety cabinet | The Baker Company | SG403A | or similar |
| Large magnetic depletion (LD) Column | Miltenyi Biotec | 130-042-901 | |
| Magentic seperator (MidiMACS separator and multistand) | Miltenyi Biotec | 130-090-329 | |
| Magnetic stir bar | Fisher Scientific | 14-512-127 | or similar |
| Magnetic streptavidin beads (Streptavidin MicroBeads) | Miltenyi Biotec | 130-048-101 | |
| Maxiprep kit | Machery-Nagel | 740414.5 | or similar |
| Media Bottles 2L with cap | Cole-Parmer | UX-34514-26 | or similar |
| MegaCD40L | Enzo | ALX-522-110-C010 | |
| MicroAmp Optical 384-well Reaction Plate | Fisher Scientific | 4309849 | |
| MicroAmp Optical Adhesive Film | Fisher Scientific | 4311971 | |
| Microcentrifuge 5424 R | Eppendorf | 5404000014 | or any other table top centrifuge for 1.5 mL tubes |
| Microwave oven | Panasonic | NN-SD987SA | or similar |
| Nikon TMS Inverted Phase Contrast Microscope | Nikon | TMS | or any other Inverted phase-contrast microscope for cell culture |
| Non-essential amino acids, 100x | Gibco | 11140-050 | |
| Nuclease-free Duplex buffer | Integrated DNA Technologies | 11-01-03-01 | |
| Nuclease-free Water | Qiagen | 129115 | |
| pH meter | Mettler Toledo | 30019028 | or similar |
| Pipetman Classic Starter Kit, 4 Pipette Kit, P2, P20, P200, P1000 and tips | Gilson | F167380 | or similar set of pipettes and tips |
| Pluronic F-68 10 % | Gibco | 24040-032 | |
| Polyethylene Glycol 8000 | Fisher Scientific | BP233-1 | |
| Polyethylenimine, Linear, MW 25000, Transfection Grade (PEI 25K | Polysciences | 23966-100 | |
| Precision Balance | Mettler Toledo | ME4001TE | or similar |
| Pre-Separation Filters (30 µm) | Miltenyi Biotec | 130-041-407 | |
| Pyrex glass beaker 2 L | Cole-Parmer | UX-34502-13 | or similar |
| Pyrex glass beaker 250 mL | Millipore Sigma | CLS1000250 | or similar |
| qPCR Instrument | Fisher Scientific | 4485691 | or similar |
| RC1 antigen randomly biotinylated | Bjorkman lab, CalTech | in house | |
| RPMI-1640 | Gibco | 11875-093 | |
| S.p. Cas9 Nuclease | Integrated DNA Technologies | 1081059 | |
| Scientific 1203 Water Bath | VWR | 24118 | or any water bath set to 37 °C |
| Sodium chloride | Sigma-Aldrich | S7653-5KG | |
| Sodium hydroxide | Sigma-Aldrich | S8045-500G | |
| Sodium Pyruvate 100 mM | Gibco | 11360-070 | |
| Sterile Disposable Filter Units with PES Membranes | Thermo Scientific Nalgene | 567-0020 | |
| Streptavidin-PE | BD Biosciences | 554061 | |
| SYBR Green Master Mix | Fisher Scientific | A25742 | |
| Tetracycline-enabled, self-silencing adenoviral vector RepCap6 | Oxgene | TESSA-RepCap6 | |
| Trypan Blue Solution, 0.4% | Gibco | 15250061 | |
| Trypsin-EDTA (0.05%), phenol red | Gibco | 25300054 | |
| Water Purification System | Millipore Sigma | ZEQ7000TR | or similar |
| Zombie-NIR | Biolegend | 423106 |
Ссылки
- Victora, G. D., Nussenzweig, M. C. Germinal centers. Annual Review of Immunology. 40, 413-442 (2022).
- Plotkin, S. A. Correlates of protection induced by vaccination. Clinical and Vaccine Immunology. 17 (7), 1055-1065 (2010).
- Brinkmann, V., Heusser, C. H. T cell-dependent differentiation of human B cells into IgM, IgG, IgA, or IgE plasma cells: High rate of antibody production by IgE plasma cells, but limited clonal expansion of IgE precursors. Cellular Immunology. 152 (2), 323-332 (1993).
- Chernecky, C. C., Berger, B. J. . Protein Electrophoresis - Serum., 6th edition. , 917-920 (2013).
- Balazs, A. B., et al. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. 481 (7379), 81-84 (2011).
- Greiner, V., et al. CRISPR-mediated editing of the B cell receptor in primary human B cells. iScience. 12, 369-378 (2019).
- Hartweger, H., et al. HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. Journal of Experimental Medicine. 216 (6), 1301-1310 (2019).
- Huang, D., et al. Vaccine elicitation of HIV broadly neutralizing antibodies from engineered B cells. Nature Communications. 11, 5850 (2020).
- Jeske, A. M., Boucher, P., Curiel, D. T., Voss, J. E. Vector strategies to actualize B cell-based gene therapies. Journal of Immunology. 207 (3), 755-764 (2021).
- Nahmad, A. D., et al. In vivo engineered B cells secrete high titers of broadly neutralizing anti-HIV antibodies in mice. Nature Biotechnology. 40 (8), 1241-1249 (2022).
- Nahmad, A. D., et al. Engineered B cells expressing an anti-HIV antibody enable memory retention, isotype switching and clonal expansion. Nature Communications. 11, 5851 (2020).
- Voss, J. E., et al. Reprogramming the antigen specificity of B cells using genome-editing technologies. eLife. 8, 42995 (2019).
- Pesch, T., et al. Molecular design, optimization, and genomic integration of chimeric B cell receptors in murine B cells. Frontiers in Immunology. 10, 2630 (2019).
- Cheong, T. C., Compagno, M., Chiarle, R. Editing of mouse and human immunoglobulin genes by CRISPR-Cas9 system. Nature Communications. 7, 10934 (2016).
- Rogers, G. L., Cannon, P. M. Genome edited B cells: A new frontier in immune cell therapies. Molecular Therapy. 29 (11), 3192-3204 (2021).
- Hung, K. L., et al. Engineering protein-secreting plasma cells by homology-directed repair in primary human B cells. Molecular Therapy. 26 (2), 456-467 (2018).
- Johnson, M. J., Laoharawee, K., Lahr, W. S., Webber, B. R., Moriarity, B. S. Engineering of primary human B cells with CRISPR/Cas9 targeted nuclease. Scientific Reports. 8, 12144 (2018).
- Wu, C. M., et al. Genetic engineering in primary human B cells with CRISPR-Cas9 ribonucleoproteins. Journal of Immunological Methods. 457, 33-40 (2018).
- Luo, B., et al. Engineering of alpha-PD-1 antibody-expressing long-lived plasma cells by CRISPR/Cas9-mediated targeted gene integration. Cell Death and Disease. 11 (11), 973 (2020).
- Laoharawee, K., et al. Genome engineering of primary human B cells using CRISPR/Cas9. Journal of Visualized Experiments. (165), e61855 (2020).
- Laoharawee, K., Johnson, M. J., Moriarity, B. S. CRISPR/Cas9-mediated genome engineering of primary human B cells. Methods in Molecular Biology. 2115, 435-444 (2020).
- Moffett, H. F., et al. B cells engineered to express pathogen-specific antibodies protect against infection. Science Immunology. 4 (35), (2019).
- Hartweger, H., Nussenzweig, M. C. CRISPR comes a-knock-in to reprogram antibodies in vivo. Nature Biotechnology. 40 (8), 1183-1184 (2022).
- Nishimura, Y., Martin, M. A. Of mice, macaques, and men: Broadly neutralizing antibody immunotherapy for HIV-1. Cell Host & Microbe. 22 (2), 207-216 (2017).
- Shedlock, D. J., Silvestri, G., Weiner, D. B. Monkeying around with HIV vaccines: Using rhesus macaques to define 'gatekeepers' for clinical trials. Nature Reviews Immunology. 9 (10), 717-728 (2009).
- Kreuser, S., et al. Efficient methods for generation and expansion of, and gene delivery to rhesus macaque plasma B cells. bioRxiv. , (2021).
- Gujer, C., Sundling, C., Seder, R. A., Karlsson Hedestam, G. B., Lore, K. Human and rhesus plasmacytoid dendritic cell and B-cell responses to Toll-like receptor stimulation. Immunology. 134 (3), 257-269 (2011).
- Kim, J. S., et al. Cell enrichment-free massive ex-vivo expansion of peripheral CD20(+) B cells via CD40-CD40L signals in non-human primates. Biochemical and Biophysical Research Communications. 473 (1), 92-98 (2016).
- Su, W., et al. Self-attenuating adenovirus enables production of recombinant adeno-associated virus for high manufacturing yield without contamination. Nature Communications. 13, 1182 (2022).
- Endo, Y., et al. Short- and long-term clinical outcomes in rhesus monkeys inoculated with a highly pathogenic chimeric simian/human immunodeficiency virus. Journal of Virology. 74 (15), 6935-6945 (2000).
- Balaphas, A., Buchs, N. C., Meyer, J., Hagen, M. E., Morel, P. Partial splenectomy in the era of minimally invasive surgery: The current laparoscopic and robotic experiences. Surgical Endoscopy. 29 (12), 3618-3627 (2015).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 6 (5), 343-345 (2009).
- Labun, K., et al. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Research. 47, 171-174 (2019).
- Strobel, B., Miller, F. D., Rist, W., Lamla, T. Comparative analysis of cesium chloride- and iodixanol-based purification of recombinant adeno-associated viral vectors for preclinical applications. Human Gene Therapy Methods. 26 (4), 147-157 (2015).
- Guo, P., et al. Rapid and simplified purification of recombinant adeno-associated virus. Journal of Virological Methods. 183 (2), 139-146 (2012).
- Vandenberghe, L. H., et al. Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Human Gene Therapy. 21 (10), 1251-1257 (2010).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Conant, D., et al. Inference of CRISPR edits from Sanger trace data. The CRISPR Journal. 5 (1), 123-130 (2022).
- Wilkinson, A. C., et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nature Communications. 12, 686 (2021).
- Wang, Z., et al. A broadly neutralizing macaque monoclonal antibody against the HIV-1 V3-Glycan patch. eLife. 9, 61991 (2020).
- Escolano, A., et al. Immunization expands B cells specific to HIV-1 V3 glycan in mice and macaques. Nature. 570 (7762), 468-473 (2019).
- Pathiraja, V., Matar, A. J., Gusha, A., Huang, C. A., Duran-Struuck, R. Leukapheresis protocol for nonhuman primates weighing less than 10 kg. Journal of the American Association for Laboratory Animal Science. 52 (1), 70-77 (2013).
- Stratigopoulos, G., De Rosa, M. C., LeDuc, C. A., Leibel, R. L., Doege, C. A. DMSO increases efficiency of genome editing at two non-coding loci. PLoS One. 13 (6), 0198637 (2018).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены