
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
DeepOmicsAE: Представление сигнальных модулей при болезни Альцгеймера с помощью анализа протеомики, метаболомики и клинических данных с помощью глубокого обучения
В этой статье
Резюме
DeepOmicsAE — это рабочий процесс, основанный на применении метода глубокого обучения (т. е. автоэнкодера) для уменьшения размерности мультиомиксных данных, обеспечивая основу для прогностических моделей и сигнальных модулей, представляющих несколько слоев омиксных данных.
Аннотация
Большие омиксные наборы данных становятся все более доступными для исследований здоровья человека. В этом документе представлен DeepOmicsAE, рабочий процесс, оптимизированный для анализа мультиомиксных наборов данных, включая протеомику, метаболомику и клинические данные. В этом рабочем процессе используется тип нейронной сети, называемый автоэнкодером, для извлечения краткого набора признаков из многомерных многоомиксных входных данных. Кроме того, рабочий процесс предоставляет метод оптимизации ключевых параметров, необходимых для реализации автоэнкодера. Чтобы продемонстрировать этот рабочий процесс, были проанализированы клинические данные когорты из 142 человек, которые были либо здоровы, либо у которых была диагностирована болезнь Альцгеймера, а также протеом и метаболом их посмертных образцов мозга. Признаки, извлеченные из латентного слоя автоэнкодера, сохраняют биологическую информацию, которая разделяет здоровых и больных пациентов. Кроме того, отдельные выделенные признаки представляют собой отдельные молекулярные сигнальные модули, каждый из которых уникально взаимодействует с клиническими особенностями индивидуумов, обеспечивая способ интеграции протеомики, метаболомики и клинических данных.
Введение
Все большая часть населения стареет, и ожидается, что вближайшие десятилетия бремя возрастных заболеваний, таких как нейродегенерация, резко возрастет1. Болезнь Альцгеймера является наиболее распространенным типом нейродегенеративного заболевания2. Прогресс в поиске лечения был медленным, учитывая наше плохое понимание фундаментальных молекулярных механизмов, управляющих возникновением и прогрессированием заболевания. Большая часть информации о болезни Альцгеймера получена посмертно при исследовании тканей головного мозга, что сделало различение причин и следствий труднойзадачей. Проект по изучению памяти и старения религиозных орденов (ROSMAP) представляет собой амбициозную попытку получить более широкое понимание нейродегенерации, которая включает в себя изучение тысяч людей, которые взяли на себя обязательство ежегодно проходить медицинские и психологические обследования и предоставлять свой мозг для исследований после своейкончины. Исследование посвящено переходу от нормального функционирования мозга к болезни Альцгеймера2. В рамках проекта посмертные образцы мозга были проанализированы с помощью множества омиксных подходов, включая геномику, эпигеномику, транскриптомику, протеомику5 и метаболомику.
Омиксные технологии, обеспечивающие функциональное считывание клеточных состояний (т.е. протеомики и метаболомики)6,7, являются ключом к интерпретации заболеваний 8,9,10,11,12 из-за прямой связи между содержанием белка и метаболитов и клеточной активностью. Белки являются первичными исполнителями клеточных процессов, а метаболиты – субстратами и продуктами биохимических реакций. Мультиомиксный анализ данных дает возможность понять сложные взаимосвязи между данными протеомики и метаболомики, а не оценивать их по отдельности. Мультиомика — это дисциплина, которая изучает несколько слоев многомерных биологических данных, включая молекулярные данные (последовательность и мутации генома, транскриптом, протеом, метаболом), данные клинической визуализации и клинические особенности. В частности, мультиомиксный анализ данных направлен на интеграцию таких слоев биологических данных, понимание динамики их взаимной регуляции и взаимодействия, а также на обеспечение целостного понимания возникновения и прогрессирования заболевания. Тем не менее, методы интеграции мультиомиксных данных остаются на ранних стадиях разработки13.
Автоэнкодеры, разновидность неконтролируемой нейронной сети14, являются мощным инструментом для интеграции мультиомиксных данных. В отличие от контролируемых нейронных сетей, автоэнкодеры не сопоставляют выборки с конкретными целевыми значениями (например, здоров или болен) и не используются для прогнозирования результатов. Одно из их основных применений заключается в уменьшении размерности. Тем не менее, автоэнкодеры имеют ряд преимуществ по сравнению с более простыми методами уменьшения размерности, такими как анализ главных компонент (PCA), t-распределенное стохастическое вложение соседей (tSNE) или однородная многообразная аппроксимация и проекция (UMAP). В отличие от PCA, автоэнкодеры могут фиксировать нелинейные зависимости в данных. В отличие от tSNE и UMAP, они могут обнаруживать иерархические и мультимодальные отношения в данных, поскольку они полагаются на несколько слоев вычислительных блоков, каждый из которых содержит нелинейные функции активации. Таким образом, они представляют собой привлекательные модели для отражения сложности мультиомиксных данных. Наконец, в то время как основное применение PCA, tSNE и UMAP заключается в кластеризации данных, автоэнкодеры сжимают входные данные в извлеченные признаки, которые хорошо подходят для последующих задач прогнозирования15,16.
Вкратце, нейронные сети состоят из нескольких слоев, каждый из которых содержит несколько вычислительных блоков или «нейронов». Первый и последний слои называются входным и выходным слоями соответственно. Автоэнкодеры — это нейронные сети со структурой «песочные часы», состоящие из входного слоя, за которым следуют от одного до трех скрытых слоев и небольшой «латентный» слой, обычно содержащий от двух до шести нейронов. Первая половина этой структуры известна как энкодер и объединена с декодером, отражающим энкодер. Декодер заканчивается выходным слоем, содержащим то же количество нейронов, что и входной слой. Автоэнкодеры принимают входные данные через узкое место и реконструируют их на выходном уровне с целью создания выходного сигнала, максимально точно отражающего исходную информацию. Это достигается путем математической минимизации параметра, называемого «потерями при восстановлении». Входные данные состоят из набора признаков, которые в демонстрируемой здесь заявке будут включать в себя содержание белка и метаболитов, а также клинические характеристики (т.е. пол, образование и возраст на момент смерти). Скрытый слой содержит сжатое и информационно насыщенное представление входных данных, которое может быть использовано для последующих приложений, таких как прогностические модели17,18.
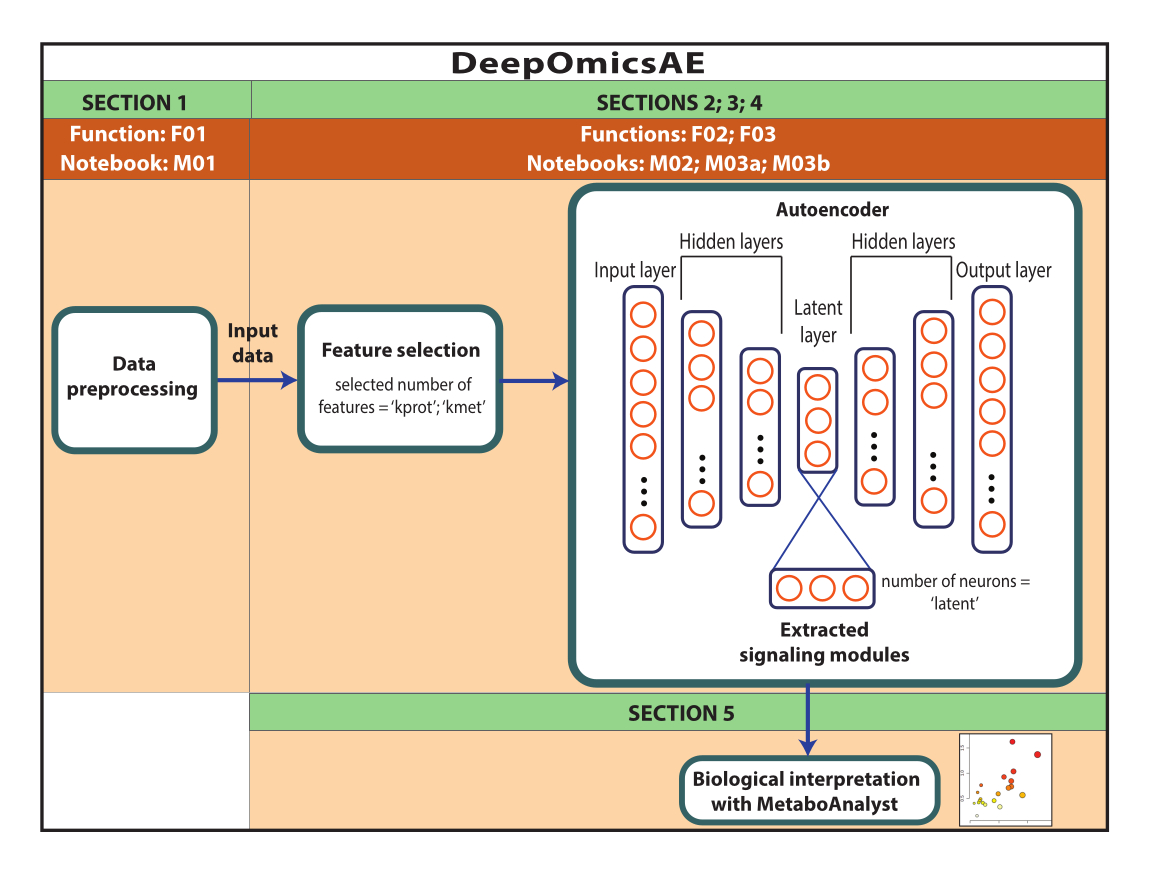
Этот протокол представляет собой рабочий процесс DeepOmicsAE, который включает в себя: 1) предварительную обработку протеомики, метаболомики и клинических данных (т. е. нормализацию, масштабирование, удаление выбросов) для получения данных с согласованным масштабом для анализа машинного обучения; 2) выбор подходящих входных характеристик автоэнкодера, так как перегрузка признаков может скрывать соответствующие паттерны заболеваний; 3) оптимизация и обучение автоэнкодера, в том числе определение оптимального количества белков и метаболитов для отбора, а также нейронов для латентного слоя; 4) извлечение признаков из латентного слоя; и 5) использование извлеченных признаков для биологической интерпретации путем идентификации молекулярных сигнальных модулей и их связи с клиническими признаками.
Этот протокол призван быть простым и применимым биологами с ограниченным опытом вычислений, которые имеют базовое понимание программирования на Python. Протокол фокусируется на анализе мультиомиксных данных, включая протеомику, метаболомику и клинические особенности, но его использование может быть расширено на другие типы данных молекулярной экспрессии, включая транскриптомику. Одним из важных новых применений, представленных этим протоколом, является отображение оценок важности исходных признаков на отдельные нейроны в латентном слое. В результате каждый нейрон в латентном слое представляет собой сигнальный модуль, детализирующий взаимодействие между конкретными молекулярными изменениями и клиническими характеристиками пациентов. Биологическая интерпретация молекулярных сигнальных модулей получена с помощью общедоступного инструмента MetaboAnalyst, который интегрирует данные о генах/белках и метаболитах для получения обогащенных метаболических и клеточных сигнальных путей17.
протокол
ПРИМЕЧАНИЕ: Здесь использованы данные РОСМАП, загруженные с портала AD Knowledge. Для загрузки и повторного использования данных информированное согласие не требуется. Протокол, представленный в настоящем документе, использует глубокое обучение для анализа мультиомиксных данных и выявления сигнальных модулей, которые различают конкретных пациентов или группы выборки, например, на основе их диагноза. Протокол также предоставляет небольшой набор извлеченных признаков, которые суммируют исходные крупномасштабные данные и могут быть использованы для дальнейшего анализа, например, для обучения прогностической модели с использованием алгоритмов машинного обучения (рис. 1). Обратитесь к Дополнительному файлу 1 и Таблице материалов для получения информации о доступе к коду и настройке вычислительной среды перед выполнением протокола. Методы следует выполнять в порядке, указанном ниже.

Рисунок 1: Схема рабочего процесса DeepOmicsAE. Схематическое изображение рабочего процесса для анализа мультиомиксных данных с помощью рабочего процесса. На изображении автоэнкодера прямоугольники представляют слои нейронной сети, а круги — нейроны внутри слоев. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
{kind=link}
1. Предварительная обработка данных
ПРИМЕЧАНИЕ: Целью этого раздела является предварительная обработка данных, включая обработку отсутствующих данных; нормализация и масштабирование протеомной, метаболомной экспрессии и клинических данных; и удаление выбросов. Протокол предназначен для набора данных, который включает данные протеомики, выраженные в виде log2 (ratio); данные метаболомики, выраженные в виде изменения складки; и клинические признаки, включая непрерывные и категориальные признаки. Пациенты или образцы должны быть сгруппированы на основе диагноза или других подобных параметров. Образцы или пациенты должны располагаться в строках, а объекты — в столбцах.
- Чтобы запустить новый экземпляр Jupyter Notebook в браузере, откройте новое окно терминала, введите следующую команду и нажмите клавишу ВВОД.
Записная книжка Jupyter - На домашней странице Jupyter в браузере щелкните записную книжку M01 — данные выражения pre-processing.ipynb, чтобы открыть ее в новой вкладке (Supplemental File 2, Step 1.1).
- Во второй ячейке записной книжки введите имя файла набора данных вместо your_dataset_name.csv.
- В последней ячейке записной книжки вместо M01_output_data.csv введите нужное имя файла выходных данных.
- В пятой ячейке записной книжки укажите положение столбцов для каждого типа данных следующим образом: данные протеомики (cols_prot), данные метаболомики (cols_met), непрерывные клинические данные (например, возраст) (cols_clin_con), бинарные клинические данные (например, пол) (cols_clin_bin). Введите индекс первого столбца для каждого типа данных вместо col_start и индекс последнего столбца вместо col_end; Например: cols_prot = slice(0, 8817). Убедитесь, что значения, указанные в объектах среза, соответствуют индексам первого и последнего столбцов, соответствующим каждому типу данных. Используйте команду в четвертой ячейке той же записной книжки (df.iloc[:, :]), чтобы определить начальную и конечную позиции для каждого типа данных (Дополнительный файл 2, шаг 1.2).
- Выбор ячейки | Запустите все из строки меню Jupyter, чтобы создать файл выходных данных в указанной папке (дополнительный файл 2, шаг 1.3).
ПРИМЕЧАНИЕ: Эти данные будут использоваться в качестве входных данных для протоколов, описанных в разделах 2, 3 или 4.
2. Кастомная оптимизация рабочего процесса (опционально)
ПРИМЕЧАНИЕ: Раздел 2 не является обязательным, так как он требует интенсивного использования компьютера. Пользователи должны сразу перейти к разделу 4, если они решают не выполнять раздел 2. Этот протокол поможет пользователю оптимизировать рабочий процесс в автоматическом режиме. В частности, метод определяет параметры, которые обеспечивают наилучшую производительность автоэнкодера с точки зрения генерации извлеченных признаков, которые хорошо разделяют группы выборок. Оптимизированные параметры, сгенерированные в качестве выходных данных, включают количество признаков, используемых для выбора признаков (k_prot и k_met), и количество нейронов в латентном слое автоэнкодера (латентном). Эти параметры затем можно использовать в протоколе, описанном в разделе 3, для создания модели.
- На домашней странице Jupyter в браузере щелкните записную книжку M02 — DeepOmicsAE model optimization.ipynb, чтобы открыть ее в новой вкладке (Supplemental File 2, Step 2.1).
- Во второй ячейке записной книжки вместо M01_output_data.csv введите имя входного файла. Входными данными для этой функции являются выходные данные из раздела 1.
- В пятой ячейке записной книжки укажите положение столбцов для каждого типа данных следующим образом: данные протеомики (cols_X_prot), данные метаболомики (cols_X_met), клинические данные (cols_clin; включает все клинические данные), все данные о молекулярной экспрессии, включая данные о протеомике и метаболомике (cols_X_expr). Введите индекс первого столбца для каждого типа данных вместо col_start и индекс последнего столбца вместо col_end; Например, cols_prot = slice(0, 8817). Убедитесь, что значения, указанные в объектах среза, соответствуют индексу первого и последнего столбцов, соответствующему каждому типу данных, и используйте команды в третьей и четвертой ячейках записной книжки для изучения данных и определения начальной и конечной позиций для каждого типа данных. Укажите имя столбца, содержащего целевую переменную, вместо y_column_name как y_label (Дополнительный файл 2, шаг 2.2).
ПРИМЕЧАНИЕ: Значения индексов, указанные в cols_X_prot, cols_X_met, cols_clin и cols_X_expr , будут отличаться от значений, используемых в разделе 1, из-за изменения формы кадра данных, происходящего во время предварительной обработки данных. - В шестой ячейке записной книжки укажите, сколько раундов оптимизации нужно выполнить, присвоив значение n_comb. Время обработки составляет примерно 4-5 минут за 10 раундов; 20 минут для 50 патронов и 40 минут для 100 патронов (Дополнительный файл 2, шаг 2.3).
- Выбор ячейки | Запустите все из строки меню Jupyter.
ПРИМЕЧАНИЕ: Выходные переменные kprot, kmet и latent будут сохранены, и к ним можно будет получить доступ из других записных книжек, которые будут использоваться для продолжения аналитического рабочего процесса. График AE_optimization_plot.pdf будет сгенерирован и сохранен в локальной папке (рисунок 2).
3. Реализация рабочего процесса с настраиваемыми оптимизированными параметрами
ПРИМЕЧАНИЕ: Выполняйте этот протокол только после оптимизации метода (раздел 2). Если пользователи не хотят выполнять оптимизацию методов, переходите непосредственно к разделу 4. Этот протокол поможет пользователю создать модель с использованием специально оптимизированных параметров, полученных из раздела 2. Автоэнкодер 1) генерирует набор извлеченных признаков, которые повторяют исходные данные, и 2) идентифицирует важные признаки, управляющие каждым нейроном в латентном слое, эффективно представляя уникальные сигнальные модули. Сигнальные модули будут интерпретированы с использованием протокола, предусмотренного в разделе 5.
- На домашней странице Jupyter в браузере щелкните записную книжку M03a — реализация DeepOmicsAE с пользовательской оптимизацией parameters.ipynb , чтобы открыть ее в новой вкладке (Supplemental File 2, Step 3.1).
- Во второй ячейке записной книжки вместо M01_output_data.csv введите имя входного файла. Входными данными для этой функции являются выходные данные из раздела 1.
- В пятой ячейке записной книжки укажите положение столбцов для каждого типа данных следующим образом: данные протеомики (cols_prot), данные метаболомики (cols_met), клинические данные (cols_clin; включает все клинические данные). Введите индекс первого столбца для каждого типа данных вместо col_start и индекс последнего столбца вместо col_end; Например: cols_prot = slice(0, 8817). Убедитесь, что значения, указанные в объектах среза, соответствуют индексам первого и последнего столбцов, соответствующим каждому типу данных, и используйте команды в третьей и четвертой ячейках записной книжки для изучения данных и определения начальной и конечной позиций для каждого типа данных. Укажите имя столбца, содержащего целевую переменную (например, 0 или 1, соответствующее healthy или diseased) вместо y_column_name как y_label.
ПРИМЕЧАНИЕ: Значения индексов, указанные в cols_X_prot, cols_X_met, cols_clin и cols_X_expr , будут отличаться от индексов, используемых в разделе 1, из-за изменения формы кадра данных, происходящего во время предварительной обработки данных. - Выбор ячейки | Запустите все из строки меню Jupyter, чтобы создать и сохранить графики PCA_initial_data.pdf, PCA_extracted_features.pdf и distribution_important_feature_scores.pdf в локальной папке (рис. 3 и дополнительный рисунок S1). Кроме того, списки важных характеристик для каждого идентифицированного модуля сигнализации будут храниться в текстовых файлах в локальной папке с именем module_n.txt, где n будет заменено номером модуля.
4. Реализация рабочего процесса с заданными параметрами
- Обратитесь к разделу 3 для получения подробных инструкций о том, как запустить этот метод (Дополнительный файл 2, шаг 4.1). Единственное различие между этими двумя протоколами заключается в том, что параметры kprot, kmet и latent (в седьмой ячейке записной книжки) получены математически на основе результатов оптимизации, выполненной так, как показано на рисунке 2.
ПРИМЕЧАНИЕ: Если в разделе 4 отсутствует разделение групп выборок, что указывает на неоптимальную производительность модели, рекомендуется выполнить оптимизацию модели (раздел 2), используя не менее 15 итераций, а по возможности и до 50.
5. Биологическая интерпретация с помощью MetaboAnalyst
- Откройте браузер и перейдите по ссылке ниже, чтобы получить доступ к функции Joint Pathway Analysis на веб-сайте MetaboAnalyst : https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml.
- Откройте папку, в которой были сохранены выходные файлы Метода 3 или Метода 4, и откройте текстовые файлы , module_n.txt для каждого модуля сигнализации n, сгенерированные Методом 3 или Методом 4.
- Найдите белки в текстовых файлах и скопируйте их.
- Вставьте список белков в окно Гены/белки с необязательными изменениями свертывания на веб-странице MetaboAnalyst.
- Повторите описанный выше шаг для метаболитов и вставьте их в окно Список соединений с необязательными изменениями сгиба на той же веб-странице.
- Выберите подходящий организм и тип идентификатора, затем нажмите «Отправить » в нижней части страницы (Дополнительный файл 2, шаг 5.1).
ПРИМЕЧАНИЕ: Убедитесь, что идентификаторы распознаются MetaboAnalyst. К распознанным идентификаторам относятся идентификатор Entrez, официальные символы генов и идентификатор Uniprot для белков; название соединения, идентификатор HMDB и идентификатор KEGG для метаболитов. Если идентификаторы отличаются от этих типов, перед анализом необходимо выполнить соответствующее преобразование. - На следующей странице проверьте сопоставление идентификаторов, прежде чем нажимать кнопку Продолжить , чтобы убедиться, что идентификаторы распознаются.
- На странице «Настройка параметров» выберите «Метаболические пути (интегрированные)» или «Все пути (интегрированные)», чтобы визуализировать соответственно вклад входных данных только в метаболические пути или во все сигнальные пути (дополнительный файл 2, шаг 5.2). На панели Выбор алгоритма выберите Анализ обогащения: Гипергеометрический тест, Мера топологии: Степень центральности и Метод интегрирования: Комбинировать значения p (уровень пути). Нажмите «Отправить» внизу страницы.
- Последняя страница — это представление результатов, в котором представлены результаты анализа обогащения. Обогащенные пути строятся на графике в зависимости от их влияния и значимости, а список путей также представлен в табличном формате.
Результаты
Чтобы продемонстрировать протокол, мы проанализировали набор данных, включающий протеом, метаболом и клиническую информацию, полученную из посмертного мозга 142 человек, которые были либо здоровы, либо у которых была диагностирована болезнь Альцгеймера.
После выполнени?...
Обсуждение
Структура набора данных имеет решающее значение для успеха протокола и должна быть тщательно проверена. Данные должны быть отформатированы в соответствии с разделом 1 протокола. Правильное назначение позиций столбцов также имеет решающее значение для успеха метода. Данные протеомики...
Раскрытие информации
Автор заявляет, что у них нет конфликта интересов.
Благодарности
Эта работа была поддержана грантом NIH CA201402 и премией Корнелльского центра геномики позвоночных (CVG) Distinguished Scholar Award. Опубликованные здесь результаты полностью или частично основаны на данных, полученных с портала знаний AD (https://adknowledgeportal.org). Данные исследования были предоставлены через Партнерство по ускорению медицины болезни Альцгеймера (U01AG046161 и U01AG061357) на основе образцов, предоставленных Центром болезни Альцгеймера Раша, Медицинский центр Университета Раш, Чикаго. Сбор данных был поддержан за счет грантов NIA P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, Департамента общественного здравоохранения штата Иллинойс и Научно-исследовательского института трансляционной геномики. Набор данных метаболомики был сгенерирован в Metabolon и предварительно обработан ADMC.
Материалы
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
Ссылки
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены