
Abstract
Protocol
Representative Results
Discussion
References
Immunology
ELISA Assays: Indirect, Sandwich, and Competitive
Published: April 30th, 2023
Enzyme-linked immunosorbent assay (ELISA) is frequently used to measure the presence and/or concentration of an antigen, antibody, peptide, protein, hormone, or other biomolecule in a biological sample. It is extremely sensitive, capable of detecting low antigen concentrations. The sensitivity of ELISA is attributed to its ability to detect the interactions between a single antigen-antibody complex (1). Moreover, the inclusion of an enzyme-conjugated antigen-specific antibody permits the conversion of a colorless substrate into a chromogenic or fluorescent product that can be detected and easily quantitated by a plate reader. When compared to the values generated by titrated amounts of a known antigen of interest, the concentration of the same antigen in the experimental samples can be determined. Different ELISA protocols have been adapted to measure antigen concentrations in a variety of experimental samples, but they all have the same basic concept (2). Choosing the type of ELISA to perform, indirect, sandwich, or competitive, depends on a number of factors, including the complexity of the samples to be tested and the antigen-specific antibodies available to use. The indirect ELISA is frequently used to determine the outcome of an immunological response, such as measuring the concentration of an antibody in a sample. The sandwich ELISA is best suited for analyzing complex samples, such as tissue culture supernatants or tissue lysates, where the analyte, or antigen of interest, is part of a mixed sample. Finally, the competitive ELISA is most often used when there is only one antibody available to detect the antigen of interest. Competitive ELISAs are also useful for detecting a small antigen with only a single antibody epitope that cannot accommodate two different antibodies due to steric hinderance. The protocol will describe the basic procedures for the indirect, sandwich, and competitive ELISA assays.
The indirect ELISA assay is commonly used to measure the amount of antibodies in serum or in the supernatant of a hybridoma culture. The general procedure for the indirect ELISA assay is:
- Coat wells with antigens
- Add serum or hybridoma culture supernatant containing antibody (primary or 1° antibody)
- Incubate and wash
- Add secondary (or 2°) enzyme-conjugated antibody
- Incubate and wash
- Add substrate
The sandwich ELISA assay differs from the indirect ELISA assay in that the method does not involve coating the plates with a purified antigen. Instead, a "capture" antibody is used to coat the wells of the plate. The antigen is "sandwiched" between the capture antibody and a second "detection" enzyme-conjugated antibody - where both antibodies are specific for the same antigen, but at different epitopes (3). By binding to the capture antibody/antigen complex, the detection antibody remains in the plate. Either monoclonal antibodies or polyclonal antisera can be used as the capture and detection antibodies. The main advantage of the sandwich ELISA is that the sample does not have to be purified before analysis. Moreover, the assay can be quite sensitive (4). Many commercially available ELISA kits are of the sandwich variety and use tested, matched pairs of antibodies. The general procedure for the sandwich ELISA assay is:
- Coat wells with capture antibody
- Add test samples containing antigen
- Incubate and wash
- Add enzyme-conjugated detection antibody.
- Incubate and wash
- Add substrate
Most commercially available sandwich ELISA kits come with enzyme-conjugated detection antibodies. In cases where an enzyme-conjugated detection antibody is not available, a secondary enzyme-conjugated antibody specific for the detection antibody can be used. The enzyme on the secondary antibody performs the same role, which is to convert the colorless substrate to a chromogenic or fluorescent product. The above-mentioned secondary enzyme-conjugated antibody would more like to be used in a "homemade" sandwich ELISA developed by an investigator who has generated their own monoclonal antibodies, for example. One drawback to using a secondary enzyme-conjugated antibody is to be sure it only binds to the detection antibody, and not the capture antibody bound to the plate. This would result in a measurable product in all wells, regardless of the presence or absence of antigen or detection antibody.
Finally, the competitive ELISA assay is used to detect soluble antigens. It is simple to perform, but it is only suitable when the purified antigen is available in a relatively large amount. The general procedure for the competitive ELISA assay is:
- Coat wells with antigen
- Incubate and wash
- Preincubate test sample with enzyme-conjugated primary antibodies
- Add mixture to well
- Incubate and wash away any unbound enzyme-conjugated primary antibody
- Add substrate
The "competition" in this assay comes from the fact that more antigen in the test sample used in step 3 will result in less antibody available to bind to the antigen coating the well. Thus, the intensity of the chromogenic/fluorogenic product in the well at the end of the assay is inversely related to the amount of antigen present in the test sample.
A key component in any type of ELISA is the titrated standards of known concentrations that will allow the user to determine the antigen concentration present in the test samples. Typically, a series of wells are designated for creating a standard curve, where known amounts of a purified recombinant protein are added to the wells in decreasing amounts. When these wells are processed at the same time as the test samples, the user can then have a reference set of absorbance values obtained from a microplate reader for known protein concentrations to go along with the absorbance values for the test samples. The user can then calculate a standard curve to which the test samples can be compared for determining the amount of protein of interest present. The standard curve can also determine the degree of precision of the user's dilution making.
Finally, the last step in each of the ELISA types listed above calls for the addition of a substrate. The degree of conversion of the substrate to product is directly related to the amount of enzyme present in the well. Horseradish peroxidase (HRP) and alkaline phosphatase (AP) are the most common enzymes found conjugated to antibodies. As expected, there are a number of substrates available specific for either enzyme that produce a chromogenic or fluorescent product. Moreover, substrates are available in a range of sensitivities that can increase the overall sensitivity of the assay. The user must also take into consideration the type of instrumentation available for reading the plate at the end of the experiment when picking the type of substrate to use, along with its corresponding enzyme-conjugated antibody.
Commonly used chromogenic substrates for HRP include 2,2'-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) and 3,3',5,5'-tetramethylbenzidine (TMB), whereas p-Nitrophenyl Phosphate (PNPP) is used for AP. ABTS and TMB produce water-soluble green and blue colored reaction products, respectively. The green ABTS product has two major absorbance peaks, 410 and 650 nm, while the blue TMB product is best detected at 370 and 652 nm. The colors of ABTS and TMB change to yellow upon the addition of an acidic stop solution, which is best read at 450 nm. Color development for ABTS is slow, while it is fast for TMB. TMB is more sensitive than ABTS, and may produce a higher background signal if the enzymatic reaction proceeds too long. PNPP produces a yellow water-soluble product after AP conversion that absorbs light at 405 nm.
1. Indirect ELISA
An indirect ELISA is one where the primary antigen-specific antibody is recognized by a secondary conjugated antibody. The following protocol is an example of an indirect ELISA method, where the serum samples of of influenza A virus (IAV)-infected mice are tested for the presence of IAV-specific IgG antibody. One strength of this example is that different secondary antibodies can be used that recognize all antibody isotypes or specific isotypes (e.g., IgG).
Coating antigen to the microplate
- Coat the wells of a 96-well ELISA plate with purified antigen by pipetting 50 µL of purified antigen (2 mg/mL of purified A/PR/8 Influenza A virus in 0.05M Tris-HCl buffer (pH 9.5)) into each well of the plate.
- Cover the plate with an adhesive cover and incubate it overnight at 4°C to allow the antigen to bind to the plate.
- Upon incubation completion, remove the coating solution by flicking the plate over a sink.
Blocking
- Block the remaining protein-binding sites in the coated wells by adding 200 µL blocking buffer, 5% donkey serum in 1X PBS is used here, per well. Alternative blocking reagents include 5% non-fat dry milk or BSA in PBS or normal serum from an animal in which the secondary antibody was generated.
- Incubate for at least 2 hours at room temperature or overnight at 4°C.
- Following the incubation, remove the blocking buffer by flicking the plate and then wash plate with PBS containing 1% Tween-20.
Incubation with the primary antibody
- Prepare a serial dilution of the serum sample, which contains the primary antibody, to obtain a dilution range of 1 to 204,800, using 1X PBS. To do this, first dilute the serum 1:12.5 and then perform a 4X dilution (dilution range - 1:12.5 to 1:204,800).
- Add 100 µL of the serially-diluted serum samples to the wells.
- Cover plate with adhesive cover and incubate at room temperature for 1-2 h.
- Following the incubation, flick the plate over a sink and wash plate with PBS containing 1% Tween-20.
Incubation with the secondary antibody
- Add 100 µL of an enzyme-conjugated secondary antibody, horseradish peroxidase, HRP-conjugated donkey anti-mouse secondary in this experiment, to each well.
- Incubate the plate for 1 hour at room temperature.
- Following the incubation, flick the plate over a sink and then wash plate with PBS containing 1% Tween-20.
Detection
- Add 100 µL of the indicator substrate (3,3',5,5'-tetramethylbenzidine (TMB)) at a concentration of 1 mg/mL to each well.
- Incubate the plate with the substrate for 5-10 min at room temperature.
- After 10 min, stop the enzymatic reaction by adding 100 µL 2N Sulfuric acid (H2SO4).
Within 30 min of adding the stop solution, read the plate using a microplate reader at 405 nm to determine the absorbance of the wells.
2. Sandwich ELISA
In this ELISA version, the experimental sample is "sandwiched" between an unconjugated capture antibody and a conjugated detection antibody, both of which are specific to the same protein but at different epitopes. In the following sandwich ELISA example, concentration of human TNFα was determined in unknown sample using a standard curve generated from 2.5X serial dilution of a known standard, recombinant human TNFα (stating at concentration of 75 pg/mL).
Coating capture antibody to the microplate
- Coat the wells of a 96-well ELISA plate with purified capture antibody by adding 100 µL of capture antibody (1-10 µg/mL range) to each well of the plate.
- Cover plate with an adhesive plate cover and incubate it overnight at 4°C.
- After incubation, remove the coating solution from plate by flicking the plate over a sink.
Blocking
- Block the remaining protein-binding sites in the antibody coated wells by adding 200 µL blocking solution, 5% nonfat dry milk containing PBS, to the wells.
- Incubate for at least 2 h at room temperature or overnight at 4°C.
- Following the incubation, remove the blocking buffer by flicking the plate and then wash plate with PBS containing 1% Tween-20.
Add antigen containing test samples
- Add 100 µL of the test sample to the wells. Seal the plate with an adhesive cover.
- Incubate for 1-2 h at room temperature or overnight at 4°C.
- After incubation, remove the samples by flicking the plate over the sink and then wash the wells with 200 µL 1X PBS containing 1% Tween-20.
Add enzyme-conjugated detection antibody
- Add 100 µL of enzyme-conjugated detection antibody to the wells at a preoptimized concentration.
- Seal the plate with an adhesive cover and incubate at room temperature for 2 h.
- Remove the unbound detection antibody by flicking the plate over a sink and wash the wells with 200 µL 1X PBS containing 1% Tween-20.
Detection
- Add 100 µL of the indicator substrate at a concentration of 1 mg/mL. Any bound enzyme-conjugated detection antibody will convert the substrate to a detectable signal.
- Incubate the plate for 5-10 min at room temperature.
- After 5-10 min, stop the enzymatic reaction by adding 100 µL 2N H2SO4 to the wells. Within 30 min of adding the stop solution, read the plate using a microplate reader to determine the absorbance of the wells.
3. Competitive ELISA
The steps of a competitive ELISA are different from those used in indirect and sandwich ELISA, with the main difference being the competitive binding step between the sample antigen and the "add-in" antigen. The sample antigen is incubated with the unlabeled primary antibody. These antibody-antigen complexes are then added to the ELISA plate, which has been pre-coated with the same antigen. After an incubation period, any unbound antibody is washed away. There is an inverse correlation between the amount of free antibody available to bind the antigen in the well and the amount of antigen in the original sample. For example, a sample with abundant antigen would have more antigen-primary antibody complexes, leaving little unbound antibody to bind to the ELISA plate. An enzyme-conjugated secondary antibody specific to the primary antibody is then added to the wells, followed by the substrate.
Coating antigen to the microplate
- Coat the wells of a 96-well ELISA plate with 100 μL of purified antigen at a concentration of 1-10 μg/mL.
- Cover plate with an adhesive plate cover and incubate the plate overnight at 4°C.
- Following incubation, remove the unbound antigen solution from the wells by flicking the plate over a sink.
Blocking
- Block the remaining protein-binding sites in the coated wells by adding 200 μL of blocking buffer to each well, which can be either 5% non-fat dry milk or BSA in PBS.
- Incubate the plate for at least 2 h at room temperature or overnight at 4°C.
Incubation sample (antigen) with the primary antibody
- While blocking the wells, prepare the antigen-antibody mixture by mixing 150 μL sample antigen and 150 μL of primary antibody for each well in the assay.
- Incubate this mixture for 1 h at 37°C.
Add antigen-antibody mixture to the well
- Now, remove the blocking buffer from the wells by flicking the plate over a sink.
- Then, wash the wells with 1X PBS containing Tween-20.
- Add 100 μL of the sample antigen-primary antibody mixture.
- Incubate the plate at 37°C for 1 h.
- Remove the sample mixture by flicking the plate over a sink.
- Then, wash the wells with 1X PBS containing 1% Tween-20 to remove any unbound antibody.
Add the secondary antibody
- Add 100 μL of an enzyme conjugated secondary antibody, which in this case is AP-conjugated antibody, to each well.
- Incubate the plate for 1 h at 37°C.
- Following incubation, wash the plate with 1X PBS containing 1% Tween-20.
Detection
- Add 100 μL of the substrate solution to each well.
- Wait for 5-10 min.
- After 10 min, stop the enzymatic reaction by adding 100 μL 2N sulfuric acid to the wells. Then, measure the absorbance in a microplate reader within 30 min of adding the stop solution
In the following example of an indirect ELISA, the presence of influenza A virus (IAV)-specific IgG in the serum of IAV-infected mice was determined. C57Bl/6 mice were infected with influenza A virus (A/PR/8; 105 PFU in 100 µL PBS i.p.) and serum was collected 28 days later. To quantitate the amount of IAV-specific IgG in the serum, 96-well ELISA plates were coated with purified A/PR/8 Influenza A virus (50 µL/well of 2 mg/ml PBS virus) overnight at 4°C. Coated plates were blocked for 1 hour at room temperature with 5% normal donkey serum in PBS, followed by incubation with diluted serum samples from IAV-challenged mice overnight at 4°C. The serum was initially diluted 1:12.5, followed by 1:4 dilutions (dilution range - 1:12.5 to 1:204,800). After washing, plates were incubated with an alkaline phosphatase (AP)-conjugated donkey anti-mouse IgG for 1 h. The plates were washed, and then p-Nitrophenyl Phosphate (PNPP; 1 mg/mL, 100 µL/well) was added. The colorless PNPP solution turns to a yellow color when AP is present. After 5-10 min, the enzymatic reaction was stopped by adding 100 µL/well 2N H2SO4. The plate was read on a microplate reader at 405 nm. The results obtained are shown in Table 1 and Figure 1.
| Sample | Wells | OD405 | Mean |
| Serum 1:12.5 | A1 | 2.163 | 2.194 |
| B1 | 2.214 | ||
| C1 | 2.204 | ||
| Serum 1:50 | A1 | 1.712 | 1.894 |
| B1 | 2.345 | ||
| C1 | 1.624 | ||
| Serum 1:200 | A1 | 1.437 | 1.541 |
| B1 | 1.73 | ||
| C1 | 1.456 | ||
| Serum 1:800 | A1 | 1.036 | 0.957 |
| B1 | 0.912 | ||
| C1 | 0.923 | ||
| Serum 1:3200 | A1 | 0.579 | 0.48 |
| B1 | 0.431 | ||
| C1 | 0.429 | ||
| Serum 1:12800 | A1 | 0.296 | 0.281 |
| B1 | 0.312 | ||
| C1 | 0.236 | ||
| Serum 1:51200 | A1 | 0.308 | 0.283 |
| B1 | 0.299 | ||
| C1 | 0.243 | ||
| Serum 1:204800 | A1 | 0.315 | 0.303 |
| B1 | 0.298 | ||
| C1 | 0.297 |
Table 1: Indirect ELISA assay data. Serum dilutions (from 1:12.5 to 1:204,800), of influenza A virus (IAV)-infected mice containing IAV-specific IgG, optical density (OD) (405 nm) values and mean OD405 values.
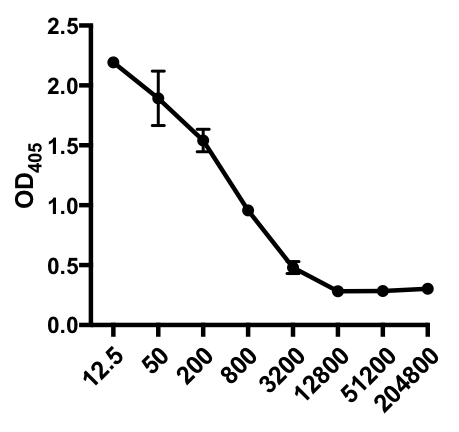
Figure 1: Indirect ELISA assay scatter plot of mean OD405 values(+ S. D.) and serum dilutions (from 1:12.5 to 1:204,800), of influenza A virus (IAV)-specific IgG in the serum of IAV-infected mice. The OD405 values can be inversely correlated to the serum dilutions.
In the following example of a sandwich ELISA, a 1:2.5 dilution of recombinant human TNFα standards (starting at a concentration of 75 pg/mL) was added to the indicated wells of a 96-well flat-bottom plate. These standards led to a corresponding 2.5-fold change in the absorbance readings.
| Sample | Concentration (pg/mL) | Wells | Values | Mean Value | Back Concentration Calculation | Average |
| Standard 1 | 75 | A1 | 1.187 | 1.169 | 76.376 | 75.01 |
| A2 | 1.152 | 73.644 | ||||
| Standard 2 | 30 | B1 | 0.534 | 0.52 | 30.827 | 29.962 |
| B2 | 0.506 | 29.098 | ||||
| Standard 3 | 12 | C1 | 0.23 | 0.217 | 12.838 | 12.105 |
| C2 | 0.204 | 11.372 | ||||
| Standard 4 | 4.8 | D1 | 0.09 | 0.084 | 5.055 | 4.726 |
| D2 | 0.078 | 4.398 | ||||
| Standard 5 | 1.92 | E1 | 0.033 | 0.031 | 1.941 | 1.86 |
| E2 | 0.03 | 1.778 | ||||
| Standard 6 | 0.768 | F1 | 0.009 | 0.011 | 0.626 | 0.764 |
| F2 | 0.014 | 0.901 | ||||
| Standard 7 | 0.307 | G1 | 0.002 | 0.004 | 0.238 | 0.377 |
| G2 | 0.007 | 0.516 |
Table 2: TNFα Sandwich ELISA standard curve data. A 1:2.5 dilution of recombinant human TNFα standards (75 to 0.3 pg/mL), OD (450 nm) values, mean OD450 values, back concentration calculations and their averages.
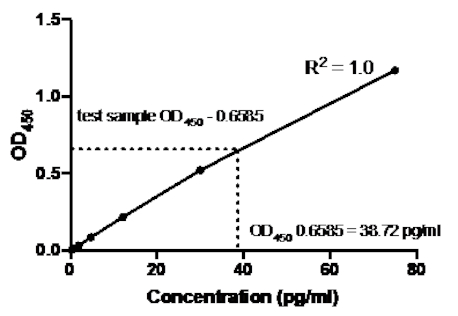
Figure 2: Standard Curve for TNFα sandwich ELISA. A 1:2.5 dilution of recombinant human TNFα standards (75 to 0.3 pg/mL) was analyzed using sandwich ELISA.The OD450 values can be directly correlated to the standard dilution concentrations. The amount of TNFα protein in the test sample was determined using the standard curve, which corresponds to a concentration of 38.72 pg/mL.
Once the standard curve was generated, the amount of TNFα protein in the test sample was determined. In this sandwich ELISA example, the test samples gave OD450 readings of 0.636 and 0.681, which give an average of 0.6585. When plotting this OD450 reading on the above chart, this corresponds to a TNFα concentration of 38.72 pg/ml.
As demonstrated, a range of immunoassays (with slight variation in protocols) fall within the ELISA technique family. Determining which version of ELISA to use depends on a number of factors, including what antigen is being detected, the monoclonal antibody available for a particular antigen, and the desired sensitivity of the assay (5). Some strengths and weaknesses of the different ELISAs described herein are:
| ELISA | Strengths | Weaknesses |
| Indirect | 1) High sensitivity due to the fact that multiple enzyme-conjugated secondary antibodies can bind to the primary antibody | 1) High background signal may occur because the coating of the antigen of interest to the plate is not specific (i.e., all proteins in the sample will coat the plate) |
| 2) Many different primary antibodies can be recognized by a single enzyme-conjugated secondary antibody giving the user the flexibility of using the same enzyme-conjugated secondary antibody in many different ELISA (regardless of the antigen being detected) | ||
| 3) Best choice when only a single antibody for the antigen of interest is available | ||
| Sandwich | 1) The use of antigen-specific capture and detection monoclonal antibody increases the sensitivity and specificity of the assay (compared to the indirect ELISA) | 1) Optimizing the concentrations of the capture and detection monoclonal antibodies can be difficult (especially for non-commercial kits) |
| 2) Best choice for detecting a large protein with multiple epitopes (such as a cytokine) | ||
| Competitive | 1) Impure samples can be used | 1) Requires a large amount of highly pure antigen to be used to coat plate |
| 2) Less sensitivity to reagent dilution effects | ||
| 3) Ideal for detecting small molecules (such as a hapten) |
Table 3: Summary. A summary of the strengths and weaknesses of the different ELISA techniques.
While a simple and useful technique, there are also some drawbacks to any ELISA. One is the uncertainty of the amount of the protein of interest in the test samples. If the amount is too high or too low, the absorbance values obtained by the microplate reader may fall above or below the limits of the standard curve, respectively. This will make it difficult to accurately determine the amount of protein present in the test samples. If the values are too high, the test sample can be diluted prior to adding to the wells of the plate. The final values would then need to be adjusted according to the dilution factor. As mentioned, homemade kits often require careful optimization of the antibody concentrations used to yield a high signal-to-noise ratio.
- Porstmann, T. and Kiessig S.T. Enzyme immunoassay techniques. An overview. Journal of Immunological Methods. 150 (1-2), 5-21 (1992).
- Suleyman Aydin. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides, 72, 4-15 (2015).
- Gan. S. D. and Patel K. R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. Journal of Investigative Dermatology, 133 (9), 1-3 (2013).
- Kohl, T. O. and Ascoli C.A. Immunometric Antibody Sandwich Enzyme-Linked Immunosorbent Assay. Cold Spring Harbor Protocols, 1 (6), (2017).
- Sakamoto, S., Putalun, W., Vimolmangkang, S., Phoolcharoen, W., Shoyama, Y., Tanaka, H., and Morimoto S. Enzyme-linked immunosorbent assay for the quantitative/qualitative analysis of plant secondary metabolites. Journal of natural medicines, 72 (1), 32-42 (2018).
Tags
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved