
Summary
Abstract
Protocol
Discussion
Acknowledgements
Materials
References
Biology
アイソフォーム特異的遺伝子標的を同定するためのチップを使用してゲノムワイド解析
ここでは、ヒストン結合ドメインが異なるタンパク質アイソフォームのゲノムワイド位置解析のためのクロマチン免疫沈降(ChIP)の手順を提示している。我々はKDM5A/JARID1A/RBP2ヒストン脱メチル化酵素の標的を同定するためのChIP - seqの分析に適用しています。
彼らのターゲットへの転写やエピジェネティックな要因の採用は、彼らの調節に重要なステップです。目立つように特定のヒストン修飾に結合するタンパク質のドメインは、募集に掲載。そのようなドメインは、いくつかのクロマチン結合タンパク質に見られる植物のホメオドメイン(PHD)、です。エピジェネティックな因子RBP2が複数のPHDドメインがある、しかし、彼らはさまざまな機能(図4)がある。特に、ヒト白血病におけるRBP2発癌性の融合に見られるC末端PHDドメインは、ヒストンH3のトリメチル化リシン4(H3K4me3)1に結合する。 C末端PHDを含むRBP2のアイソフォームに対応する転写産物は、単球2にpromonocytic、リンパ腫由来、U937細胞の分化の間に蓄積されます。両方のデータセットと一致し、ゲノムワイドな解析は、分化したU937細胞では、RBP2タンパク質が非常にH3K4me3 3に富むゲノム領域に局在化されることを示した。そのターゲットにRBP2の局在はRBP2ヒストン脱メチル化酵素活性と転写活性の減少により、H3K4me3の減少と相関している。対照的に、RBP2の二つの博士号は、H3K4me3をバインドすることはできません。特に、RBP2のC末端ドメインPHDは小さなRBP2アイソフォーム4の不在である。それはH3K4me3との相互作用を欠いているRBP2、、の小さなアイソフォームは、ゲノムの位置に大きなアイソフォームとは異なることが考えられる。 RBP2アイソフォームの遺伝子位置の違いはRBP2機能で観測された多様性を占める可能性があります。具体的には、RBP2は、網膜芽細胞腫タンパク質(pRBの)により媒介される細胞の分化において重要なプレーヤーです。これらのデータ、以前のゲノムワイドな解析と一致し、アイソフォームを区別することなく、RBP2標的遺伝子の2つのグループ識別:1)差別化とは独立した方法でRBP2に拘束される遺伝子;でRBP2に拘束される2)遺伝子の分化依存的。
アイソフォーム間で局在の違いを識別するために我々はChIP - seqでゲノムワイド位置解析を行った。我々はすべてのRBP2のターゲットの位置を確認したRBP2アイソフォームの両方を検出する抗体を使用する。さらに我々は唯一の大規模な、そして小さなはなくRBP2アイソフォーム(図4)に結合する抗体を持っている。大規模なアイソフォームのターゲットを特定した後、一つは、小さなアイソフォームのターゲットを明らかにするために、すべてのRBP2ターゲットからそれらを差し引くことができます。これらのデータは、ゲノム中のその結合部位へのタンパク質補充にクロマチンと相互作用するドメインの寄与を示しています。
プロトコルは、もともとB.漣、2001年から適応されました。それは、オドムらによるプロトコルのわずかな修正を表します。で見つけることができる5 http://jura.wi.mit.edu/cgi-bin/young_public/navframe.cgi?s=22&f=appendices_downloads
1。事前にブロックし、磁気ビーズへの抗体の結合は、(次のステップの前に夜を実行する必要があります)
- 新鮮BSA / PBS溶液(50 mgを10 mLのBSA PBS -このソリューションは、一週間続く)。1mLにダイナビーズプロテインGを100μLを(IPごとに、複数のIPに対して結合)ウォッシュ
- 磁気スタンドを使用してビーズを収集し、洗浄操作をさらに2回繰り返す。
- 次に、PBS / BSA溶液中でのビーズのスラリー(IP毎)の250μlのために抗体10μgを追加し、4℃で回転プラットフォーム上で一晩インキュベート℃の
- 完了すると、PBS / BSA溶液1mlにビーズを3回洗浄し、PBS / BSA溶液(IP毎)10μlに再懸濁します。
2。細胞架橋
- この手順を開始するには、各免疫沈降、またはIPのために約10 8細胞を成長させる。ここでは、びまん性組織球性リンパ腫U937細胞を96時間のTPAによる単球分化のために使用され、誘導されています。
- 細胞増殖に続いて、1%の最終濃度に直接メディアにホルムアルデヒド溶液を加える。その後、スワールフラスコの簡潔にし、それらを室温で10分間座ってすることができます。その後、培地を吸引除去し、氷冷PBS 15 mLで細胞をリンス。一度この洗浄を繰り返します。
- 次に、それぞれに溶解バッファー1(プロテアーゼ阻害剤を含む50mM HEPES - KOH、pH7.5の、140mMのNaCl、1mMのEDTA、10%グリセロール、0.5%NP - 40、0.25%トリトンX - 100、)6 mLを加え氷の上にフラスコの。その後、4℃で20分間フラスコを揺らし℃に
- 最後に、セルスクレーパーを用いて細胞を収穫し、15 mLコニカルチューブに移す。この時点で、細胞は-80℃で保存することができます
3。細胞の超音波処理
- 細胞が凍結されていた場合、それらを解凍。融解後、4℃で10分間、3,000 rpmで細胞をスピンダウンし、上清を捨てる。その後、溶解バッファー2(10mMトリス- HCl、pH8.0の、200 mMのNaCl、1mMのEDTA、0.5mMのEGTA、プロテアーゼ阻害剤を含む)の6 mLで細胞を懸濁します。室温で10分間穏やかにチューブを揺らし。
- 遠心分離を繰り返して、溶解バッファー3(10mMトリス- HCl、pH8.0の、100mMのNaCl、1mMのEDTA、0.5mMのEGTA、0.1%デオキシコール酸ナトリウム、0.5%N -ラウロイルサルコシン、プロテアーゼ阻害剤を含む)2.5 mLで細胞を再懸濁。
- 次に、50〜60%の振幅の間に設定さブランソン450超音波処理のマイクロチップを氷水のビーカーに置くことで超音波処理のために懸濁液を調製。
- 1分間氷上で30秒間一定バーストと涼しいのためのソリューションを超音波処理してください。これらのパルスを10〜15回繰り返します。その後、上下チューブの内容物をピペットで新しいチューブに移す。さらに5回パルスソリューションを冷却し続ける。
- 超音波処理後、溶液の体積のトリトンX - 100〜1 / 10 10%を追加。 1.5 mLの遠心チューブにライセートを移し、マイクロ遠心機でゴミをスピンアウト。次に、新しいチューブに細胞溶解液を移す。
4。クロマチン免疫沈降
- 前のクロマチン免疫沈降法、またはチップに、入力のサンプルとして、細胞溶解物50μLを保存します。その後、ダイナビーズ上述のように事前に準備されている抗体に事前にバインドされて清澄化細胞ライセートを組み合わせる。 4℃で一晩、50μLの入力サンプルに加えて、混合物を揺らし。
- 洗浄緩衝液1 mL(50mMのHEPES - KOH、pHは7.6、0.5 M塩化リチウム、1mMのEDTA、0.7%デオキシコール酸ナトリウム、1%NP - 40)でビーズを洗浄する。その後、ビーズを収集し、上清を除去する磁気スタンドを使用してください。この洗浄を6〜8回を繰り返します。
- 1 mLのTE -プラス50mMのNaCl(10mMトリス- HCl、pH8.0の、50mMのNaCl、1mMのEDTA)で最終洗浄を行います。 4℃で2分間3000rpmで遠心でビーズをスピン℃に遠心分離に続いて、任意の残留TEバッファーを吸引除去する。
- その後、試料に溶出バッファーを100μL(50mMのTris - HCl、pH8.0の、10mMのEDTA、1%SDS)を追加します。直後に、65でサンプルをインキュベート℃で10分間から15分間。懸濁液中にビーズを保つために1.5 mlチューブをラックごとに2分に対して、チューブに傷を付けない。
- 遠心分離と磁気スタンドを使用してビーズを収集し、そしてPCRチューブに上清を移す。その後、以前に保存した入力サンプルを取得し、溶出バッファーの3つのボリュームを追加します。
- 最後に、65℃一晩サーマルサイクラーにIPと入力サンプルを配置° Cにクロスリンケージを逆。
- 翌日、試料をTE緩衝液の1ボリュームを追加します。その後、0.2μg/μLの終濃度にRNase Aを追加。 37℃で1〜2時間サーマルサイクラーにサンプルをインキュベート℃に
- インキュベーション後、0.2μg/μLの終濃度にプロテイナーゼKを追加します。その後、2時間55℃でサーマルサイクラーにサンプルをインキュベートする。
- 次に、フェノールの一冊で一回のサンプルを抽出する。クロロホルム:イソアミルアルコール、フェノールの一冊でサンプルをもう一度抽出する。イソアミルアルコール:最後に、クロロホルムの一冊で一回より多くのサンプルを抽出する。
- その後、各サンプルにグリコーゲンの30μgのを追加。また、サンプルにエタノールの0.2モルと2つのボリュームの最終濃度にNaClを加える。 -80 30分のためにそれらをインキュベート℃にインキュベーション後、サンプルをスピンし、上清をデカントする。 75%エタノール500μLでペレットを洗浄してください。その後、ペレットを乾燥し、それらは水40μLで再懸濁する。
- 超音波処理の手順によって生成されるDNA断片のサイズを確認するには、1.8%アガロースゲルで精製された入力サンプルの5μgをロードし、ゲルを実行します。超音波処理の手順は、150から350塩基対の範囲内でフラグメントを生成する必要があります。フラグメントがこの範囲に該当しない場合は、、超音波処理の手順は、バーストと振幅の数を変えることによって調整されるべきである。
- IPサンプルと入力サンプルの希釈系列の1μlと遺伝子特異的PCRを行うことにより、制御結合領域の濃縮は、チップの堅牢性と特異性を確認するには。 2〜3"バウンド"の領域と非連結コントロールの領域は、IPと入力サンプルの品質をテストするために選択する必要があります。
5。ゲノムDNAの増幅
- ゲノムDNAサンプル調製キットを使用してDNA断片へのアダプターの'テールに加え、ライゲーション、最後の修復を実行、IPサンプル34μlまたは、入力サンプルの200 ngのを皮切りにhttp://www.illumina.com/systems/をgenome_analyzer.ilmn (イルミナ社、サンディエゴ、CA)。
- アプライPCR精製キットを用いてDNAを精製し、その後50℃にあらかじめ温めておいたれているバッファEB(10 mMトリスClを、pH8.5)にそれを溶出させる例えば、それぞれ、IPと入力のDNAサンプルの最終的な溶出に23μLと46μLを使用してください。
- アダプタを搭載したDNAの23μL、およびキットから25μLPhusion DNAポリメラーゼ、20μMのSol_PCR_1、および20μMSol_PCR_2の1μLの1μLを用いてPCR反応を行う。次のようにPCR反応実行:、2)10秒98℃、3)65 ° Cで30秒、℃、98℃1)30秒4)30秒、72℃で、繰り返しのステップステップ2-4 18最終的に4℃で保持して時間と72℃で5分° C、° C.
- PCR増幅に続いて、QIAGENアプライPCR精製キットでDNAを精製。 50〜バッファのEBのプレ暖かい15μL℃、DNAを溶出する。サンプル1:4の0.5μLを希釈し、光度計の読み取りを行います。
6。増幅産物のゲル精製
- 増幅産物を精製するために、HPLCグレードの水を用いて50 mLの1.8%アガロースゲルを準備する。アガロースが溶解した後、TAEとエチジウムブロマイドを追加。その後、ゲルを注ぐ。入力とIPサンプルにローディングバッファーの4μLを追加した後ゲルをロードします。その後、45分間120ボルトで泳動する。
- それが実行された後のゲルを解析する。ゲルは、150および350塩基対の間の範囲のフラグメントが生成されていることを明らかにする必要があります。彼らは長さと、アダプタで50〜250塩基対の間のゲノムDNA断片を表しています。
- メスを持つ150 350塩基対の範囲内の物質を含むゲルの物品の領域。約120塩基対で動作するアダプタ - アダプタのバンドを、切り出すように注意してください。メーカーの指示ごとQIAクイックゲル抽出キットを用いてDNAを回収する。具体的には、400 mgまたは以下のゲルスライスのQIAクイックゲル抽出キットから1つの列を使用してください。抽出プロセスを開始するには、ゲルの1ボリュームにQGバッファの3つのボリュームを追加します。イソプロパノールの1ボリュームを追加し、カラムに混合物をロードする。キット取扱説明書に記載されている標準の手順に続いて、カラムを洗浄するQGを0.5 mlのを使用してください。 H 2 Oを使用して、予め温めておいた50℃DNAを溶出する。それをスピンダウンする前に37℃で5分間、H 2 O30μlの° Cでカラムをインキュベートします。
- 熱のないSpeedvacでダウン正確に11μLのサンプルを乾燥させる。サンプルの1μLを削除し、光度計分光光度計を用いてDNA濃度を測定する。
- 最後に、で説明したイルミナGenome AnalyzerIIe使用して濃縮された遺伝子産物を識別http://genesdev.cshlp.org/content/supplを/ 2008/12/15/22.24.3403.DC1/GuentherSuppMat.pdf 6。
7。代表的な結果
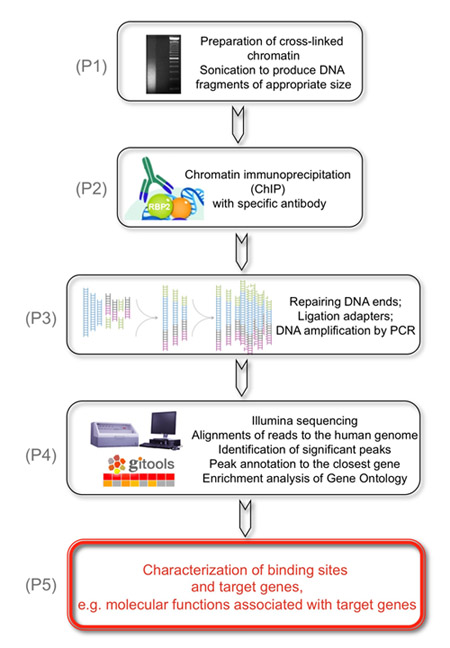
図1は、次の実験の全体的な目標は、KDM5A/JARID1A/RBP2ヒストン脱メチル化酵素のゲノム標的を同定することです。
(P1)これは、適当な大きさのDNA断片を生成するために超音波処理され、クロスリンクされたクロマチンの調製により達成されます。 (P2)第2段階として、RBP2結合したDNA断片は、RBP2抗体で免疫沈降されています。 (P3)次に、回収したDNA断片は、両端に修復され、アダプタは、イルミナClusterの駅でフローセルに分析のためにゲノムDNAのライブラリーを準備するためにゲノムDNAの末端に連結されている。 DNAライブラリーは、サイクル数が少ないを使用してPCRにより増幅されています。 (P4)最後に、イルミナシーケンス短いが一意にヒトゲノムに整列されている読み取り、有意なピークがRBP2濃縮領域を識別するために、同定され、最も近い遺伝子にアノテートされています。 (P5)の結果はRBP2濃縮領域のゲノムワイドな同定に基づくRBP2標的遺伝子と関連分子の機能を示すことが得られる。
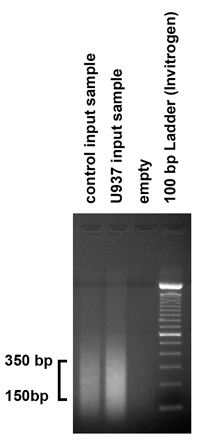
図2。超音波処理の手順によって生成されるDNA断片のサイズを確認するにはクロマチン超音波処理の結果をチェックし 、精製された入力サンプルの5μgの(1 / 10)1.8%アガロースゲルにロードされました。予想通り、私たちの超音波処理の手順は、150から350 bpの範囲内のフラグメントを作り出した。フラグメントがこの範囲に該当しない場合は、、超音波処理の手順は、バーストと振幅の数を変えることによって、それに応じて調整する必要があります。
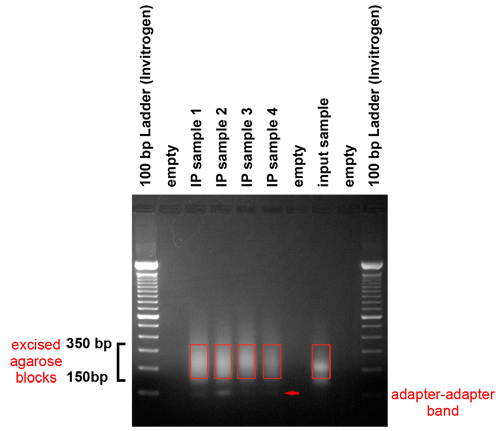
図3。増幅産物のゲル精製。PCR産物は、アダプタを削除し、クラスタ生成プラットフォーム用のテンプレートのサイズ範囲を選択するにはゲル上で実行されています。このゲルは、150および350塩基対の間の範囲内のフラグメントが作り出されたことを示しています。彼らは長さと、アダプタで50〜250塩基対の間のゲノムDNA断片を表しています。メスを用いてゲルの我々は、物品税、選択された地域。ケアは、約120塩基対で動作するアダプタ - アダプタのバンドを、避けるように注意してください。
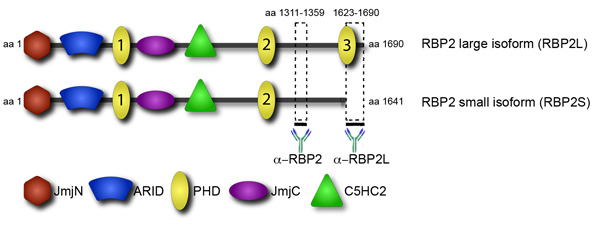
図4。アイソフォーム特異的抗体は、RBP2の大小のアイソフォームを区別することができます。RBP2タンパク質の構造がドメインのビューで提供される。触媒ヒストン脱メチル化JmjCドメインと関連付けられているJmjNドメイン、配列特異的DNA結合、潜在的にDNAや他のタンパク質と相互作用する可能性C5HC2ジンクフィンガー、および複数のPHDドメインが可能な乾燥ドメイン:RBP2は、いくつかのドメインが含まれています。 RBP2アイソフォームを区別するための2つの抗RBP2抗体は太い線で示されるRBP2断片に対して誘導された。
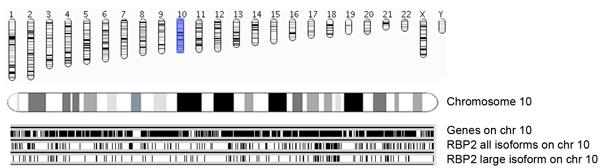
図5a。 (全てのアイソフォームと大きなアイソフォーム)濃縮RBP2を識別結合領域の染色体とEnsemblの遺伝子と一緒にRBP2結合領域の概要。ゲノム座標は、染色体が賢明表示されます。 Ensemblの遺伝子のOccupances(バージョン54、HG18)染色体では(この写真では10番染色体)、黒いバー(上部パネル)として表示されます。各RBP2結合領域は、中央のパネルはすべてRBP2アイソフォームの10番染色体、および底部パネルの結合領域を示す垂直線として表されますRBP2大きなアイソフォームの結合領域を示しています。中央と下部のパネルは、10番染色体上の重複やRBP2アイソフォームの特異的占有の概要を示します。
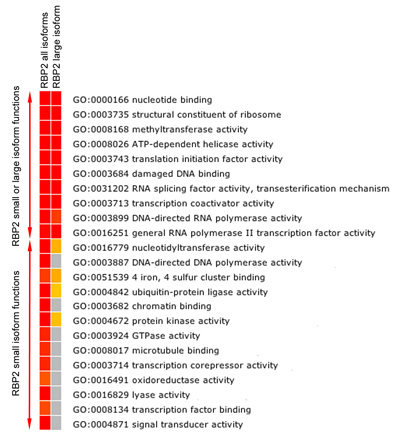
図5b。 RBP2標的遺伝子の機能的な濃縮分析。ヒートマップFDRが大幅に修正を示す(p値≤0.05)濃縮はRBP2(すべてと大きなアイソフォーム)で(RBP2結合領域に最も近い遺伝子)バインドされた遺伝子の中で分子機能のカテゴリを移動します。赤色に向かって色が高い統計的有意性を示すため、黄色は低い統計的有意性を示し、灰色は統計的有意性がないことを示します。濃縮分析はRBP2標的遺伝子の重複とアイソフォーム特異的な分子の機能を示しています。
RBP2アイソフォームの機能的な違いが決定されていない。我々はRBP2アイソフォームがバインドされているゲノム領域を特定し、これらの地域が表す機能的なカテゴリを定義する包括的なアプローチを使用している。これは、得られた配列を読み取るのバイオインフォマティクス解析に続いてのChIP - seqの分析によって達成されました。
RBP2はヒストンテール上のメチル化リジン残基を変更します。我々はRBP2大きなアイソフォームは、ヒトゲノムのさまざまな地域(図5A)に、このモジュールのバインドを欠いているメチル化ヒストンリジンとRBP2小さなアイソフォームの認識モジュールが含まれていることがわかった。重要なのは、アイソフォーム固有の領域と重複する領域が異なる分子の機能(図5B)を持つ遺伝子に属します。例えば、"クロマチン結合"と"転写因子結合"関数がRBP2小さなアイソフォームの遺伝子標的に帰することができますが、ないのRBP2大きなアイソフォーム(図5B)。実際の遺伝子を比較することによって、すべてのアイソフォームとRBP2大きなアイソフォーム(データは示さず)に対して生成されたセットに大きなアイソフォームは、特に、特定の遺伝子に募集されている場合、我々はまた、定義することができます。
この作品は、NIHから115347 - RSG - 08 - 271 - 01 - GMC ACSから、とCA138631からの助成金によって支えられている。
オリゴヌクレオチド配列2006イルミナ株式会社すべての権利を保有。
- Sol_PCR_1
配列5' - AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC T - 3' - Sol_PCR_2
配列5' - CAA GCA GAA GAC GGC ATA CGA GCT CTT CCG ATC T - 3'
- Wang, G. G. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 459 (7248), (2009).
- Benevolenskaya, E. V. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell. 18 (6), 623-623 (2005).
- Lopez-Bigas, N. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol Cell. 31 (4), 520-520 (2008).
- Benevolenskaya, E. V. Histone H3K4 demethylases are essential in development and differentiation. Biochem Cell Biol. 85 (4), 435-435 (2007).
- Odom, D. T. Control of pancreas and liver gene expression by HNF transcription factors. Science. 303 (5662), 1378-1378 (2004).
- Guenther, M. G. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 22 (24), 3403-3403 (2008).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved