蛋白质 - 配体相互作用采用差示扫描荧光法测定
In This Article
Summary
Differential scanning fluorimetry is a widely used method for screening libraries of small molecules for interactions with proteins. Here, we present a straightforward method to extend these analyses to provide an estimate of the dissociation constant between a small molecule and its protein partner.
Abstract
范围广泛的方法是当前可用于确定所述解离的蛋白质之间的恒定和相互作用的小分子。然而,大多数的这些需要访问的专门设备,并且常常需要一定程度的专门知识,以有效地建立可靠的实验和分析数据。差示扫描荧光(DSF),正在越来越多地被作为一种可靠的方法用于蛋白质的相互作用的小分子,无论是对生理确定合作伙伴或打的发现初步筛选。这种技术具有需要只适合于定量PCR,并因此合适的仪器是在大多数机构可用PCR仪的优点;一个优秀的一系列协议已经上市;并且有在文献中所述方法的多种用途强先例。以前的工作已提出了计算从DSF数据的解离常数的若干装置,但这些在数学上是苛刻的。这里,我们数字高程模型onstrate用于从DSF的实验数据有中等量估算解离常数的方法。这些数据通常可以被收集,并在一天内进行分析。我们表明模型是如何不同,可以用来拟合,从简单的结合事件收集的数据,并且其中协同结合或独立的结合位点存在。最后,我们提出在标准模型不适用的情况下的数据分析的实例。这些方法被示出具有收集在市售的对照蛋白的数据,并从我们的研究计划的两种蛋白质。总的来说,我们的方法提供了一种简单的方法为研究人员能够迅速获得进一步的洞察使用DSF的蛋白质 - 配体相互作用。
Introduction
所有的蛋白质会结合,具有不同的亲和力,从简单的离子等大分子其他分子的多元化。在许多情况下,蛋白质结合的小分子伙伴作为其正常功能的一部分( 例如,激酶结合ATP)。其它相互作用可能是无关的功能,但是是作为工具( 例如,小分子,稳定蛋白质,以提高结晶成功,或协助维持在溶液中的蛋白质)实验有用;同时结合于活性位点和蛋白的变构位点可以作为抑制剂,并因此调节酶的活性的小分子。
有多种可用于确定蛋白质的伴侣分子的亲和技术。等温滴定量热1被广泛看作是“黄金标准”,因为它提供了丰富的反应信息,为无标记,并在有限的机会实验rtifacts。然而,尽管在仪表和实验设置自动化的灵敏度最近的改进,它仍然是在蛋白质方面要求相对昂贵的,已经在最佳的低到中等的吞吐量,并且最适合于相互作用的中度至高亲和力(10纳米至100微米杀敌 )2。其他无标签的方法,如表面等离子体共振或双层干涉3提供更高的吞吐量,并取得了灵敏度探测更小的分子,低至100道尔顿。然而,高通量的仪器,这些方法都是比较昂贵的,是正义的地方会有一个持续的吞吐量相关的项目,因此很可能是无法进入的许多学术实验室。
差示扫描荧光(DSF或thermofluor)于2001年4首次被描述为用于药物开发的方法。在这种实现方法具D,蛋白质是培养用荧光染料(最初萘磺酸的染料被使用),以改变其在结合到蛋白质的疏水区的荧光。蛋白质 - 染料样品,然后加热,并在荧光监测的热量增加。蛋白质的折叠和蛋白的疏水部分暴露,产生了一个特征图案中的荧光作为温度( 图1A)的函数。该实验可以在小体积中进行的任何商业定量PCR仪,并因此在一个实验中,大量的样品,可同时测试(通常是48,96或384个样品,这取决于仪器型号)。实验通常可以执行在围绕一个小时,提供的样品5的高通量分析的可能性。
该方法的进一步改进已经看到采用的染料具有较好的光谱特性6,7 ,进行数据分析的通用工具,并建议方案进行初步筛选8,9。方法的应用范围已经扩展,并特别侧重于建立编制和贮藏蛋白10,最佳的条件和确定潜在的具有约束力的合作伙伴,以帮助结晶11。相对高的吞吐量的方法的,在蛋白质的成本相对较低(〜每反应2微克),并适用于学习弱结合分子已取得的DSF基于片段的药物设计的一个非常有价值的工具,特别是在学术环境12-14。
尽管DSF的广泛应用,以研究蛋白质 - 配体相互作用,很少有研究描述的决心,从这些研究中解离常数。然而,这些往往产生描述该蛋白质的展开,具有许多参数的详细方程式必须被装配到稀疏数据或在某些情况下ëstimated 7,15-17。这些方法是特别重要的挑战性的情况下,如紧密结合化合物或蛋白质显示不寻常的过渡。然而,对于许多实验室中,这些详细的分析过于繁琐常规使用。因此,我们建议替代疗法用于不同的场景,并展示如何将这些可用于适应不同的蛋白质 - 配体的相互作用产生的数据。我们的方法使用在StepOne定量PCR仪,为此定制的数据分析软件是可用的;而这加速了数据分析,从其它仪器的结果可以使用以前发表的方法9进行处理,并且在同一下游分析可以被执行。
Protocol
1,确定的解离常数的近似值( 即在一个数量级)
- 制备表1中详述的混合物。
- 制备目标配体的库存在可用的最高浓度,以及本然后在6点10倍稀释液。其中一个近似杀敌从现有数据已知的,目的是具有至少两个浓度高于和低于杀敌 。
- 分装18μL混合成8口井的定量PCR板。加入2微升的溶剂中,在第一井。加入2微升的配体的稀释系列(步骤1.2)中的每个成员的一个剩下的七个孔以及每一个。
- 将一个密封的qPCR在板上。为了实现板的良好密封,放置一个手涂布器(见特定试剂的表),在板的中部。平滑向下的密封件一侧,然后重复上的另一半该板。
- 离心反应板在500×g离心2分钟以除去气泡。
- 将板StepOne扩增定量PCR仪。选择“解链曲线”选项,ROX过滤器,并选择快爬坡速率(这提供了一个2分钟的停顿,在25℃,随后的升温到99℃,经40分钟,然后2分钟的停顿)。运行热变性。
在脚本文件执行运行可在网上通过http:注意// www.exeter.ac.uk/biosciences/capsular。 - 在仪器运行结束后,点击屏幕上的“分析”按钮。保存结果文件。
- 打开蛋白的热位移的软件。
- 创建一个新的研究;在属性选项卡,给这一个名字,并在条件选项卡,详细的配体。
- 移动到实验文件选项卡,并导入保存的结果文件(XXX.eds),并设定每口井(FIL模板的内容ES可从作者)。
- 移动分析选项卡,然后按“分析”按钮。
注意:这将分析结果。它可以导出结果作进一步调查与使用Excel导出选项卡。结果导出选项卡划定的格式。最好是在Excel中打开导出的文件,并立即保存为Excel格式。
- 检查中的单独溶剂的存在下,蛋白质提供了类似于图1A中所示的结果。接下来,检查结果发现,在“复制”窗格中的熔化温度。确保这说明随着配体浓度明显增加的熔化温度。
请注意:在理想情况下,这将提供一个明确的最大熔解温度(假定蛋白是完全配体结合),以及一个近似杀敌其中熔融温度为配位体的无蛋白和半之间的最大值。
2,实验装置测定解离常数
- 准备详细的表2作为主结构的混合物。
- 制备的配位体的库存为十五个不同浓度,这将在稀释10倍,在最终的实验。理想情况下,包括浓度高于和低于所估计的杀敌大小的至少两个数量级,并且围绕所估计杀敌的浓度。着眼于七个推定杀敌的一个数量级范围内的点,与在此两边另外四个点;如果有选择,有更多的点,那是饱和值。
注:如果需要的话,它是改变实验条件,使得该配体股是在试验浓度加倍,其中配位体的溶解度的限制是可行的。 - 添加120μL的高手搭配八口井在U形底96孔板,充当水库的主结构方便配药。使用8道移液器免除18微升到PCR板的一列。重复,另外5个栏目,给一共有48口井填在盘上6×8模式。
- 加入20μl的配位体的股票,或溶剂,以便在U形底96孔板分开的孔中。使用8道移液器,吸2微升的八个不同配体的股票(或溶剂)。这些加入的PCR板是充满了预混步骤2.3的一列。重复使用相同的8配体/溶剂股的另外两个列。吸2微升其余八配位体或溶剂的股票,而这些添加在板中的第四列。重复此为另外两个列。这会给一式三份样品的全部16个配体和溶剂的样品。
- 将一个密封的qPCR在板(见步骤1.4)。
- 离心反应板在500×g离心2分钟。
- 将平板中的定量PCR仪。运行使用在步骤1.6中指定的参数的热变性。
- 在仪器运行结束后,点击屏幕上的“分析”按钮。保存结果文件。
- 打开蛋白的热位移的软件。创建一个新的研究;在属性选项卡,给这一个名字,并在条件选项卡,详细的配体。
- 移动到实验文件选项卡,并导入保存的结果文件(XXX.eds),并设置每个内容很好。
注:模板文件可在网上通过http:// www.exeter.ac.uk/biosciences/capsular。 - 移动分析选项卡,然后按“分析”按钮。
- 选择在屏幕的左手侧上的菜单中选择“重复数”标签来显示结果为一式三份。评估的数据的基础上,一式三份如何紧是可靠性。年满D中的一式三份表明重复性差,仔细检查原始数据。
- 同时使用玻尔兹曼或衍生物的方法来评估的熔融温度分析数据。选择“复制结果”选项卡,在“复制结果暗算”,“以剧情:”切换按钮之间的“以旧换新 - 玻耳兹曼”和“以旧换新 - 衍生”。选择给出了样品的更大可重复性的方法。导出的结果作进一步调查与使用Excel导出选项卡。
注意:对于显示多个样品的转换,它几乎总是最好用多个熔体模式的衍生方法。结果导出选项卡划定的格式。最好是在Excel中打开导出的文件,并立即保存为Excel格式。 - 重复实验至少两次,其中包括在一个单独的一天重复,以保证结果的可重复性。如果数据分析(见下文第3步)表明杀敌的值显著不同于原先的估计,相应地改变了配体浓度(见步骤2.2),以保证良好的范围内围绕杀敌值。
3,数据分析,以确定下解离热变性恒
- 在Excel中创建的配体浓度的表和熔化温度。
- 打开的GraphPad Prism软件,并创建一个XY工作台。输入数据,使用的配体浓度和熔化温度结果在Y列在X列。
注:图中所示的例子是图1B。用公式预装,并使用SPSS统计软件包替代指令的脚本,可在网上通过http:// www.exeter.ac.uk/biosciences/capsular。 - 在分析选项卡,选择该选项来改变分析参数S(Ctrl + T键)。输入正确的模型,选择“新建”,“创建新的公式”。将详述于表3为“单点配体结合”的公式。
注意:这些步骤的一个例子示于图1C中 。当使用脚本用方程预装,相关方程可被引导从列表中而不是输入的选择。这个方程的推导在附录中提供。 - 选择“规则的初始值”框中,输入初始值的规则如表3中详细说明。
- 约束的参数P,作为“恒等于”,并输入蛋白的终浓度(在同一个单元作为配位体中给出)。
- 选择确定以进行分析。
注意:这些步骤的一个例子示于图1D。的绘图软件产生一个数字表示的数据,并在适合的模型。吨的例子灰褐色的分析示于代表性数据。
4,数据拟合,以合作模式
为了适应数据合作模式,无论是简单的合作模式,或在两个不同的解离常数定义的模型之间进行选择。第一种方法是优选的负协同的情况下,或者作为初始调查。然而,原则上它是在正协同箱子更好模拟两种不同的解离常数18。在这种情况下,造型可以继续假定任一连续的结合配体,或独立的配体的结合。
- 遵循相同的初始步骤,在协议3,然而,在步骤3.3,在表3中被列为“简单合作模式”中插入一个方程“的两个配位体序列的结合”,或18“独立的两个配体结合”。
- 选择初始值的相关规则与每个这些方程表3相关联。
- 检查模型与数据的拟合。如果数据符合不佳,可以考虑另一种模式。
注意:这也是很重要的仔细研究的熔融温度,以由蛋白质的热转移软件(步骤2.9)中的数据拟合:有时有必要在这里改变的参数,以获得最佳的结果。另一个要考虑的是数据点的范围是否理想,是否有任何异常点:要么一组数据,在有限杀敌的两侧,或单点异常(特别是在最高配体浓度),可以显著影响结果。 - 重复实验至少两次(见步骤2.12),以确保重现性。
5,数据拟合为曲线显示在熔融温度二进制班次
偶尔,而不是配体分级响应,蛋白质已观察采用二进制响应,其中必然样本显然与未结合的样品中分离。一个例子是,在代表性的结果( 图4)提供的。在这种情况下,嵌合的熔化温度也不会提供良好的适合杀敌 。
- 导出由蛋白质的热转移软件输出的原始数据。对于每个温度点,计算平均荧光的零配体和配体的最高浓度。制表从明年这些每个数据点的结果。
注:此处产生的误差小于在嵌合熔融温度的误差。 - 打开SPSS统计软件包。复制的温度下,这两个平均值的数据集,以及数据对于每个实验,在SPSS数据窗口。在变量标签中,设置平均数据集没有配体为“低”,且平均数据集的最高配位体浓度为“高”。
- 可下载的语法文件在网上在http:// www.exeter.ac.uk/biosciences/capsular 。选择“运行→运行所有”。
- 必然结果复制比例到一个新的Excel工作簿,与相关的配体浓度。
- 打开GRAPHPAD软件,并创建一个XY工作台。输入数据,使用的配体浓度和熔化温度结果在Y列在X列。在分析选项卡,选择“改变分析参数”。输入正确的模型,选择“新建”,“创建新的公式”。输入在表3中给出的方程,列为“分析中的熔融温度的二进制移位”。
- 选择“规则的初始值”框中,输入表3中详述的初始值的规则。约束的参数P,如“恒等于”,并输入蛋白质的最终浓度(单位同西甲第二是给出)。
注意:在完成这些方块为在第3协议的例子示于图1C,D。 - 如果有一个良好的配合,其结果可以外推到无限大的配体浓度的预期结果得到改善。从约束在每个配体浓度的比例的模型,检查为配体浓度的最高值的值。如果这是0.99或更大,进一步的分析是不可能的,以改善结果。
- 如果该比例低于0.99,附加的步骤是必需的,以校正最高配体浓度的样品中的未结合蛋白的效果。在步骤5.2中,写的配体结合的,在最高配体浓度点(从步骤5.7)的比例为单元R2(可以使用不同的小区,和R2适当地替换等式表3)。创造最高配体浓度结果的平均值后,一个额外的列。在冷杉T细胞中,复制在表3中列出的方程为“外推到无限大的配体浓度”。这个公式复制到剩余的细胞在此列。
注:此计算中移除未结合的蛋白在最高配体浓度的影响。该配体游离蛋白和最高配体浓度之间的差值乘以绑定在最高配体浓度,以提供在各温度点的充分结合和未结合的蛋白状态之间的预期差的比例的倒数。这种差异被添加或从该未结合状态中减去,得到了充分的配体结合的蛋白质的预期荧光。 - 替换在SPSS数据与此新列最高配体浓度列,重复数据拟合。
注意:步骤5.7 - 5.9,可能需要重复,如果模型显示在绑定在最大配体concentrati的比例进一步显著变化(如果是这样的话,它很可能是理想的,以重复实验与包括较高的配体浓度点)。 - 在情况下,蛋白显示出的二进制移位和显示器合作的行为,该方程在步骤5.5建议应该与那些来自步骤4.1被取代。的“顶”和“底”的参数应当由1和0分别替换。
- 重复实验至少两次(见步骤2.12),以确保重现性。
Representative Results
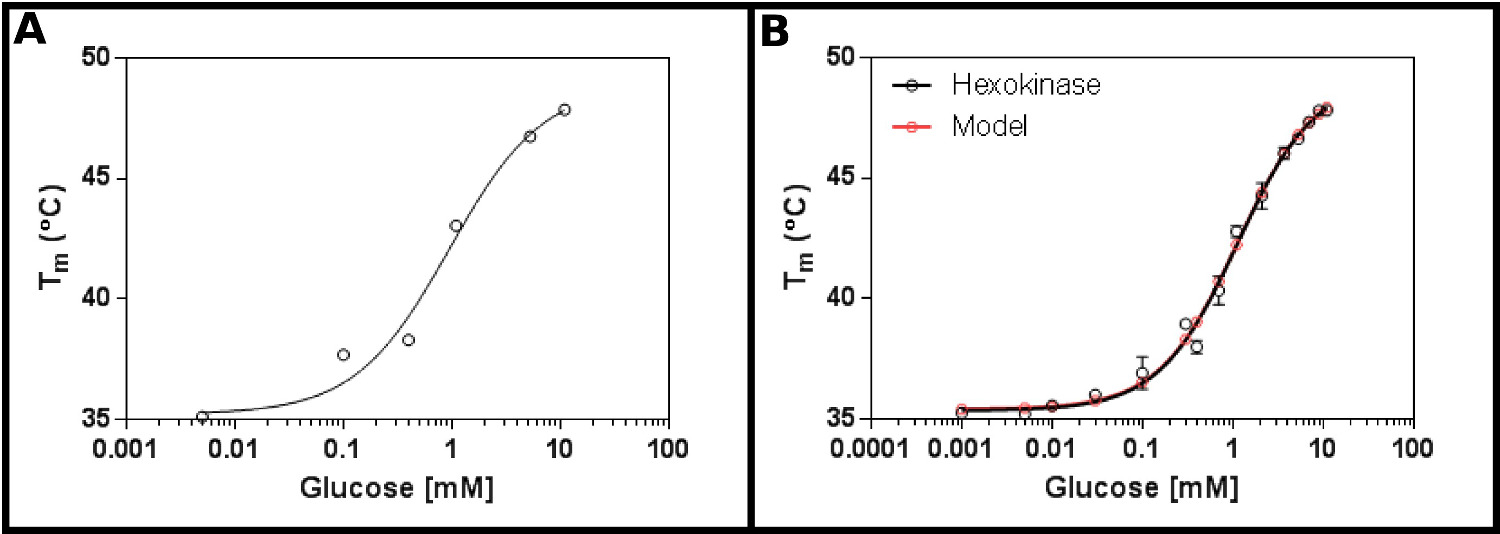
此方法的一个极好的试验基体是己糖激酶。这具有可容易地商购获得,并且具有被发现在大多数实验室的,并且提供在测定明确的,可重复的结果两个衬底的优点。的初始浓度屏(方案1),采用己糖激酶和葡萄糖( 图2A),表明可能杀敌将在范围从0.2到1.7毫米。因此,更大的屏幕(协议2)进行,使用表4的结果( 图2B)所示的浓度表现出良好的适合单点配体结合式[9](协议第3.3节),并给A K ð1.2±0.1毫米。
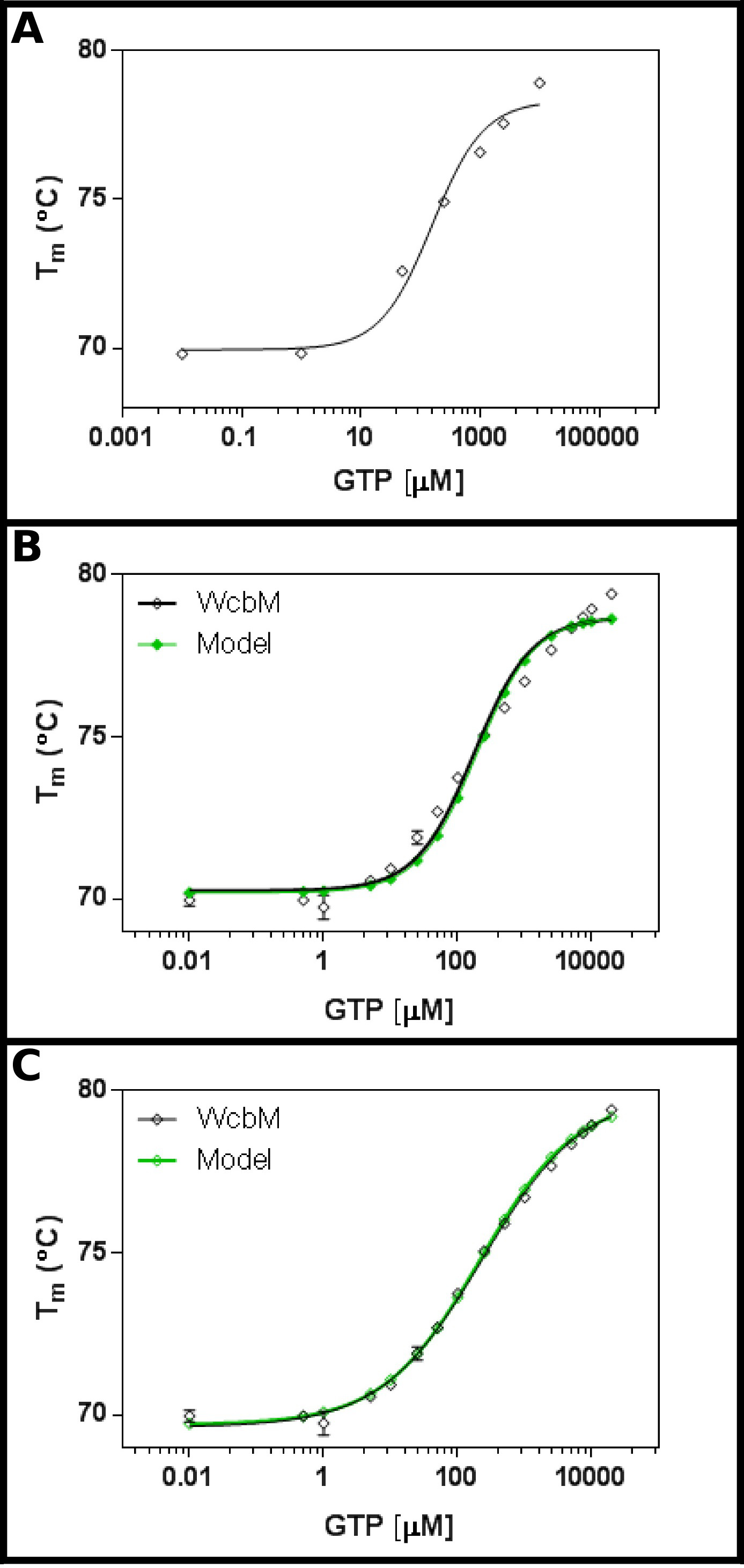
假定庚糖脒基转移酶WcbM 19,20显示了结合GTP( 图3A)强大的热转变。初始屏幕提示第kD是在大约100微米的范围内。因此,一个完整的屏幕被设置,使用表5中所示的浓度的拟合结果,以公式3.3的结果显示在合理的拟合(0.981 R 2;图3B)。。然而,存在的数据,并在之间具有明显的区别模型,这表明不同的方程是必要的。检索蛋白质数据库21与WcbM序列表明,它的结构已被确定形成二聚体最接近的同源物。该数据是因此使用三个方程的合作,顺序和独立的两个配体(协议4)的结合分析。的拟合统计的合作模式给了0.998的R2值和0.215残差(Sy.x)的标准差,而顺序和独立的具有约束力的模型给出了0.992的R2值和0.480和0.461ÅSy.x分别。这表明,该模型给出最佳拟合该数据是在合作模式:在这里,A K½230±10μM的观察,为0.52±0.02( 图3C)的n的值。这表明一个负向协同的结合。注意的是,K½被用在这种情况下,而于K D,因为在单位中K D会是相当令人不满意μM0.52。
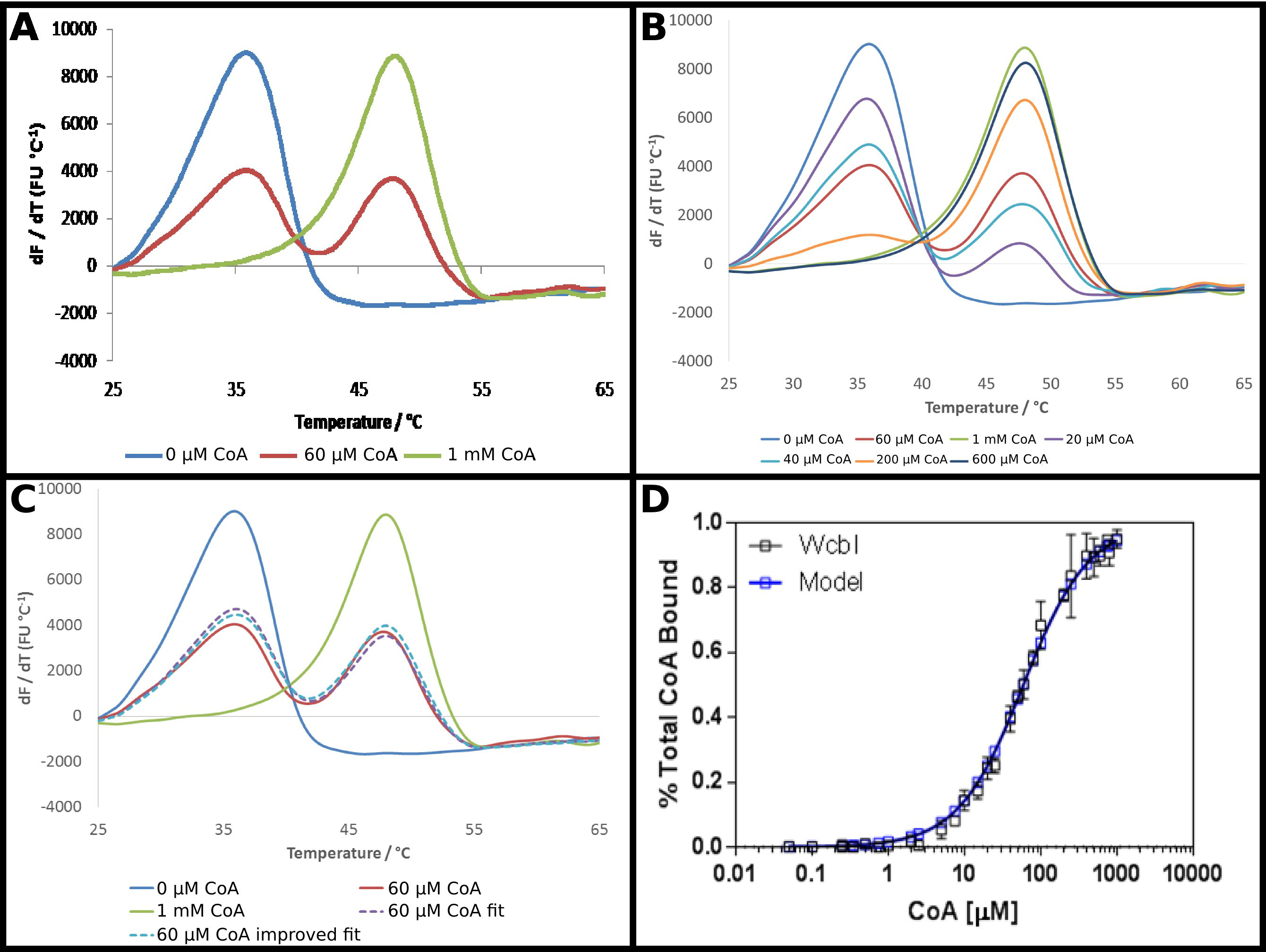
假定GDP-6-脱氧-β-D- 甘露 -heptopyranose 2Ø-acetylase,WcbI 22,显示了一个不寻常的结果差示扫描荧光。在没有任何配位体,它示出了一个简单明了的变性( 图4A)。戊二酰辅酶A(辅酶A)被确定为一个配位体使用DSF的这种蛋白质,并且该蛋白质为这个伙伴的亲和力如在协议中所述进行了研究。在高浓度的辅酶,强大的S的存在HIFT至较高温度时观察到的,与在15℃熔化温度的变化。然而,在中间浓度,而不是转移到单相的熔点的中间熔融温度,WcbI显示出双相熔化,与蛋白质出现融化在任一配体的无温度,或者完全结合的解链温度( 图4A) 。改变以剂量依赖的方式,随着底物浓度增加,在较高温度下( 图4B)熔化的比例在两个物种的比例。这些数据的直接分析是具有挑战性的:嵌合于波尔兹曼方程给出了非常差的配合,而衍生物的方法强调了两个熔点事件正在发生,但没有协助证明随配体浓度的变化。
因此,分析这些数据一个较传统的方法是通过(协议5)。荧光ð没有配体和在最高配体浓度erivative结果作为代表,基本上所有的蛋白质在较低的熔融温度,或更高的熔融温度的状态。剩余的衍生物数据拟合在每个点处,因为每个在这两种状态中的比例的总和,与比例相加,以统一( 图4C)。所获得的数据,然后安装为前获得的表观杀敌 ,使用相同的方程如前。这强调了“高”配体点很可能是只有95%的配体结合的。然后将数据外推到的结果为100%结合蛋白的预测,和安装重复数据给出的58±2微米的明显杀敌 。这提供了实验结果的一个非常适合于结合模型( 图4D)。
FO:内容宽度=“5英寸”SRC =“/文件/ ftp_upload / 51809 / 51809fig1highres.jpg”WIDTH =“500”/>
图的实验设置与分析1。例子。 (A)中的热变性曲线的期望的形状的实施例(取自数据为酵母己糖激酶)。原始数据的特征形状示出了渐进的上升的荧光为最大,其次为浅下降(更详细地在9所讨论的)。这是伴随着荧光的一阶导数的单峰。数据进入的GraphPad(B)的实施例。配位体浓度给出的X轴,并且观察到熔融温度,在Y-轴。中的GraphPad方程定义(C)的实施例(D)的实施例正确地设定的变量和固定的蛋白质浓度,初始值,让正确的决心解离常数。米/文件/ ftp_upload / 51809 / 51809fig1highres.jpg“目标=”_ blank将“>请点击这里查看该图的放大版本。

, 己糖激酶和葡萄糖利用差示扫描荧光测定的图2相互作用(A)中的初始实验中测试了广泛的葡萄糖浓度表明在K d为可能是在0.2的范围内- 1.7毫米(B)的一个详细试验,检测葡萄糖浓度16,可以测定表观杀敌为1.12±0.05毫米。该数据符合得非常好,以该模型为单个结合事件(与底面(T1)和顶部(T2)的温度下嵌合到35.4±0.2ºC和49.3±0.5℃)。注意这些数据被收集在10mM MgCl 2的存在下进行。这些图像是使用GraphPad准备。 请点击这里查看该图的放大版本。
{kind=link}

WcbM与GTP的图3。互动透着一股反协同结合。 (一)初步的实验测试范围广泛的GTP浓度表明, 杀敌很可能是在200〜 - 500微米(B)详细的实验,检测16浓度的GTP,提出了一个价值明显杀敌 120±20微米。然而,当以对数标度,用于x轴,有一个显著discre模型和数据之间pancy。与合作模式相同的数据(三)分析表明,非常适合到一个简单的合作模式正在使用的数据。这里,K值为230±20μM½测定,与协同指数n = 0.52±0.02(与底面(T1)和顶部(T2)的温度下嵌合到69.63±0.06ºC和79.9±0.1ºC分别)。如WcbM似乎是二聚体,这意味着该酶是在其结合到GTP完美anticooperative。这些图像是使用GraphPad准备。 请点击这里查看该图的放大版本。
{kind=link}

图4 WcbI显示双相融模式在其配体辅酶A(COA)的存在。 (一)WcbI,在没有配体(蓝色),显示了一个简单的单相熔的模式。在高配体浓度(1毫米;绿线),类似的模式观察。然而,在中间的配体的浓度(60μM;红线),两个不同的熔融峰,对应于无配体和配体结合的状态被观察到(B)中的两组峰之间的过渡是剂量依赖性的在整个浓度范围。双相熔为配体自由和高配结果的比例之和(C)的建模提供了一个很好的拟合数据(虚线紫线,红色线相比)。这种配合是通过外推观察到高浓度的配体(如该模型表明〜95%的占用率),以充分占用(蓝色虚线)的结果得到改善。(D)为WcbI的绑定辅酶昭比例中得到的数据 ws的一个非常适合于简单的结合模型,用58±2微米A K D(基于所述第一组结果,这些数据代表收集在两个不同天的数据,以选择用于在第二天稍微不同的配体浓度)。面板(A - C)使用Excel准备和面板(D)采用GRAPHPAD 请点击这里查看该图的放大版本。
{kind=link}
表1配方的初步实验。
| 试剂 | 在混合量(μl) |
| 蛋白质 | 到0.11毫克/毫升的最终浓度 |
| 0.3 | |
| 0.5 M羟乙基pH值7.0 | 3.7 |
| 5 M氯化钠 | 5.6 |
| 水 | 180微升 |
这说明了“主混合物”蛋白,检测试剂和缓冲液的初始侦察实验提供杀敌的估计值,如在协议部分1,该缓冲器的混合物很适合用作通用的蛋白质进行说明。在以前的研究结果表明其它缓冲液应使用,这些应被取代。如果蛋白质的库存是在低浓度下( 即,小于0.3毫克/毫升),这可能是必要的,以减少额外缓冲液的加入量,以补偿缓冲器已经存在的蛋白质样品中。
表2配方测定杀敌的。| 试剂 | 在混合量(μl) |
| 蛋白质 | 到0.11毫克/毫升的最终浓度 |
| 5000倍SYPRO橙 | 1.78 |
| 0.5 M羟乙基pH值7.0 | 22.2 |
| 5 M氯化钠 | 33.3 |
| 水 | 180微升 |
这说明了“母液”蛋白质,检测试剂和Buffer用于蛋白质样品的完整的决心杀敌 ,如在协议第2节描述这个缓冲区的混合物适用于一般蛋白质。在以前的研究结果表明其它缓冲液应使用,这些应被取代。如果蛋白质的库存是在低浓度下( 即,小于0.3毫克/毫升),这可能是必要的,以减少额外缓冲液的加入量,以补偿缓冲器已经存在的蛋白质样品中。
表3方程,并进行数据分析参数 。
D| 在实验方案步骤 | 所需的公式 | 所需的参数 | 变量和参数说明 |
| 3.3 | |||
| 单点配体结合 | Y =底+((上下)*(1 - ((PKÐ-X + SQRT(((P + X + K D)^ 2) - (4 * P * X)))/(2 * P )))) | 病人:蛋白质浓度。的Kd:解离常数。 P和Kd值中给出了用于配位体的浓度,相同的单元。顶部,底部:熔融温度在无限的配体浓度分别为无配体浓度。 | |
| 3.4 | 底= * YMIN | YMIN:Y的(最低试验蛋白的Tm,在此情况下)的最低值 | |
| TOP = * YMAX | YMAX:Y的最大值(最高试验蛋白的Tm) | ||
| 杀敌 * X在YMID = | YMID:对应于YMIN与YMAX的平均值的Y值。 X是相应的X的值(这里,相关的配体浓度) | ||
| P =(初始值,为配合) | |||
| 4.1 | |||
| 简单的合作模式 | Y =底+((上下)*(((X / K D)^ N)/(1 +((X / K D)^ N)))) | Ñ:H生病系数。这说明了协同性,或其它生化性质,该蛋白质的,并且不一定在蛋白质配体结合位点的数目的估计值。一希尔系数表示没有协同性;值低于1表示负协同性,和值大于一个正协同性。 | |
| 底= * YMIN | |||
| TOP = * YMAX | |||
| 杀敌 * X在YMID = | |||
| P =(初始值,是适合 ) | |||
| N =(初始值,为配合) | |||
| 连续两个配体的结合 | Y =底+((上下)*((x ^ 2)/(K D * K2))/(1 +(X / K D)+((x ^ 2)/(K D * K2))) ) | K2:解离常数第二个结合事件。 | |
| 底= * YMIN | |||
| TOP = * YMAX | |||
| K2 = *在YMID点¯x | |||
| P =(初始值,为配合) | |||
| 两个独立的配体结合 | Y =底+((上下)*((x ^ 2)/(K D * K2))/(1 +(2 * X / K D)+((x ^ 2)/(K D * K2) ))) | ||
| 底= * YMIN | |||
| 杀敌 * X在YMID = | |||
| K2 = *在YMID点¯x | |||
| P =(初始值,为配合) | |||
| 5.5 | |||
| 在熔融温度二进制移分析 | Y = 1 - ((PKð-X + SQRT(((P + X + K D)^ 2) - (4 * P * X)))/(2×P)) | ||
| 底= * YMIN | |||
| TOP = * YMAX | |||
| 杀敌 * X在YMID = | |||
| P =(初始值,为配合) | |||
| 5.8 | |||
| 外推至无限的配体浓度 | (C 2 - ((1 - $ R $ 2)* B2))/ $ R2美元 | B2:包含结果与没有配体的细胞。 C2:包含结果与最大的配体的细胞。 $ R $ 2:含约束最大配体浓度的比例细胞。 |
步骤3,4和5所需要的另外的详细方程分解为分析软件,并开始对数据进行分析的参数的精确定义。方程式为每个相关步骤被示出,与参数的正确选择。提供了一种用于参考的变量和参数的含义进行说明。
表4浓度为己糖激酶和葡萄糖的相互作用的筛选。
| 采样点 | 配体(glucosE)浓度(MM) |
| 1 | 0 |
| 2 | 0.001 |
| 3 | 0.005 |
| 4 | 0.01 |
| 5 | 0.03 |
| 6 | 0.1 |
| 7 | 0.3 |
| 8 | 0.4 |
| 9 | 0.7 |
| 10 | 1.1 |
| 11 | 2.1 |
| 12 | 3.7 |
| 13 | 5.3 |
| 14 | 7 |
| 15 | 9 |
| 16 | 11 |
从芽殖酵母酿酒酵母己糖激酶被添加到主结构在协议中说明,并辅以10毫米氯化镁镁是一种已知的辅助因子。第k D的初始估计为0.5和2毫米之间。实验设置,以提供所指示的终浓度的葡萄糖。
表5浓度WcbM与GDP的相互作用筛选。
| 采样点 | 配位体(GTP)的浓度(μM) |
| 1 | 0 |
| 2 | 0.5 |
| 3 | 1 |
| 4 | 5 |
| 5 | 10 |
| 6 | 25 |
| 7 | 50 |
| 8 | 100 |
| 9 | 250 |
| 10 | |
| 11 | 千 |
| 12 | 2500 |
| 13 | 5000 |
| 14 | 7500 |
| 15 | 万 |
| 16 | 20000 |
由类鼻疽伯克氏菌 WcbM被添加到主结构在协议中说明。第k D的初始估计为大约100微米。实验设置,以提供所指示的终浓度GTP的,目的在于涵盖上方和下方杀敌大小的至少两个数量级。
Discussion
差示扫描荧光显示了其力量的强大和通用的方法表征蛋白质和识别潜在的蛋白配体。在加快蛋白质的稳定性,药物发现(尤其是在欠资金充足的实验室)和结晶10,23-25 有据可查的成就使其成为了化合物的初步筛选一个有吸引力的方法。加入到蛋白的化合物在表观熔融温度-7,9明显的剂量依赖性增加。然而,有过一些尝试,以使用来自这些实验的结果,以确定表观结合常数中排名化合物其亲和力来帮助。这里,我们提出了对系统确定的表观解离常数为蛋白质的配体的存在的方法。
这里给出的结果表明,DSF能迅速,鲁棒提供的解离常数的估计值蛋白质 - 配体结合。所观察到的数据可以用市售的工具来操作,以提供快速测定杀敌的,而不需要做出关于参数的可能值的假设。该方法具有显著优势的是简约中蛋白质和时间需要一些可比方法。这里描述的实验将消耗0.13毫克蛋白每实验(约0.4毫克实验以一式三份重复的)。这与等温滴定量热法(ITC),其中一个实验中,平均40 kDa的蛋白质会消耗同样数量相比,毫不逊色。全套本议定书所需的实验将消耗约4小时,包括准备,为单组实验。再次,这是很可能比方法如ITC或表面等离子共振,这同时有力常常需要相当大的优化以实现最佳的数据相当快。
我们的结果表明,仍然存在着需求仔细研究的原始数据,这些数据的拟合来确定熔融温度和熔融温度数据的拟合,以确定解离常数。第一个挑战是在蛋白融化所产生的原始数据的形状。在某些情况下,该形状可能不是近似于图1A中观察到。常见的问题包括对配体的低温转变结合,高背景荧光,且温度异常多的过渡。低温度的变化被认为具有约束力的一些配体。对于这个方法,最关键的参数是在T m测定的误差,相对于温度的漂移。数据通常可装相当好时三次测量的标准偏差不超过未结合和完全结合的蛋白质之间的熔化温度转变的10%。我们的经验是,如果这样的温度erature位移仅2℃,这可能是足够的,用于安装的数据,如果各个数据点是高度精确的。第二个问题是不寻常的形状的曲线。这些通常是免费的蛋白质和配体结合的形式有所不同,作为配体结合会影响展开的蛋白质模式。在这些情况下,用户必须考虑该数据是否可以适当考虑模型被用来被用于确定熔融温度和离解常数。另一个常见的问题是,除了辅因子的蛋白( 例如,MgCl 2的在我们的例子中与己糖激酶)是必需的,以获得最可靠的数据。我们的经验是,仔细地考虑所有可能的因素在实验中,在服用初期读数必须获得最好的结果的阶段。此外,替代疗法的理论可以揭示这些数据15,17的特点。最后,这是不寻常的某些蛋白质将contain本身暴露的疏水区域,显示高背景荧光。有许多解决这些问题,它已被广泛地审查别处6,9。
特别是,用户必须考虑是否使用玻尔兹曼或衍生物的模型( 例如, 图4),并在使用的衍生物的情况下,无论是多个熔体必须进行建模。造型的热去折叠的两种方法的区别在于玻尔兹曼方法的实验数据拟合至波尔兹曼方程,假设一个正常的S形形状的解折叠曲线。与此相反,该衍生物的方法开出的实验数据的一阶导数在每个点处( 图1A中的下图),并认为该熔化温度是最高的一次导数的点。 3℃ - 导数方法一般约2返回一个较高的熔融温度。大多数蛋白质会返回一个更一致的结果( 即,熔融温度为三次重复实验的标准误差是低级)的两种方法之一。这通常是密切相关的蛋白质解折叠曲线的精确形状,并且根据经验确定在每一种情况下,最好的方法是必要的。其中衍生模型的情况下,同样重要的是要考虑多种熔化事件。一些数据清楚地表明依据多个过渡,并且在这些情况下,结果可能是更容易解释,如果这些多个熔化事件建模。在此协议的上下文中,它通常是,加入配位体的可引起蛋白质从具有多个熔化转变到单过渡转移( 例如,通过稳定的热最脆弱的子域),或反之亦然的情况。因此,我们主张,才考虑这做法将是最好用的原始数据进行检查起来。
继米个人熔化温度odelling,进一步的问题也出现在这些配件在协议部分介绍的车型。当务之急是要仔细检查用对数刻度的配合,以解离常数公式,因为这种分析往往突出了观测数据和模型之间的差异(E·G。, 图3)。而获得的结果通常是健壮的,小心的解释提供了机会,以提取更好的结果,而最含义,从内容
通过这些数据提出的一个具体问题是,应该放在这表明协同蛋白质或多个绑定的事件,在DSF的解释。我们,迄今为止,仅观察到了这种现象在蛋白质,预计将有多个特异性结合事件( 例如,WcbM,一种蛋白质,其最好的同源物是一种多聚体26,和它作为一种多聚体上的大小排阻层析[数据不所示])。它是不完全清楚,在DSF变性中观察到的负协同表明酶将最终呈现负协同性:更确切地说,这可能是复杂的结合的指示必须使用更大范围的方法来研究更彻底。但这不过建议给我们,这种蛋白质的更广泛的研究,有可能识别有趣的效果。
使用这种方法给出的解离常数的值通常的顺序相同的那些用其它方法,如等温滴定量热法和表面等离子体共振提供。然而,观察到的绝对值经常比用这些方法观察到的高。这是至少部分地一个事实,即解离常数是在含有配体的蛋白质的熔融温度观察到的结果。此杀敌一般比在生理温度更高。该dissociatioÑ常数是相关于由方程式反应的温度:
 [1]
[1]
 [2]
[2]
(其中cθ的标准参考浓度,ΔR G是该反应的吉布斯自由能的变化中,R是摩 尔气体常数,ΔH是在反应的焓变,并且ΔS是在反应的熵变。)
以解离常数的反应在该方法的测量范围通常具有负ΔR G,和因此增加了温度对方程的影响[1]是增加的解离常数。二者构成的吉布斯自由能的ΔH和Δ 的条款(方程[2])是temperaturë依赖性27,并在解离常数的影响将依赖于这些温度相依性的大小和符号,并且将一定的相互作用有关。因此,它是难以避免的,通过这种方法所确定的解离常数,有时比由在RT操作方法确定更大。温度依赖性的,当然,也对许多其他方法,这些方法倾向于提供的解离常数在温度低于生理温度下一个警告。
DSF的方法的另一个需要注意的是,它是一种标记方法,不像美国国际贸易委员会。使用(SYPRO橙)的荧光标记物是疏水性的,因此,在某些情况下,可以与疏水性配体的蛋白质的结合竞争。因此,很可能在一些情况下,所得到的解离常数将被人为地由于与标签竞争提高。然而,对于不同的配位体的比较中,(主用DSF),差异不太可能充分显著通过亲和力影响化合物的排名。
这种方法的一个潜在缺点是检测限是可以实现的。原则上,它不应该是能够精确地测量用于杀敌即蛋白质浓度低于50%,且在此范围内,即使值很可能是可疑的精度的值。同时检测的范围的这一端的期限可以延长一点通过减少蛋白质和染料的浓度,该仪器的灵敏度会防止进一步降低蛋白质浓度。类似地,灵敏度的上端部将通过所述配体中的溶解度来确定。以获得数学稳健估计中K D,用90%的蛋白质中存在的配体结合的形式,这就要求配位体浓度是近似的获得数据,是最重要的LY 10次杀敌 (假设没有协同)。因此,检测限必然是在有关缓冲区中的配位体的溶解度的十分之一。这意味着,检测方法的限制将在1微米和1至100毫米的范围通常,根据不同的蛋白质和配体。
总之,差示扫描荧光,适用范围广的蛋白质的一种通用技术。用这里介绍的方法,可以迅速且廉价地确定为不同的配体的蛋白质的亲和力。这对于在蛋白质纯化和稳定化的应用程序,阐明从宏基因组的酶的功能或特异性的潜力很大,并且在药物发现,特别是在小的实验室。
Disclosures
The authors declare that they have nothing to disclose.
Acknowledgements
This work was funded by grant from the BBSRC (grant number BB/H019685/1 and BB/E527663/1) to the University of Exeter.
Materials
| Name | Company | Catalog Number | Comments |
| StepOne real time PCR instrument | Life Technologies | 4376357 | DSF can be performed with many other instruments. The StepOne instrument has very convenient software for data analysis. |
| Protein thermal shift software v1.0 | Life Technologies | 4466037 | |
| MicroAmp Fast optical 48-well plates | Life Technologies | 4375816 | |
| Optical sealing tape | Life Technologies | 4375323 | Bio-rad part no. 223-9444 is an alternative supplier |
| U-bottomed 96-well plates | Fisher | 11521943 | |
| SYPRO Orange | Life Technologies | S6650 | For a smaller volume supplier, use Sigma part no. S5692 |
| SPSS statistics version 20 | IBM | N/A | Other statistics packages will provide similar functionality |
| GraphPad Prism 6.02 | GraphPad | N/A | Other statistics packages will provide similar functionality |
| Hand applicator (PA1) | 3M | 75-3454-4264-6 | |
| Hexokinase from Saccharomyces cerevisiae | Sigma-Aldrich | H5000 | |
| Glucose | Fisher scientific | 10141520 |
References
- Freyer, M. W., Lewis, E. A. Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 84, 79-113 (2008).
- Ladbury, J. E. Calorimetry as a tool for understanding biomolecular interactions and an aid to drug design. Biochem Soc Trans. 38, 888-893 (2010).
- Abdiche, Y., Malashock, D., Pinkerton, A., Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal Biochem. 377, 209-217 (2008).
- Pantoliano, M. W., et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 6, 429-440 (2001).
- Senisterra, G., Chau, I., Vedadi, M. Thermal denaturation assays in chemical biology. Assay Drug Dev Technol. 10, 128-136 (2012).
- Ericsson, U. B., Hallberg, B. M., Detitta, G. T., Dekker, N., Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 357, 289-298 (2006).
- Lo, M. C., et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 332, 153-159 (2004).
- Nettleship, J. E., Brown, J., Groves, M. R., Geerlof, A. Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol Biol. 426, 299-318 (2008).
- Niesen, F. H., Berglund, H., Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2, 2212-2221 (2007).
- Geders, T. W., Gustafson, K., Finzel, B. C. Use of differential scanning fluorimetry to optimize the purification and crystallization of PLP-dependent enzymes. Acta Crystallogr Sect F Struct Biol Cryst Commun. 68, 596-600 (2012).
- Vedadi, M., et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA. 103, 15835-15840 (2006).
- Davis, B. J., Erlanson, D. A. Learning from our mistakes: the 'unknown knowns' in fragment screening. Bioorg Med Chem Lett. 23, 2844-2852 (2013).
- Larsson, A., Jansson, A., Aberg, A., Nordlund, P. Efficiency of hit generation and structural characterization in fragment-based ligand discovery. Curr Opin Chem Biol. 15, 482-488 (2011).
- Scott, D. E., et al. Using a fragment-based approach to target protein-protein interactions. Chembiochem. 14, 332-342 (2013).
- Cimmperman, P., et al. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J. 95, 3222-3231 (2008).
- Matulis, D., Kranz, J. K., Salemme, F. R., Todd, M. J. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 44, 5258-5266 (2005).
- Zubriene, A., et al. Measurement of nanomolar dissociation constants by titration calorimetry and thermal shift assay - radicicol binding to Hsp90 and ethoxzolamide binding to CAII. Int J Mol Sci. 10, 2662-2680 (2009).
- Weiss, J. N. The Hill equation revisited: uses and misuses. FASEB J. 11, 835-841 (1997).
- Cuccui, J., et al. Characterization of the Burkholderia pseudomallei K96243 capsular polysaccharide I coding region. Infect Immun. 80, 1209-1221 (2012).
- DeShazer, D., Waag, D. M., Fritz, D. L., Woods, D. E. Identification of a Burkholderia mallei polysaccharide gene cluster by subtractive hybridization and demonstration that the encoded capsule is an essential virulence determinant. Microb Pathog. 30, 253-269 (2001).
- Berman, H., Henrick, K., Nakamura, H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 10, 980 (2003).
- Vivoli, M., Ayres, E., Beaumont, E., Isupov, M., Harmer, N. Structural insights into WcbI, a novel polysaccharide biosynthesis enzyme. IUCr Journal. 1 (1), 28-38 (2014).
- Sorrell, F. J., Greenwood, G. K., Birchall, K., Chen, B. Development of a differential scanning fluorimetry based high throughput screening assay for the discovery of affinity binders against an anthrax protein. J Pharm Biomed Anal. 52, 802-808 (2010).
- Uniewicz, K. A., et al. Differential scanning fluorimetry measurement of protein stability changes upon binding to glycosaminoglycans: a screening test for binding specificity. Anal Chem. 82, 3796-3802 (2010).
- Wan, K. F., et al. Differential scanning fluorimetry as secondary screening platform for small molecule inhibitors of Bcl-XL. Cell Cycle. 8, 3943-3952 (2009).
- Koropatkin, N. M., Holden, H. M. Molecular structure of alpha-D-glucose-1-phosphate cytidylyltransferase from Salmonella typhi. J Biol Chem. 279, 44023-44029 (2004).
- Paleskava, A., Konevega, A. L., Rodnina, M. V. Thermodynamics of the GTP-GDP-operated conformational switch of selenocysteine-specific translation factor SelB. J Biol Chem. 287, 27906-27912 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved