
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biology
Phage-vermittelte Lieferung von Targeted sRNA Konstruiert Down-Gene nach Knock Expression in
We describe a method to knock down gene expression in a growing population of E. coli cells using sequence-targeted sRNA expression cassettes delivered by an M13 phagemid vector.
RNA-vermittelte Niederschläge werden weithin zur Kontrolle der Genexpression verwendet. Diese vielseitige Familie von Techniken macht Gebrauch von kurzen RNA (sRNA), die mit jeder Sequenz synthetisiert werden kann, und gestaltet jedes Gen für silencing gezielt zu ergänzen. Weil sRNA Konstrukte können direkt auf vielen Zelltypen eingeführt werden, oder eine Vielzahl von Vektoren verwenden, können die Genexpression in lebenden Zellen ohne aufwendige genetische Veränderung unterdrückt werden. Die häufigste RNA Knockdown-Technologie, RNA-Interferenz (RNAi), nutzt die endogene RNA-induzierten Silencing-Komplex (RISC) -Sequenz Erkennung und Spaltung der Ziel-mRNA zu vermitteln. Anwendungen dieser Technik sind daher auf RISC-exprimierenden Organismen beschränkt ist, in erster Linie Eukaryoten. Vor kurzem haben eine neue Generation von RNA Biotechnologen alternative Mechanismen entwickelt, die für die Genexpression durch RNA steuern und so möglich, die RNA-vermittelte Gen in Bakterien hergestellt Niederschlägen. Hier beschreiben wir ein Verfahren zum Schweigen zu bringen Gen Expression in E. coli , die funktionell ähnlich RNAi. In diesem System wird ein synthetisches Phagemid wurde entwickelt sRNA auszudrücken, die entworfen können eine beliebige Sequenz abzuzielen. Das Expressionskonstrukt ist auf eine Population von E. zuge coli - Zellen mit nicht-lytische Phage M13, wonach es in der Lage ist , in stabiler Weise als ein Plasmid replizieren. Antisense - Ansatz und silencing der Ziel - mRNA durch die Hfq Protein endogen E. vermittelter coli. Dieses Protokoll umfasst Methoden für die Gestaltung der Antisense sRNA, die Konstruktion des Phagemid-Vektor, Verpackung des Phagemid in M13-Bakteriophagen, eine Live-Zellpopulation für eine Infektion der Vorbereitung und Durchführung der Infektion selbst. Das fluoreszierende Protein mKate2 und das Antibiotikaresistenz-Gen Chloramphenicol-Acetyltransferase (CAT) sind gezielte repräsentative Daten zu generieren und Zuschlags Wirksamkeit zu quantifizieren.
RNA-vermittelte Gen-Knockdowns gehen in zwei Stufen. Zunächst wird ein RNA-Molekül zu einer Zelllinie oder eines Organismus der Studie eingeführt. Zweitens endogene RNA-bindende Proteine RNA-Zielerkennung erleichtern und die Dämpfungswirkung erzeugen. Alle RNA-Technologien profitieren Knockdown von der anpassbaren Natur des synthetischen sRNAs, die leicht ein bestimmtes Ziel von Interesse übereinstimmen hergestellt werden können. Allerdings variieren die molekularen Details der RNA-Aufnahme und silencing weit über Modellsystem beschränke wo und wie RNA Niederschlägen angewendet werden kann.
In Nematoden, doppelsträngige Moleküle RNA (dsRNA) in den Medien direkt oder durch die Würmer mit einer Bevölkerung von dsRNA-exprimierenden E. Fütterung coli - Zellen 1,2. In Drosophila kann RNAi 3 durch Mikroinjizieren Embryonen mit dsRNA erreicht werden, oder in Zelllinien durchgeführt , indem einfach dsRNA zu dem Kulturmedium 4 hinzugefügt wird . In Säugetierzelllinien,synthetischen small interfering RNAs (siRNAs) an lebenden Zellen durch Elektroporation 1,2,5, verpackt in Liposomen 3,6, oder ausgedrückt von DNA - Plasmid - Vektoren 4,7 geliefert werden. Sobald die RNA-Spezies in das Cytosol erreicht, stützt sich die RNAi-Weg auf der RISC-Komplex dsRNA zu verarbeiten, erleichtern Antisense Erkennung des Ziels und katalysieren translationale Repression Abbau mRNA oder Heterochromatinbildung, auf dem Host abhängt.
Aufgrund dieser Anforderungen können klassische RNAi nur in Organismen durchgeführt werden, die effizient exogene RNA aufnehmen und exprimieren RISC oder eine RISC-ähnliche Aktivität. Bemerkenswert ist , schließt dies das Modell Bakterium E. coli, die das RNAi - Weg fehlt. die jüngsten Fortschritte in der synthetischen Biologie bieten jedoch die Werkzeuge sowohl das Lieferproblem und das Silencing Problem zu lösen.
In diesem Protokoll werden sRNA - Konstrukte in E. ausgedrückt coli aus einer DNA - Vektor geliefert living Zellen des M13 Phagemid / Helfer-System. Ein Phagemid ist jedes Plasmid mit einem Phagen abgeleitete f1-Replikationsursprung. Ein Helferplasmid, in diesem Fall M13KO, trägt die alle Maschinen Viruspartikel zu produzieren erforderlich, ist aber selbst nicht zuständig für die Replikation und Verpackung. Wenn ein Phagemid und Helferplasmid werden co-transformiert, haftet allein der Phagemid an der f1-Ursprung repliziert, verpackt und ausgeschieden werden. Die vektorisiert Phagemid ist dann zuständig Live E. zu infizieren coli über das F - Pilus.
In diesem System wird der Schalldämpfungseffekt durch individuelle sRNA Kassetten Kombinieren einer Gerüstsequenz mit einem Target-Bindungssequenz erzeugt wird. Die Zielbindungssequenz ist 24 Basenpaare Antisense-mRNA zu einem Ziel, typischerweise an der Ribosomenbindungsstelle (RBS). Die Gerüstsequenz, entwickelt von Na und Kollegen 8 enthält eine Hfq-Bindungsmotiv aus micc extrahiert, eine kleine regulatorische RNA endogenen E. coli. Die Hfq Protein stimuliert RNA-RNA Binding und mRNA Degradation, eine Rolle in diesem System ähnlich RISC in RNAi dient. 1 zeigt ein komplettes System für den Phagen-vermittelter sRNA Niederschläge, inklusive der sRNA Kassettenstruktur, Phagemid Vektorisierung und Silencing - Mechanismus.
Als ein Verfahren der Genexpression in E. modulieren coli, ist sRNA Silencing einfach, schnell und vielseitig. Die gezielte E. coli ist nicht über Ausbreiten des Phagemid und Expression des sRNA belastet. Dies kann im Rahmen der synthetischen Biologie oder der Grundlagenforschung in denen die Expression von größeren heterologe Konstrukte belasten können zelluläre Ressourcen 9 relevant sein. Phagemide mit neuen Ziele können mit einem einzigen PCR und geerntet einen Tag nach Phagemid Transformation hergestellt werden. Schließlich kann nahezu jede mRNA gerichtet werden. Die sRNA - Regulationskassette (auf einem Standard - Plasmid) wurde auf einer Vielzahl von Zielen in den Stoffwechsel mit typischen Repression Ebenen> 90% 8 zu arbeiten , gezeigt.
10 Kassetten. Zuerst wird eine verpackte Phagemid zu einer Batch - Kultur von E. eingeführt coli Zellen und verwendete Ausdruck des fluoreszierenden Proteins mKate2 zum Schweigen zu bringen. Nachfolgende Fluoreszenzänderungen werden in Echtzeit überwacht. Zweitens Klopfen nach unten das CAT-Gen wird gezeigt, auf Agarplatten phänotypischen Resistenz gegen Chloramphenicol zu reduzieren. In beiden Fällen führt das Phagemid selbst ein GFP-Marker, so dass Infektionsrate unabhängig von der Zuschlagseffizienz gemessen werden.
1. Planung und Bau von Phagemidvektoren Bearing sRNA Silencing Cassetten
- De - novo - Design von sRNA Silencing Kassetten 8
- Identifizieren die vollständige Sequenz der mRNA eine DNA-Sequenz-Datenbank zum Schweigen gebracht werden. Um die Zielsequenz erzeugen, notieren Sie die ersten 24 bp der kodierenden Sequenz von Position 1 bis 24 , beginnend mit dem Start - Codon (zB ATG).
Hinweis: Silencing ist weniger effizient , wenn andere Seiten oder Abschnitten der mRNA 8 ausgerichtet sind. - Nehmen Sie die reverse Komplement der Zielsequenz des GUIDE-Sequenz für die sRNA Kassette zu erzeugen. Siehe Tabelle 1 für Beispiele von TARGET und GUIDE Sequenzen für Chloramphenicolacetyltransferase (CAT).
- Zur Gestaltung des 292 bp komplette sRNA Expressionskassette, ordnen Sie die pR - Promotor, GUIDE Sequenz, Hfq Protein - Bindungsdomäne und T1 / TE Transkriptionsterminatorsequenzen in Serie (Tabelle 2).
- Fügen Sie zusätzliche Klonierungsstellen der Wahl Klonierung der sRNA Kassette in den Zielvektor zu erleichtern.
- Beziehen Sie die komplette sRNA Kassette durch kommerzielle Gensynthese oder ein ähnliches Verfahren und klonen Sie es in jeden Phagemid - Vektor mit einem funktionellen f1 - Replikationsursprung 11. Siehe die vollständige Sequenz des endgültigen Phagemid-Vektor Hintergrundinformationen.
- Identifizieren die vollständige Sequenz der mRNA eine DNA-Sequenz-Datenbank zum Schweigen gebracht werden. Um die Zielsequenz erzeugen, notieren Sie die ersten 24 bp der kodierenden Sequenz von Position 1 bis 24 , beginnend mit dem Start - Codon (zB ATG).
- Das Ändern der von einer vorhandenen sRNA - Expressionskassette Zielsequenz unter Verwendung von PCR-basierten Website - Directed Mutagenesis 12
- Identifizieren Sie die 24 bp GUIDE-Sequenz in einer bestehenden sRNA-Expressionskassette. Hinweis: Die kommentierte pAB.001 Plasmid, in dieser Arbeit verwendet wird, als eine zusätzliche Sequenz-Datei zur Verfügung steht.
- Design-Vorwärts- und Rückwärtsprimer mit kurzen Regionen mit Homologie zu den bestehenden sRNA Kassette die neue 24 bp GUIDE Sequenz flankieren. Besorgen Sie sich die Primer durch kommerzielle Oligonukleotidsynthese.
Hinweis: Primer-Design für die ortsgerichtete Mutagenese ist depicted in Abbildung 2. Genaue Templatsequenzen Primer sind in Tabelle 3 vorgesehen. - Bereiten Sie eine 5 - ml - Kultur von E. coli Durchführung der sRNA Ausdruck Phagemid Vorlage. Wachsen die Zellen über Nacht bei 37 ° C unter Schütteln in LB-Medium mit geeigneten Antibiotika.
- Extrahieren und reinigen Sie die Vorlage sRNA Ausdruck Phagemid aus dem 5 ml Bakterienkultur eine DNA - Minipräparationskit oder ähnliches Verfahren unter Verwendung von 12.
- Bereiten zwei PCR - Reaktionen unter Verwendung der Schablone sRNA Expression Phagemid und High - Fidelity - Polymerase, eine mit dem vorderen und eine mit dem Rückwärts - Primer (Tabelle 4). Verwendung der PCR - Bedingungen , wie durch die Polymerase - Lieferanten (Tabelle 5) empfohlen. Erhöhen Sie die Vorlage Konzentration auf 10-50x höher als eine Standardreaktion für die Tatsache zu berücksichtigen, dass die einzelnen Primer Reaktion nicht exponentielle Amplifikation erzeugt.
- Kombinieren Sie die beiden oben genannten PCR-Reaktionen in einem Reaktionsgefäß. Anneal die Produkte durch Erhitzen auf 98 ° C in einem kochenden Wasserbad. Unmittelbar nach dem Reaktionsgefäß im Wasserbad platziert, die Wärmequelle entfernen und das Bad erlauben langsam über 1-2 h auf Raumtemperatur zurückzukehren.
- Zur Beseitigung der nicht-mutierten Vorlage sRNA Ausdruck Phagemid, fügen 1 ul. DpnI-Restriktionsenzym zur Mischung und für 1 h bei 37 ° C inkubieren, oder die vom Hersteller für die vollständige Verdauung empfohlene Zeit.
Hinweis: DpnI verdaut nur methylierte Zielstellen, die auf dem Host-replizierten Phagemide vorhanden sind, aber nicht PCR-Produkte. - Trans gekauft oder 13 chemisch kompetente E. vorbereitet coli mit 1-5 & mgr; l des PCR - Produkts geglüht. Isolieren Sie einzelne Kolonien des transformierten Stammes durch selektive Beschichtung auf LB-Agar-Platten, die entsprechenden Antibiotika.
- Um den Einbau des richtigen Leitsequenz, Screenen der resultierenden Kolonien durch Kolonie-PCR verifizieren. Mit einer 200 ul Pipettenspitze, collect eine geringe Menge an Zellen, die aus einer einzigen transformierten Kolonie. Mark und die ursprüngliche Kolonie für nachgeschaltete Verwendung nach Überprüfung bewahren.
- Fügen Sie die gesammelten Zellen zu 50 ul Nuklease-freies Wasser in einem Reaktionsgefäß. Mischen durch Pipettieren von oben und unten.
- Verwendung einer Benchtop-Thermocycler oder ein siedendes Wasserbad, lysieren die Zellen durch Erhitzen auf 95 ° C für 2 min.
- PCR-Amplifizierung der Phagemid-Region unter Verwendung von 1 ul wärme lysierten Zellen als ein DNA-Templat. PCR - Bedingungen und Thermozykler - Protokolle sind in den Tabellen 6 und 7 vorgesehen. Siehe die zusätzliche pAB.001 Sequenzdatei zur Überprüfung Primersequenzen.
- Sequenz, die die PCR-Produkt Einbau des richtigen GUIDE-Sequenz zu überprüfen.
- Impfen eine 5 - ml - Kultur des E. coli Klon , der die Sequenz-verifiziert sRNA Ausdruck Phagemid trägt. Wachsen die Zellen über Nacht bei 37 ° C unter Schütteln in LB selektiven Medien.
- Bereiten Sie einen Glycerolstammlösungder Sequenz-verifiziert Klon. In 750 ul der Übernachtkultur auf 250 & mgr; l von 60% Glycerin in einem Schraubverschluss Kryo Röhrchen.
- Lagern Sie das Glycerolstammlösung bei -80 ° C auf unbestimmte Zeit. Der Rest der Übernachtkultur kann als eine Quelle für die sRNA Expression Phagemid in Schritt 2 verwendet werden.
2. Produktion und Ernte von Phagemid Stocks-M13 verpackt
- Bereiten Sie eine 5 - ml - Kultur von E. coli Durchführung des sRNA Ausdruck Phagemid. Wachsen die Zellen über Nacht bei 37 ° C unter Schütteln in LB-Medium mit geeigneten Antibiotika. Hinweis: Die sRNA Ausdruck Phagemid kann , wie in Schritt 1.1.5 oder modifiziert werden aus einem bestehenden Phagemid und geerntet in Schritt 1.2.16 beschrieben durch de novo - Klonen erhalten werden.
- Ebenso bereiten eine 5 - ml - Kultur von E. coli Durchführung des M13KO7 Helferplasmid. Wachsen die Zellen über Nacht bei 37 ° C unter Schütteln in LB selektiven Medien.
- Extrahieren und reinigen die sRNA expression Phagemid und Helferplasmid eine DNA - Extraktions - Kits oder ein ähnliches Verfahren unter Verwendung von 12.
- Cotransform gekauft oder 13 hergestellten chemisch kompetente E. coli mit 1 ul jeweils von sRNA Ausdruck Phagemid und Helferplasmid. Wählen Sie für Kotransformanten durch Plattierung auf LB-Agar mit selektiven Antibiotika für beide Konstrukte.
- Bereiten Sie eine 10-ml-Kultur aus einer einzelnen Kolonie des cotransformiert Stamm in LB mit selektiven Antibiotika. bei 37 ° C bebrütet 8-12 h oder über Nacht unter Schütteln.
- Zentrifugieren der Kultur bei 3.300 × g für 10 min. Sammeln Sie den Überstand und filtriert durch ein 0,2 um Filter. Achtung: Bei Medien verschütten, reinigen Sie den Bereich mit verdünnter Bleiche (0,5%) infektiöse Phagenpartikel zu zerstören.
- Lagern Sie das verpackte Phagemid Filtrat bei 4 ° C. Anmerkung: Die Proben können für Tage bis Wochen ohne Aktivitätsverlust aufrechterhalten werden.
3. Herstellung von F + Zellen Ziel für Silencing
- Bestimmen Sie, ob die Zellen zum Schweigen zu bringen , die F - Pilus 14 ausdrücken gezielt werden. Wenn die F-Pilus bereits vorhanden ist, gehen Sie zu Schritt 4.
Hinweis: Allgemeine Laborstämme von E. coli werden als F + oder F 'kommentierten die Anwesenheit des F - Pilus in ihrem Genom , um anzuzeigen , oder auf einem Plasmid. - Erhalten Sie F + Stamm von E. coli wie TOP10F '.
Hinweis: Stellen Sie sicher, dass die Zielstamm trägt einen einzigartigen Resistenzmarker, um aus dem F-Plasmid Spender nach der Konjugation getrennt werden. - F pilus durch Konjugation mit einem F + Stamm einzuführen, bereiten 5 ml Kulturen sowohl der Zielstamm und F-Plasmid Spender 14. Wachsen die Zellen über Nacht bei 37 ° C unter Schütteln in LB-Medium mit geeigneten Antibiotika.
- Am folgenden Tag verdünnen beide Stämme 1: 100 in 5 ml LB selektiven und weiter Kultivieren bei 37 ° C unter Schütteln.
- Bestimmen Sie die Wachstumsphase der Zellen durch die Opt Messsche Dichte der Kultur bei 600 nm (OD 600) ein benchtop Spektralphotometer. Kultur für die Zellen etwa 2 h , bis eine OD 600 von 0,3 erreicht ist, was anzeigt , log-Phasenwachstum 15.
- Vorbereitung 3 Konjugationsreaktionen in Mikrozentrifugenröhrchen 0,5 ml F-Plasmid-Donor + 0,5 ml Zielstamm, 0,5 ml F-Plasmid-Donor + 0,5 ml LB-Medium (negative Kontrolle) und 0,5 ml Zielstamm + 0,5 ml LB-Medium (negative Kontrolle). Lassen Sie die Konjugation für 2 Stunden bei 37 ° C ablaufen unter Schütteln.
- Platte 100 ul jeder Konjugationsreaktion auf selektive LB-Agar mit Antibiotika spezifisch für das F-Plasmid (typischerweise Tetracyclin) und Zielstamm. Platte, die negativen Kontrollreaktionen bestätigen, dass weder Spender oder Empfängerstamm beide Antibiotikaresistenzen auszudrücken.
4. Eine Infektion mit Packaged Phagemide für Silencing
- Beimpfen einer einzelnen Kolonie von F + Zielzellen in einen 5 - ml - Kultur von LB medieine mit geeigneten Antibiotika. über Nacht bei 37 ° C inkubieren unter Schütteln.
- Am folgenden Tag, verdünne die F + Zielzellen von 1: 100 in 5 ml LB selektiven Medien und zur Kultur bei 37 ° C unter Schütteln fortgesetzt.
- Bestimmung der Wachstumsphase der Zellen , die durch die optische Dichte der Kultur bei 600 nm gemessen (OD 600) ein benchtop Spektralphotometer. Kultur für die Zellen etwa 2 h , bis eine OD 600 von 0,3 erreicht ist, was anzeigt , log-Phasenwachstum 15. Hinweis: Die Expression des F-Pilus und Infektionseffizienz ist in der log-Phase am höchsten.
- Fügen Sie die M13-verpackten Phagemide (aus Schritt 2.6) an die Zielzellen in einem Volumenverhältnis von 1: 100 fast 99% Befall der Zielpopulation zu erreichen. Lassen Sie die Infektion bei 37 ° C ablaufen 30-60 Minuten unter Schütteln.
- Assay, um die sRNA-Silencing-Phänotyp nach der Methode der Wahl.
Hinweis: für ein fluoreszierendes Protein Target kann der Schalldämpfungseffekt zu quantifizierendirekt durch Fluorometrie 10. Alternativ kann phänotypischen Assays verwendet werden , um die phänotypischen Folgen Knock-Down 8 zu beobachten. - Bereiten Sie einen Glycerolstammlösung für die sRNA-exprimierenden Phagemid Host folgende Schritte 1.2.14-1.2.16. Hinweis: Die Phagemid auf unbestimmte Zeit im Wirtsstamm ausbreitet und kann mit Antibiotika ähnlich einem herkömmlichen Plasmid beibehalten werden.
Silencing von mKate2 Fluoreszenz in flüssigen Medien
Abbildung 1 zeigt das Schema für sRNA-vermittelten Knockdowns in dieser Arbeit beschrieben, einschließlich der sRNA Kassettenentwurf, Phagemid Vektorisierung und Silencing - Mechanismus. Nach Protokoll 1.2 wurde die sRNA-Silencing-Kassette von Plasmid pAB.001 verändert mKate Ziel. Die sRNA - Kassette wurde synthetisiert und kloniert in Phagemid Litmus28i_J23115-B0032-GFP, ein Geschenk von Monica Ortiz und Drew Endy 11. Dieses Phagemid trägt Marker für die GFP-Expression und Kanamycin-Resistenz, wodurch erfolgreiche Infektionen zu verfolgen. Verpackt Phagemid Bestände wurden folgende Protokoll 2 hergestellt.
Ein Derivat von E. coli K12 MG1655 eine konstitutiv exprimiert, chromosomal integriert mKate2 Marker trägt , wurde für Phageninfektion durch Konjugation vorbereitet mit einem F-Stamm Plasmid Spender folgende Protokoll 3. Die Zellen folgende Protokoll 4. Nach Phagemid Infektion bis Mitte der log-Phase und dem Phagemid eingeführt wurden gezüchtet wurden 200 & mgr; l Kulturen auf einem Fluoreszenz-Plattenlesegerät übertragen und Fluoreszenz wurde kontinuierlich für 24 Stunden überwacht.
Abbildung 3 zeigt die Wirkung von sRNA-vermitteltes Silencing auf mKate2 Expression. Der Stamm infiziert mit einem Anti-mKate2 Phagemid zeigte keine nachweisbare mKate2 Fluoreszenz gegenüber dem Hintergrund. Im Gegensatz dazu hat dieser Stamm das GFP-Marker exprimieren, zeigen damit die erfolgreiche Aufnahme des Phagemid. Nicht infizierte Kontrollzellen mKate2 Fluoreszenz produziert, aber nicht GFP. Eine zusätzliche Steuerung, in dem die anti-mKate2 Targeting-Domäne mit einer Sequenz Targeting CAT ersetzt wurde, hatte keine Wirkung auf mKate2 Fluoreszenz.
Silencing von Chloramphenicol-Resistenz auf Agar-Platten
Inhalt "fo: keep-together.within-page =" 1 "> Nach Protokoll 1.1, eine sRNA - Silencing - Kassette wurde hergestellt CAT Ziel ein Derivat von E. coli K12 MG1655 ein konstitutiv exprimiert, chromosomal integrierte CAT Marker trägt , wurde vorbereitet. Phageninfektion durch Konjugation mit einem F-Plasmid Donorstamm folgende Protokoll 3. die Zellen bis mittleren log-Phase gezüchtet worden waren, und die Phagemid bei 37 ° C folgende Protokoll 4. nach 1 Stunde Inkubation eingeführt wurden infizierte Zellen seriell verdünnt und unter einer plattierten Bereich von Chloramphenicol-Konzentrationen. die Platten wurden durch Zählen von koloniebildenden Einheiten (KBE) am nächsten Tag über Nacht, und der Anteil der resistenten Zellen bei jeder Konzentration wurde bestimmt Chloramphenicol inkubiert.Figur 4 zeigt die Wirkung von sRNA-vermitteltes Silencing auf dem Chloramphenicol - Resistenz - Phänotyp. Nicht-infizierte Zellen oder Zellen infiziert mit Phagemid mKate2 Targeting, waren resistent gegenChloramphenicol bei allen getesteten Konzentrationen. Im Gegensatz dazu mit Phagemid Targeting CAT-infizierten Zellen zeigten Überleben bei niedrigen Konzentrationen Chloramphenicol reduziert und nahezu 99% bei höheren Konzentrationen zu töten. Die Zugabe von Ampicillin, für nur Bakterien zur Auswahl der Phagemid trägt, reduziert Chloramphenicol Überleben auf ein nicht nachweisbares Niveau. Dies steht im Einklang mit früheren Arbeiten , dass die Flucht aus dem Phagen - Infektion zeigt eine gemeinsame Route 10 zu entkommen zum Schweigen zu bringen.
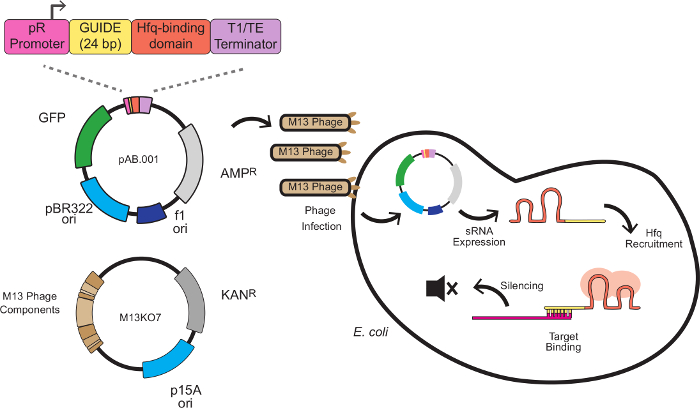
Abbildung 1: Gene Silencing in E. coli mit sRNA - Expressionskassetten Lieferung von M13 Phage Die sRNA Kassette aus 4 Modulen zusammengesetzt ist. Die PR - Promotor (ein konstitutiver Promotor abgeleitet aus dem Bakteriophagen Lambda), eine 24 bp Targeting - Domäne, eine Hfq Bindungsdomäne von micc extrahiert und einen Transkriptionsterminator 8 E. zu infizieren coli Expression des F - Pilus, wo sRNA Ausdruck beginnt. Die sRNA rekrutiert dann das Hfq Protein (in rot dargestellt) und bindet ein Antisense - mRNA Ziel in der Nähe der Ribosomen - Bindungsstelle, in translationale Repression und mRNA - Abbau führt. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
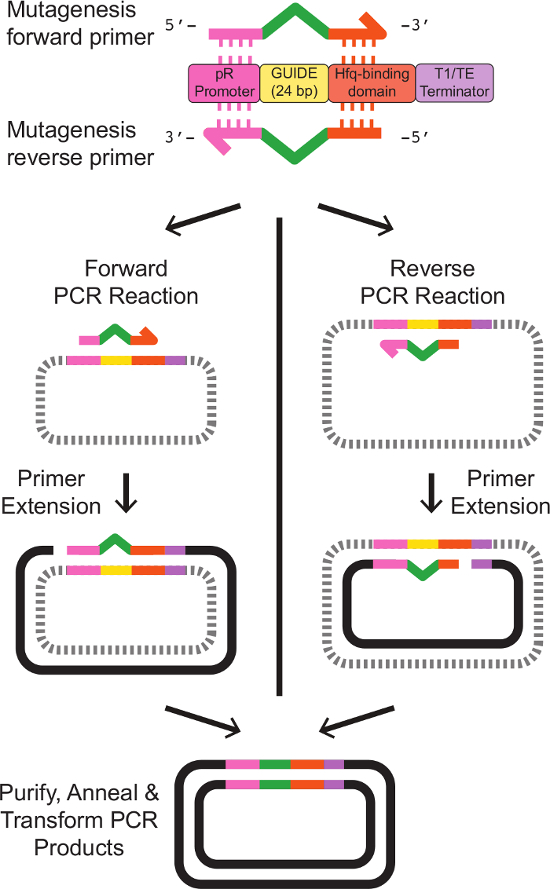
Abbildung 2:. Primer - Design und zielgerichtete Mutagenese des sRNA Zielsite werden zwei Primer mit teilweise Homologie zu einer vorhandenen sRNA Kassette entworfen. Der Vorwärtsprimer enthält 20 bp homolog zu der pR-Promotor am 5'-Ende, gefolgt von24 bp die neue Führungssequenz darstellt, dann 18 bp homolog zur Hfq-Bindungsdomäne an das 3'-Ende. Der Reverse-Primer ist die genaue reversen Komplement des Vorwärtsprimers, mit Regionen der Homologie zu den bestehenden sRNA Kassette das reverse Komplement der neuen Führungssequenz flankieren. Exact Primersequenzen sind in Tabelle 3. Separate Einzelprimer - PCR - Reaktionen mit den Vorwärts- und den Rückwärtsprimer erzeugen lineare, einzelsträngige DNA mit der gewünschten modifizierten Sequenz. Das Glühen der Vorwärts- und Reaktionsprodukte umzukehren, nach Clean-up wie im Protokoll beschrieben, resultiert in doppelsträngiger Plasmid - DNA trägt die gewünschte modifizierte sRNA Kassette. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
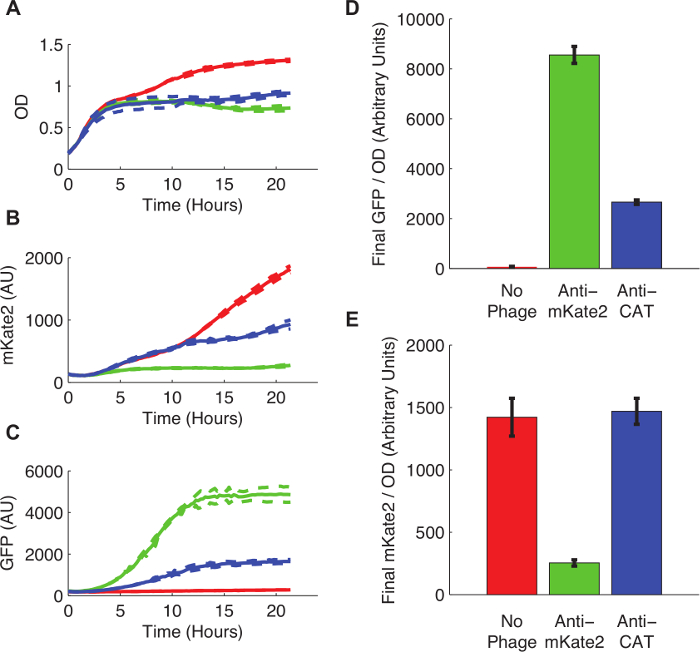
Abbildung 3: Knockdown eines chromosomal integriert mKate2 Fluorescent Reporter. E. coli MG1655 K12 mKate2 exprimieren , wurden entweder unbehandelt gelassen (rote Linien und Balken), infiziert mit einem Anti-mKate2 Phagemid (grüne Linien und Balken) oder mit einer Steuer Phagemid Targeting CAT (blaue Linien und Balken) infiziert. (A) Unbehandelte E. coli stieg auf höhere Sättigungsdichte, eine metabolische Kosten Phageninfektion anzeigt. Die gepunkteten Linien zeigen die Standardabweichungen von drei Replikaten. (B) Das mKate2 Signal wurde reduziert , um in der Nähe von Hintergrundwerte in der Anti-mKate2 behandelten Stamm, aber nicht in Kontrollstämmen. (C) GFP - Fluoreszenz, die ebenfalls von dem Phagemid getragen nachweisbar war nur in Phagemid-behandelten Kontrollen. (D, E) Abschlussfluoreszenzmesswerte nach 24 h Wachstum OD normalisiert wurden. GFP-Signal, Phagemid Infektion anzeigt, war in unbehandelten Kontrollen fehlen, aber auch anti-CAT p folgende erheblich reduzierthagemid Behandlung. Dies kann Nebeneffekte des Phagemid zeigen. Das mKate2 Signal wurde von anti-mKate2 Phagemid Behandlung reduziert im Vergleich zu unbehandelten Kontrollen. Die CAT-gezielte Steuerung Phagemid zeigte keine Wirkung auf mKate2 Fluoreszenz. Die Fehlerbalken stellen die Standardabweichung von 3 Replikaten. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
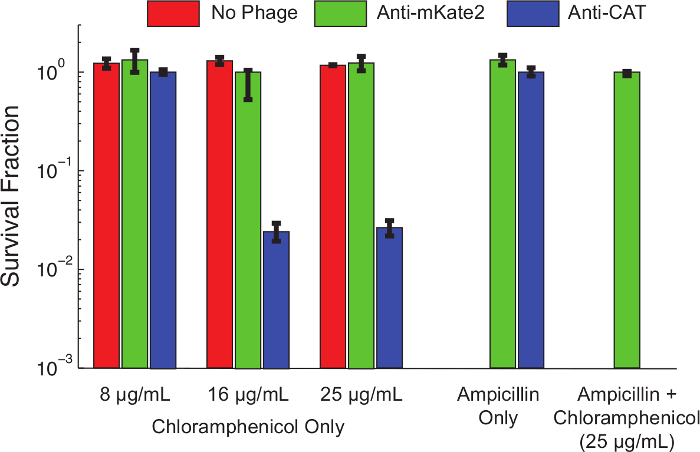
Abbildung 4: Preissturz von CAT Stellt Chloramphenicol - Empfindlichkeit auf einen genetisch resistenten Population E.. coli MG1655 K12 eine chromosomal integrierte CAT - Gen exprimieren , wurden unbehandelt gelassen (rote Balken), mit einer Steuer Phagemid behandelt mKate2 (grüne Balken) Targeting oder mit einem Phagemid behandelt , um eine anti-CAT Srna (blaue Balken) ausdrückt. Nach 1 Stunde der Infektion, die Lebensfähigkeit auf der angezeigten antibiotics wurde durch serielle Verdünnungen und Galvanisieren bewertet. Stämme mit anti-CAT Phagemid behandelt wurden signifikant (> 90%) von Chloramphenicol bei höheren Konzentrationen getötet, während Kontrollbehandlungen unbeeinflusst waren. Zugabe von Ampicillin zu dem Kulturplatten wählt positiv für Phagemid-Infektion und beseitigt nicht-infizierten Zellen. Unter diesen Bedingungen wurde beobachtet, keine Chloramphenicol-resistenten Kolonien nach der anti-CAT-Behandlung. Dies zeigt, dass die meisten Überlebenden ein Versagen der Infektion darstellen, sondern als ein Ausfall silencing. Die Fehlerbalken stellen die Standardabweichung von 3 Replikaten. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
| CAT TARGET-Sequenz | 5 '- ATGGAGAAAAAAATCACTGGATAT - 3' |
| CAT Leitsequenz | 5 '- ATATCCAGTGATTTTTTTCTCCAT - 3' |
Tabelle 1:. Ein Beispiel TARGET und Führungssequenz für das CAT - Gen Beachten Sie die reverse Komplement Beziehung.
| pR-Promotor | TAACACCGTGCGTGTTGACTATTTTACCTCTGGCGGTGATAATGGTTGC | ||||
| Leitsequenz | ATATCCAGTGATTTTTTTCTCCAT | ||||
| Hfq Bindungsdomäne | TTTCTGTTGGGCCATTGCATTGCCACTGATTTTCCAACATATAAAAAGACAAGCCCGAACAGTCGTCCGGGCTTTTTT TCTCGAG | ||||
| T1 / TE terminator | CTCGAGCCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTTTTGTCGGTGAA CGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA | ||||
Tabelle 2:. Sequence Komponenten der sRNA Cassette Jede Sequenz geschrieben 5'-3 '. Der komplette Kassette ist die Verkettung dieser vier Elemente in Ordnung und umfasst 292 bp.
| Forward Primer | 5 '- CTGGCGGTGATAATGGTTGC [GUIDE] TTTCTGTTGGGCCATTGC - 3' |
| Reverse Primer | 5 '- GCAATGGCCCAACAGAAA [TARGET] GCAACCATTATCACCGCCAG - 3' |
Tabelle 3: Primer Entwurf zu ändern Bestehende Leitelemente Der Vorwärts - Primer enthält die letzten 20 bp des pR - Promotor, die neue GUIDE - Sequenz und die ersten 18 bp des Hfq-Bindungsdomäne.. Die Zielsequenz ist das genaue Gegenteil Ergänzung des GUIDE-Sequenz. Der Reverse-Primer ist die genaue reversen Komplement des Vorwärtsprimers.
Tabelle 4: Empfohlene Bedingungen für die Single-Primer mutagene PCR.
| Schritt | Temp | Zeit |
| anfängliche Denaturierung | 98 ° C | 30 sec |
| 30 Zyklen | 98 ° C | 10 sec |
| 55 ° C | 30 sec | |
| 72 ° C | 120 Sekunden | |
| Finale Extension | 72 ° C | 300 sec |
| Lagerung | 10 ° C |
Tabelle 5: Empfohlene Thermocycler - Protokoll für die Einzel-Primer mutagene PCR.
| Komponente | Volumen |
| Template-DNA | 1 ul |
| 10 & mgr; M Forward Primer | 0,5 ul |
| 10 & mgr; M Reverse Primer | 0,5 ul |
| Taq 2X Master Mix | 25 ul |
| Nuklease-freies water | 23 & mgr; l |
| Volle Lautstärke | 50 ul |
Tabelle 6: Empfohlene Bedingungen für die Sequenzüberprüfung PCR.
| Schritt | Temp | Zeit |
| anfängliche Denaturierung | 95 ° C | 30 sec |
| 30 Zyklen | 95 ° C | 30 sec |
| 55 ° C | 30 sec | |
| 68 ° C | 30 sec | |
| Finale Extension | 68 ° C | 300 sec |
| Lagerung | 10 ° C |
Tabelle 7: Empfohlene Thermocycler Prokoll für die Sequenzüberprüfung PCR.
Das vorliegende Verfahren erreicht 80% ige Reduktion der mKate Fluoreszenzwerte im Vergleich zu ungezielte Kontrollen. Dies steht im Einklang mit anderen RNA - Knockdown - Methoden, bei denen das gesamte Silencing wird nicht beobachtet und 50-90% Wirkungsgrad ist typisch 16,17. Auf der phänotypischen Ebene CAT-bezogene Niederschläge konnten deutlich Chloramphenicol-Resistenz zu dämpfen, und beseitigen es unter bestimmten Bedingungen.
Der Zuschlag Phänotyp war nach nur wenigen Stunden nach der Infektion (3B) auf Bevölkerungsebene nachweisbar. Dies stellt ein wichtiges Merkmal des Phagen-basierten Delivery: kann eine hohe Zuschlags Frequenz ohne vorherige genetische Modifikation direkt in Batch-Kultur erhalten werden. Im Gegensatz zu herkömmlichen Modifikationen genetischen Plasmidtransformation oder genomische Integration verwenden, wird Phageninfektion nicht verlangen, dass eine Population von einer einzelnen isolierten Kolonie wieder angebaut werden. Dadurch können die Auswirkungen von Phageninfektion zu sein explored in Populationen mit komplexen räumlichen Dynamik 11, mit bereits bestehenden räumlichen Strukturen wie Biofilme 18 oder in genetisch gemischten natürlichen Populationen 19.
Ein wichtiger Schritt in diesem Verfahren ist die Herstellung von verpackten Phagemiden bei hohen Titer. Die metabolische Belastung mit Phagenteilchens Produktion verbunden sind, können in dem Phagemid Produktionsstamm zu hohen Mutationsraten oder Plasmid-Verlust führen. Es wird empfohlen, die Phagemid Produktionsstamm direkt aus einer einzigen cotransformiert Kolonie und nicht gekühlt, gefroren oder Unter kultiviert Phagen Ernte vor kultiviert werden. Eine geringe Effizienz der Co-Transformation kann auch beobachtet werden , wenn die Phagemid und Helferplasmid E. Einführung gleichzeitig coli. In diesem Fall können höhere Wirkungsgrade durch Transformieren des ersten Helferplasmid erhalten werden, dann kompetente Zellen Herstellung des Helferplasmid für die nachfolgende Transformation mit dem Phagemid trägt.
Phagemid Infektion oder sRNA Expression erlegt auch eine nachweisbare metabolische Belastung auf den Zielzellen und in einigen phänotypischen Störungs führen kann. Zum Beispiel wurde eine Verringerung der mKate2 Fluoreszenz beobachtet , selbst wenn die Zellen mit einem Phagemid - Targeting - CAT (Abbildung 3) infiziert waren. Eine Infektion mit M13 ist nicht daran gedacht , systemische Stressreaktionen in E. auslösen 20 coli, kann aber indirekt Transkriptionsmuster verändern. Alternativ kann das GFP oder Ampicillin - Resistenzmarker auf dem Phagemid enthalten für zellulare Ressourcen konkurrieren, wodurch mKate2 Expression und das Wachstum 9. Schließlich Genexpressionsprofile verändern die sRNA Kassette selbst global durch das Hfq Protein Titrieren oder durch Off-Target-mRNA-Silencing. Aus Zielwirkungen sind häufig in in vivo RNAi - Targeting 21-23, aber sie haben noch systematisch für dieses System untersucht werden.
Eine Einschränkung dieses Verfahrens ist, dass die Infektion effizienz ist weniger als 100%, einige nicht-infizierten Bakterien ermöglicht in der Bevölkerung zu persistieren. Die Ergebnisse dieser Arbeit und frühere Arbeit 10 deuten darauf hin , dass nicht infizierte Zellen 1-10% der Gesamtbevölkerung ausmachen, und sind verantwortlich für die meisten der nonsilenced Phänotypen beobachtet. Eine Vielzahl von Routen zu M13-Resistenz sind bekannt, mit dem die gängigste Mutations - Verlust von Pilus Ausdruck 24. In Anbetracht dieser Beschränkungen sollten Kontrollen verwendet werden, um hohe Infektionsraten und Zuschlags Effizienz zu bestätigen.
Eine weitere potentielle Beschränkung für einige Anwendungen ist die gelegentliche Übertragung von Helferphagen zu kontaminieren. Obwohl M13K07 ein mutiertes Verpackungssignal enthält, kann es auf infizierte Populationen bei niedriger Frequenz und übertragen verpackt in Phagen Kapsid werden, was in Zellen kompetent für die Phagenproduktion und die fortgesetzte Ausbreitung der Phagen über die anfängliche Infektionsereignis 25. Änderungen an der Helferphage haben sich bewährtbei unspezifischen Verpackungs Verringerung, wenn auch manchmal auf Kosten einer reduzierten Phagenproduktion 26.
Ausgeführt Bakteriophagen sind zu einem unverzichtbaren Werkzeug für E. geworden coli synthetischen Biologie ermöglicht die schnelle Bereitstellung neuer Gene zu einer wachsenden Bevölkerung. Neuere Arbeiten haben 11 interzellulären Kommunikationsschaltungen erzeugt oder Transkriptionsfaktoren ausgedrückt Antibiotika - Resistenz zu unterdrücken Pfade 27. Das Protokoll vorgestellt fügt hier zu einer wachsenden Sammlung von Werkzeugen, die durch programmierte RNAs Kontrolle der bakteriellen Physiologie ermöglichen. CRISPR-Cas Nukleasen, wenn sie mutiert Nukleaseaktivität zu beseitigen, wurden Transkription 17,28 an RNA-geführten Genziele zu verdrängen gezeigt. Im Gegensatz dazu arbeitet sRNA silencing auf der Translationsebene und erfordert keine exogenen Protein-Expression. Next-Generation Biotechnologien können Transkriptions- und Translationskontrolle mit dem Phagen-vermittelte Abgabe vereinen komplexe Phänotypen zu programmierenTypen in Echtzeit.
The authors have nothing to disclose.
Die Finanzierung für diese Arbeit wurde von der Fondation Bettencourt Schueller zur Unterstützung der Pariser Bettencourt iGEM-Team zur Verfügung gestellt. Wir danken dem INSERM U1001 Forschungseinheit und Chantal Lotton für technische Hilfe und Beratung. Phagemid Litmus28i_J23115-B0032-GFP wurde von Monica Ortiz und Drew Endy von der Stanford zur Verfügung gestellt.
| Name | Company | Catalog Number | Comments |
| Plasmid Miniprep Kit | Qiagen | 27104 | |
| DpnI Enzyme | NEB | R0176S | |
| Phusion High Fidelity Polymerase | NEB | M0530S | |
| Taq 2x Master Mix | NEB | M0270L | |
| M13KO7 Helper Phage | NEB | N0315S | |
| DH5α Competent Cells | Life Technologies | 18265-017 | |
| TOP10F' Cells | Life Technologies | C3030-03 | |
| LB Broth | Sigma | L3022-250G | |
| Ampicillin | Sigma | A9393-5G | |
| Kanamycin | Sigma | 60615-5G | |
| Chloramphenicol | Sigma | C0378-5G | |
| Tetracycline | Sigma | 87128-25G |
- Ohkumo, T., Masutani, C., Eki, T., Hanaoka, F. Use of RNAi in C. elegans. RNAi. , 129-137 (2008).
- Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., Mello, C. C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 391 (6669), 806-811 (1998).
- Iordanou, E., Chandran, R. R., Blackstone, N., Jiang, L. RNAi interference by dsRNA injection into Drosophila embryos. J Vis Exp. (50), e2477 (2011).
- Ramadan, N., Flockhart, I., Booker, M., Perrimon, N., Mathey-Prevot, B. Design and implementation of high-throughput RNAi screens in cultured Drosophila cells. Nat Protoc. 2 (9), 2245-2264 (2007).
- Tsong, T. Y. Electroporation of cell membranes. Biophys J. 60 (2), 297-306 (1991).
- Kim, W. J., Chang, C. -. W., Lee, M., Kim, S. W. Efficient siRNA delivery using water soluble lipopolymer for anti-angiogenic gene therapy. J Control Release. 118 (3), 357-363 (2007).
- Shi, Y. Mammalian RNAi for the masses. Trends Genet. 19 (1), 9-12 (2003).
- Na, D., Yoo, S. M., Chung, H., Park, H., Park, J. H., Lee, S. Y. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol. 31 (2), 170-174 (2013).
- Ceroni, F., Algar, R., Stan, G. -. B., Ellis, T. Quantifying cellular capacity identifies gene expression designs with reduced burden. Nat Methods. 12 (5), 415-418 (2015).
- Libis, V. K., Bernheim, A. G., et al. Silencing of Antibiotic Resistance in E. coli with Engineered Phage Bearing Small Regulatory RNAs. ACS Synth Biol. 3 (12), 1003-1006 (2014).
- Ortiz, M. E., Endy, D. Engineered cell-cell communication via DNA messaging. J Biol Eng. 6 (1), 16 (2012).
- Edelheit, O., Hanukoglu, A., Hanukoglu, I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9 (1), 61 (2009).
- Chung, C. T., Miller, R. H. Preparation and storage of competent Escherichia coli cells. Recombinant DNA Part I. , 621-627 (1993).
- Phornphisutthimas, S., Thamchaipenet, A., Panijpan, B. Conjugation in Escherichia coli. Biochem Mol Biol Educ. 35 (6), 440-445 (2007).
- Sezonov, G., Joseleau-Petit, D., D'Ari, R. Escherichia coli Physiology in Luria-Bertani Broth. J Bacteriol. 189 (23), 8746-8749 (2007).
- Mittal, V. Improving the efficiency of RNA interference in mammals. Nat Rev Genet. 5 (5), 355-365 (2004).
- Qi, L. S., Larson, M. H., et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 152 (5), 1173-1183 (2013).
- Lu, T. K., Collins, J. J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 104 (27), 11197-11202 (2007).
- Yosef, I., Manor, M., Kiro, R., Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci USA. 112 (23), 7267-7272 (2015).
- Karlsson, F., Malmborg-Hager, A. -. C., Albrekt, A. -. S., Borrebaeck, C. A. K. Genome-wide comparison of phage M13-infected vs. uninfected Escherichia coli. Can J Microbiol. 51 (1), 29-35 (2005).
- Senthil-Kumar, M., Mysore, K. S. Caveat of RNAi in plants: the off-target effect. Methods in molecular biology. 744, 13-25 (2011).
- Jackson, A. L., Linsley, P. S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 9 (1), 57-67 (2010).
- Cho, S. W., Kim, S., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Hagens, S., Blasi, U. Genetically modified filamentous phage as bactericidal agents: a pilot study. Lett Appl Microbiol. 37 (4), 318-323 (2003).
- Kasman, L. M., Kasman, A., Westwater, C., Dolan, J., Schmidt, M. G., Norris, J. S. Overcoming the phage replication threshold: a mathematical model with implications for phage therapy. J Virol. 76 (11), 5557-5564 (2002).
- Chasteen, L., Ayriss, J., Pavlik, P., Bradbury, A. R. M. Eliminating helper phage from phage display. Nucleic Acids Res. 34 (21), e145 (2006).
- Lu, T. K., Collins, J. J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci USA. 106 (12), 4629-4634 (2009).
- Bikard, D., Jiang, W., Samai, P., Hochschild, A., Zhang, F., Marraffini, L. A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41 (15), 7429-7437 (2013).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved