
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biology
Fagos mediada por la entrega de apuntado sRNA Construye para derribar la expresión génica en
We describe a method to knock down gene expression in a growing population of E. coli cells using sequence-targeted sRNA expression cassettes delivered by an M13 phagemid vector.
caídas de ARN mediada se utilizan ampliamente para controlar la expresión génica. Esta familia versátil de técnicas hace uso de RNA corto (sRNA) que se puede sintetizar con cualquier secuencia y diseñado para complementar cualquier gen diana para silenciamiento. Debido sRNA construcciones pueden introducirse a muchos tipos de células directamente o utilizando una variedad de vectores, la expresión del gen puede ser reprimida en las células vivas sin modificación genética laborioso. tecnología desmontables El ARN más común, RNA de interferencia (RNAi), hace uso de la secuencia de reconocimiento de ARN inducida por silenciar complejo (RISC) para mediar endógeno y la escisión del ARNm diana. Las aplicaciones de esta técnica son, por tanto, limitadas a los organismos RISC que expresan, principalmente eucariotas. Recientemente, una nueva generación de los biotecnólogos de ARN han desarrollado mecanismos alternativos para controlar la expresión génica a través de RNA, y así hecho posibles caídas de genes de ARN mediada en bacterias. Aquí se describe un método para silenciar genes expressión en E. coli que funcionalmente se asemeja a RNAi. En este sistema un fagémido sintético está diseñado para expresar sRNA, que puede diseñado para dirigirse a cualquier secuencia. La construcción de expresión se suministra a una población de E. células de E. coli con el fago no lítico M13, después de lo cual es capaz de replicarse de forma estable como un plásmido. Reconocimiento antisentido y el silenciamiento de ARNm diana está mediada por la proteína Hfq, endógena a E. coli. Este protocolo incluye métodos para el diseño de la sRNA antisentido, construir el vector fagémido, el envasado del fagémido en bacteriófago M13, la preparación de una población de células vivas para la infección, y la realización de la propia infección. La proteína fluorescente mKate2 y el cloranfenicol acetiltransferasa de genes de resistencia a antibióticos (CAT) se orientan a generar datos representativos y cuantificar la eficacia desmontables.
caídas de genes de ARN mediada por proceder en dos etapas. En primer lugar, una molécula de ARN se introduce en una línea de célula u organismo de estudio. En segundo lugar, las proteínas de unión a ARN endógenos facilitar el reconocimiento de ARN diana y producen el efecto silenciador. Todas las tecnologías de ARN desmontables se benefician de la naturaleza adaptable de sRNAs sintéticos, que puede ser producido fácilmente para que coincida con un objetivo específico de interés. Sin embargo, los detalles moleculares de la absorción de ARN y silenciamiento varían ampliamente a través de sistema de modelo, lo que limita dónde y cómo caídas de ARN se pueden aplicar.
En los nematodos, el ARN de doble cadena (dsRNA) moléculas se puede introducir directamente en los medios o mediante la alimentación de los gusanos con una población de dsRNA-expresión de E. células de E. coli de 1,2. En Drosophila, el ARNi se puede lograr mediante la microinyección de embriones con dsRNA 3, o implementadas en líneas celulares por simple adición de dsRNA al medio de cultivo 4. En líneas celulares de mamíferos,ARN de interferencia pequeños sintéticos (siRNAs) se pueden suministrar a las células vivas mediante electroporación 1,2,5, empaquetado en liposomas 3,6, o expresarse a partir de vectores de plásmidos de ADN de 4,7. Una vez que las especies de ARN alcanza el citosol, la vía de RNAi se basa en el complejo RISC para procesar dsRNA, facilitar el reconocimiento antisentido de la diana, y catalizar la represión de traducción, la degradación de mRNA, o la formación de heterocromatina, dependiendo del huésped.
Debido a estos requisitos, clásica ARNi puede ser realizado solamente en los organismos que ocupan ARN exógeno de manera eficiente y expresan RISC o una actividad similar a RISC. En particular, esto excluye el modelo bacteria E. coli, que carece de la vía de RNAi. Sin embargo, los recientes avances en la biología sintética proporcionan las herramientas para resolver tanto el problema de la entrega y el problema silenciamiento.
En este protocolo, las construcciones de sRNA se expresan en E. coli a partir de un vector de ADN entregado a living células utilizando el sistema de fagémido / M13 ayudante. Un fagémido es cualquier plásmido con un origen f1 de fago derivados de la replicación. Un plásmido auxiliar, en este caso M13KO, lleva toda la maquinaria necesaria para producir partículas virales, pero no es en sí competente para la replicación y el empaquetamiento. Cuando se co-transformaron un fagémido y el plásmido auxiliar, el fagémido solo se replica en el origen f1, envasa y se secreta. El fagémido vectorizado entonces será competente para infectar E. vivo coli mediante el pilus F.
En este sistema, el efecto de silenciamiento es producido por cassettes sRNA cliente combinando una secuencia andamio con una secuencia de unión a la diana. La secuencia de unión a diana es de 24 pares de bases antisentido a un ARNm diana, típicamente en el sitio de unión de ribosomas (RBS). La secuencia andamio, desarrollado por Na y colegas 8, contiene un motivo de unión Hfq extraída del MICC, un pequeño ARN regulador endógeno de E. coli. La proteína Hfq estimula RNA-RNA binding y la degradación del ARNm, que sirve una función en este sistema similar al RISC en RNAi. La Figura 1 muestra un esquema completo para caídas sRNA de fagos mediada, incluyendo la estructura sRNA casete, vectorización fagémido, y el mecanismo de silenciamiento.
Como un método para modular la expresión génica en E. coli, sRNA silenciamiento es simple, rápido y versátil. El E. dirigida coli no está cargado más allá de la propagación de la fagémido y expresando la sRNA. Esto puede ser relevante en el contexto de la biología sintética o de investigación básica, donde la expresión de las construcciones más grandes heterólogos puede tensar los recursos celulares 9. Los fagémidos con nuevos objetivos se pueden producir con una sola PCR y se recogieron un día después de la transformación de fagémido. Por último, casi cualquier ARNm puede ser objetivo. El casete de regulación sRNA (en un plásmido estándar) se ha demostrado que trabajar en una variedad de objetivos en el metabolismo con los niveles típicos de represión> 90% 8.
10. En primer lugar, un fagémido empaquetado se introduce en un cultivo discontinuo de E. células de E. coli y se utiliza para silenciar la expresión de la proteína fluorescente mKate2. cambios de fluorescencia posteriores se controlan en tiempo real. En segundo lugar, derribando el gen CAT se muestra para reducir la resistencia fenotípica al cloranfenicol en placas de agar. En ambos casos, el fagémido en sí lleva un marcador GFP, permitiendo que la tasa de infección a medirse independientemente de la eficiencia de caída.
1. Diseño y construcción de vectores fagémidos Teniendo sRNA silenciamiento Casetes
- De Novo Diseño de sRNA silencios en 8 Casetes
- Identificar la secuencia completa del mRNA para silenciar utilizando una base de datos secuencia de ADN. Para generar la secuencia diana, tenga en cuenta el primero 24 pb de la secuencia de codificación, de la posición 1 a 24 que comienza con el codón de inicio (por ejemplo, ATG).
Nota: El silenciamiento es menos eficiente cuando otros sitios o segmentos del ARNm se dirigen 8. - Tome el complemento inverso de la secuencia diana para producir la secuencia guía para el casete sRNA. Ver Tabla 1 para ejemplos de secuencias diana y guías para cloranfenicol acetiltransferasa (CAT).
- Para el diseño del 292 pb casete de expresión sRNA completa, organizar el promotor pR, secuencia GUÍA, Hfq dominio de unión a proteínas y las secuencias de terminación de la transcripción T1 / TE en serie (Tabla 2).
- Añadir sitios de clonación adicionales de elección para facilitar la clonación del casete sRNA en el vector objetivo.
- Obtener la completa casete sRNA a través de la síntesis de genes comercial o un método similar y clonar en cualquier vector fagémido con un origen de replicación f1 funcional 11. Ver información de apoyo para la secuencia completa del vector fagémido final.
- Identificar la secuencia completa del mRNA para silenciar utilizando una base de datos secuencia de ADN. Para generar la secuencia diana, tenga en cuenta el primero 24 pb de la secuencia de codificación, de la posición 1 a 24 que comienza con el codón de inicio (por ejemplo, ATG).
- La alteración de la secuencia diana por un casete de expresión sRNA existente usando sitio basada en PCR de mutagénesis dirigida al 12
- Identificar la secuencia de guía 24 pb en un casete de expresión sRNA existente. Nota: El plásmido pAB.001 anotada, utilizado en este trabajo, está disponible como un archivo de secuencia complementaria.
- Diseño cebadores directos e inversos con regiones cortas de homología con el casete sRNA existente que flanquean la nueva secuencia GUIDE 24 pb. Obtener los cebadores a través de la síntesis de oligonucleótidos comercial.
Nota: Diseño de cebadores para la mutagénesis dirigida al sitio es depicTed en la figura 2. plantilla de secuencias de cebadores exacta se proporcionan en la Tabla 3. - Preparar un cultivo de 5 ml de E. coli que lleva la plantilla de expresión fagémido sRNA. Cultivar las células durante la noche a 37 ° C con agitación, en medio LB con los antibióticos apropiados.
- Extraer y purificar la expresión fagémido plantilla sRNA de los 5 ml de cultivo bacteriano utilizando un kit miniprep de ADN o método similar 12.
- Preparar dos reacciones de PCR utilizando el fagémido plantilla de expresión sRNA y polimerasa de alta fidelidad, una con el delantero y una con el cebador inverso (Tabla 4). Utilice condiciones de PCR como se recomienda por el suministrador de la polimerasa (Tabla 5). Aumentar la concentración de plantilla para 10-50x más alta que una reacción estándar para tener en cuenta el hecho de que la reacción de un único cebador no produce la amplificación exponencial.
- Combinar las dos reacciones de PCR anteriores en un tubo de microcentrífuga. Anneal los productos por calentamiento a 98 ° C en un baño de agua hirviendo. Inmediatamente después de colocar el tubo de microcentrífuga en el baño de agua, retire la fuente de calor y permitir que el baño para volver lentamente a temperatura ambiente durante 1-2 horas.
- Para eliminar la plantilla no mutada sRNA expresión fagémido, añadir 1 l. enzima de restricción DpnI a la mezcla y se incuba a 37 ° C durante 1 hora, o el tiempo recomendado por el fabricante para la digestión completa.
Nota: DpnI digiere sitios diana solamente metilados, que están presentes en fagémidos huésped-replicado pero no los productos de PCR. - Transform comprados o preparados químicamente competentes E. 13 coli con 1-5 l del producto de PCR recocido. Aislar colonias individuales de la cepa transformada por siembra en medio selectivo en placas de agar LB que contenían antibióticos apropiados.
- Para verificar la incorporación de la secuencia de GUÍA correcta, la pantalla de las colonias resultantes por PCR de colonias. Utilizando una punta de pipeta de 200 l, collect una pequeña cantidad de células a partir de una sola colonia transformada. Mark y preservar la colonia original para su uso aguas abajo después de la verificación.
- Añadir las células recogidas a 50 l de agua libre de nucleasa en un tubo de microcentrífuga. Mezclar pipeteando arriba y abajo.
- El uso de un termociclador de sobremesa o un baño de agua hirviendo, lisar las células por calentamiento a 95 ° C durante 2 min.
- PCR-amplificar la región fagémido usando 1 l de células lisadas con el calor como un molde de ADN. Las condiciones de PCR y protocolos termociclador se proporcionan en las Tablas 6 y 7. Vea el archivo de secuencia de pAB.001 suplementario para las secuencias de cebador de verificación.
- Secuenciar el producto de PCR para verificar la incorporación de la secuencia correcta guía.
- Inocular un cultivo de 5 ml de la E. clon de E. coli que lleva el fagémido expresión sRNA secuencia verificada. Cultivar las células durante la noche a 37 ° C con agitación, en medios LB selectivo.
- Preparar un stock de gliceroldel clon de secuencia verificada. Añadir 750 l de cultivo de la noche a 250 l de glicerol al 60% en un tubo criogénico con tapón de rosca.
- Almacenar las existencias de glicerol a -80 ° C indefinidamente. El resto del cultivo de la noche se puede usar como una fuente para el fagémido expresión sRNA en el paso 2.
2. Producción y Cosecha de las poblaciones de fagémidos empaquetados-M13
- Preparar un cultivo de 5 ml de E. coli que lleva la expresión fagémido sRNA. Cultivar las células durante la noche a 37 ° C con agitación, en medio LB con los antibióticos apropiados. Nota: La expresión fagémido sRNA se puede obtener a través de novo de clonación tal como se describe en el paso 1.1.5, o modificada a partir de un fagémido existente y se cosecha en el paso 1.2.16.
- Del mismo modo preparar un cultivo de 5 ml de E. coli que lleva el plásmido ayudante M13KO7. Cultivar las células durante la noche a 37 ° C con agitación, en medios LB selectivo.
- Extraer y purificar el expressio sRNAn fagémido y plásmido auxiliar usando un kit de extracción de ADN o método similar 12.
- Cotransform comprado o preparado 13 E. coli químicamente competentes con 1 l de cada uno de sRNA fagémido expresión y plásmido auxiliar. Seleccionar para cotransformants en placas sobre agar LB con antibióticos selectivos para ambas construcciones.
- Preparar un cultivo de 10 ml de una sola colonia de la cepa cotransformed en LB con los antibióticos selectivos. Incubar a 37 ° C con agitación durante 8-12 hr o durante la noche.
- Se centrifuga el cultivo a 3.300 xg durante 10 min. Recoger el sobrenadante y filtrar a través de un filtro de 0,2 micras. Precaución: En caso de derrame de los medios de comunicación, limpie el área con lejía diluida (0,5%) para destruir las partículas de fago infecciosas.
- Conservar el filtrado de fagémidos empaquetados a 4 ° C. Nota: Las muestras se pueden mantener durante días a semanas sin pérdida de actividad.
3. Preparación de las células F + objetivo para el silenciamiento
- Determinar si las células que se van dirigidas a silenciar expresan el pilus F 14. Si el pilus F es ya presente, continúe con el paso 4.
Nota: las cepas comunes de laboratorio de E. coli anotados como F + o F 'para indicar la presencia de la pilus F en su genoma o en un plásmido. - Obtener una cepa de E. F + coli, tal como TOP10F '.
Nota: asegúrese de que la cepa diana lleva un marcador de resistencia único con el fin de ser separado del donante F-plásmido después de la conjugación. - Para introducir F pilus por conjugación con una cepa F +, preparar 5 ml de cultivos tanto de la cepa diana y donantes F-14 plásmido. Cultivar las células durante la noche a 37 ° C con agitación, en medio LB con los antibióticos apropiados.
- El día siguiente, se diluye ambas cepas 1: 100 en 5 ml de LB selectiva y continuar el cultivo a 37 ° C con agitación.
- Determinar la fase de crecimiento de las células mediante la medición de la optdensidad ical del cultivo a 600 nm (OD 600) usando un espectrofotómetro de mesa. Cultivar las células durante aproximadamente 2 horas hasta una DO600 de 0,3 se alcanza, lo que indica un crecimiento de fase log 15.
- Preparar reacciones 3 Conjugación en tubos de microcentrífuga: 0,5 ml F-plásmido cepa donante + 0,5 ml de destino, 0,5 ml F-plásmido donante + 0,5 ml de medio LB (control negativo) y 0,5 ml cepa diana + 0,5 ml de medio LB (control negativo). Deje que la conjugación transcurriera durante 2 horas a 37 ° C con agitación.
- Placa 100 l de cada reacción de conjugación en agar LB selectivo con antibióticos específicos para el F-plásmido (típicamente tetraciclina) y la tensión de destino. La placa de las reacciones de control negativo para confirmar que ni el donante o cepa receptora expresan ambas resistencias a antibióticos.
4. La infección con fagémidos empaquetados para el silenciamiento
- Se inocula una sola colonia de células diana F + en un cultivo de 5 ml de LB mediuna con los antibióticos apropiados. Incubar toda la noche a 37 ° C con agitación.
- El día siguiente, diluir las células diana F + 1: 100 en 5 ml de medio LB selectivas y seguir de cultivo a 37 ° C con agitación.
- Determinar la fase de crecimiento de las células mediante la medición de la densidad óptica del cultivo a 600 nm (OD 600) usando un espectrofotómetro de mesa. Cultivar las células durante aproximadamente 2 horas hasta una DO600 de 0,3 se alcanza, lo que indica un crecimiento de fase log 15. Nota: La expresión de la eficiencia y la infección pilus F es más alta en la fase de registro.
- Añadir los fagémidos empaquetados-M13 (de la etapa 2.6) a las células diana en una relación en volumen de 1: 100 para lograr la infección casi 99% de la población objetivo. Permitir que la infección de proceder a 37 ° C con agitación durante 30 a 60 min.
- Ensayo, el fenotipo sRNA-silenciamiento de acuerdo con el método de elección.
Nota: Para un objetivo de la proteína fluorescente, el efecto de silenciamiento puede cuantificarsedirectamente por fluorometría 10. Alternativamente, los ensayos fenotípicos pueden ser usados para observar las consecuencias fenotípicas de gen desmontables 8. - Preparar un stock de glicerol para el anfitrión fagémido sRNA expresar siguientes pasos 1.2.14-1.2.16. Nota: el fagémido se propagará en la cepa huésped indefinidamente y puede ser mantenida con antibiótico similar a un plásmido convencional.
El silenciamiento de mKate2 fluorescencia en medios líquidos
La figura 1 representa el esquema de caídas sRNA mediada descritos en este trabajo, incluyendo el diseño sRNA casete, vectorización fagémido, y el mecanismo de silenciamiento. Siguiendo el protocolo 1.2, el casete de silenciar sRNA del plásmido pAB.001 fue alterado para apuntar mKate. El casete sRNA fue sintetizado y clonado en fagémido Litmus28i_J23115-B0032-GFP, un regalo de Mónica Ortiz y Drew Endy 11. Este fagémido lleva marcadores de expresión de GFP y la resistencia a la kanamicina, permitiendo infecciones exitosas para ser rastreados. las existencias de fagémidos empaquetados se prepararon siguiendo el protocolo 2.
Un derivado de E. coli K12 MG1655 que lleva un constitutivamente expresado, marcador mKate2 cromosómicamente integrado se preparó para la infección de fagos mediante conjugación con una cepa de plásmido donante F siguiente protocolo 3. Las células fueron cultivadas a mediados de la fase de registro y el fagémido introducido siguiente protocolo 4. Después de la infección de fagémido, cultivos de 200 l se transfirieron a un lector de placas de fluorescencia y la fluorescencia se controló de forma continua durante 24 hr.
La Figura 3 muestra el efecto de silenciamiento sRNA mediada por la expresión mKate2. La cepa infectada con un fagémido anti-mKate2 no mostró fluorescencia mKate2 detectable respecto. Por el contrario, esta cepa hizo expresar el marcador de GFP, indicando la absorción exitosa del fagémido. las células de control no infectadas producen mKate2 fluorescencia pero no GFP. Un control adicional, en la que el dominio de dirección anti-mKate2 fue sustituida por una secuencia de direccionamiento CAT, no tuvo efecto sobre la fluorescencia mKate2.
El silenciamiento de la resistencia a cloranfenicol en placas de agar
contenido "fo: keep-together.within-page =" 1 "> Siguiendo el protocolo 1.1, un casete de silenciar sRNA fue producido para apuntar CAT Un derivado de E. coli K12 MG1655 que lleva un expresado constitutivamente, cromosómicamente integrada marcador CAT estaba preparado. la infección por fagos por conjugación con una cepa donante F-plásmido siguiendo el protocolo 3. las células se cultivaron hasta la fase logarítmica media y el fagémido introdujo siguiente protocolo 4. Después de 1 h de incubación a 37 ° C, las células infectadas se diluyeron en serie y se sembraron a una intervalo de concentraciones de cloranfenicol. las placas se incubaron durante la noche, y la proporción de células resistentes a cada concentración de cloranfenicol se determinó contando unidades formadoras de colonias (UFC) al día siguiente.La Figura 4 muestra el efecto de silenciamiento mediado por sRNA en el fenotipo de resistencia a cloranfenicol. Las células no infectadas, o células infectadas con el fagémido de orientación mKate2, fueron resistentes a lacloranfenicol a todas las concentraciones ensayadas. En contraste, las células infectadas con el fagémido de orientación CAT mostraron reducción de la supervivencia a bajas concentraciones de cloranfenicol, y casi 99% de destrucción a concentraciones más altas. La adición de ampicilina, para seleccionar sólo las bacterias que portan el fagémido, redujo la supervivencia cloranfenicol a niveles indetectables. Esto es consistente con los trabajos anteriores que muestran que escape de la infección por fagos es una ruta común para escapar de silenciamiento 10.
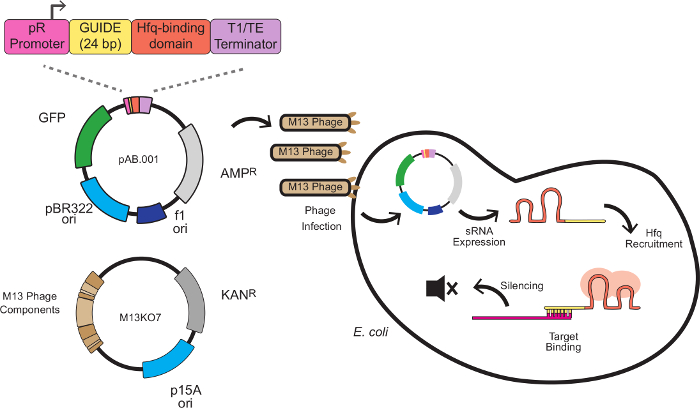
Figura 1: el silenciamiento de genes en E. coli con sRNA casetes de expresión Entregado por el fago M13 El casete sRNA se compone de 4 módulos:. el promotor P (un promotor constitutivo derivado de bacteriófago lambda), un dominio de dirección de 24 pb, un dominio de unión Hfq extrae de MICC y un terminador transcripcional 8 E. coli que expresa el pilus F, donde comienza la expresión sRNA. El sRNA, se movilizan la proteína Hfq (representado en rojo) y se une a un objetivo ARNm antisentido cerca del sitio de unión al ribosoma, lo que resulta en la represión de traducción y de la degradación del ARNm. Por favor, haga clic aquí para ver una versión más grande de esta figura.
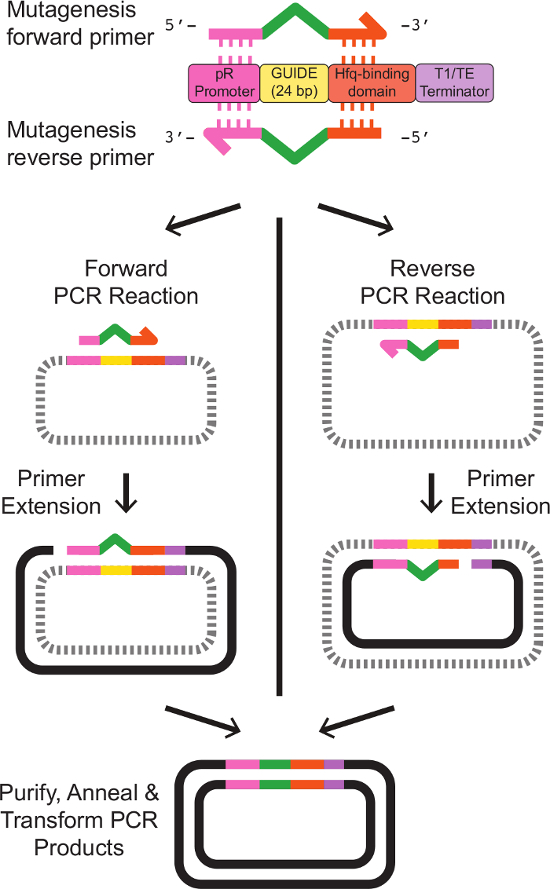
Figura 2:. Diseño de cebadores y mutagénesis dirigida de sitio de destino sRNA dos cebadores están diseñados con una homología parcial a un casete de sRNA existente. El cebador directo contiene 20 pb homóloga al promotor pR en el extremo 5 ', seguido de24 pares de bases que representa la nueva secuencia guía, y luego 18 pb homóloga al dominio de unión Hfq-en el extremo 3 '. El cebador inverso es el complemento inverso exacto del cebador directo, con regiones de homología con el casete sRNA existente que flanquea el complemento inverso de la nueva secuencia GUIDE. Primer secuencias exactas se dan en la Tabla 3.-Cebador único reacciones de PCR separadas con el avance y los cebadores inversos producen lineal, ADN de cadena sencilla con la secuencia modificada deseada. Recocer el avance y retroceso de los productos de reacción, después de la limpieza como se describe en el protocolo, los resultados en el ADN de doble cadena plásmido que lleva el casete sRNA modificado deseado. Por favor, haga clic aquí para ver una versión más grande de esta figura.
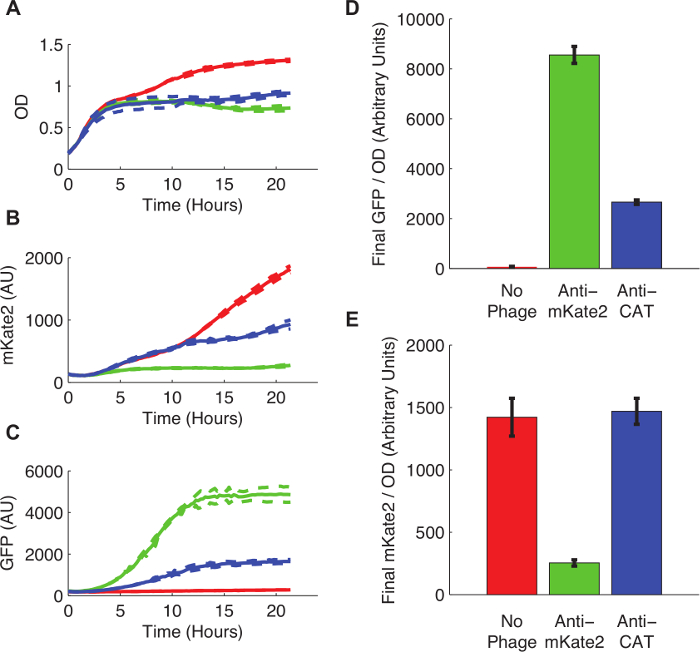
Figura 3: Knockdown de un reportero integrado cromosómicamente mKate2 fluorescente. E. coli K12 MG1655 expresar mKate2 se dejaron sin tratar (líneas rojas) y las barras de izquierda, infectado con un fagémido anti-mKate2 (líneas verdes y bares), o se infecta con un control fagémido CAT orientación (líneas azules y bares). (A) Sin tratamiento E. coli creció hasta densidades de saturación más altos, lo que indica un coste metabólico para la infección por fagos. Las líneas de puntos indican las desviaciones estándar de 3 repeticiones. (B) La señal mKate2 se redujo a niveles de fondo cerca de la anti-mKate2 cepa tratados, pero no en las cepas de control. Fluorescencia (C) GFP, también transportado por el fagémido, era detectable sólo en los controles de fagémidos tratada. (D, E) lecturas de fluorescencia final después de 24 horas de crecimiento se normalizaron a OD. GFP señal, que indica infección fagémido, estuvo ausente en los controles no tratados, sino que también reduce sustancialmente tras anti-CAT phagemid tratamiento. Esto puede indicar fuera de objetivo efectos del fagémido. La señal mKate2 se redujo mediante el tratamiento fagémido anti-mKate2 comparación con los controles no tratados. El fagémido de control CAT-dirigida mostró ningún efecto sobre la fluorescencia mKate2. Las barras de error representan la desviación estándar de 3 repeticiones. Por favor, haga clic aquí para ver una versión más grande de esta figura.
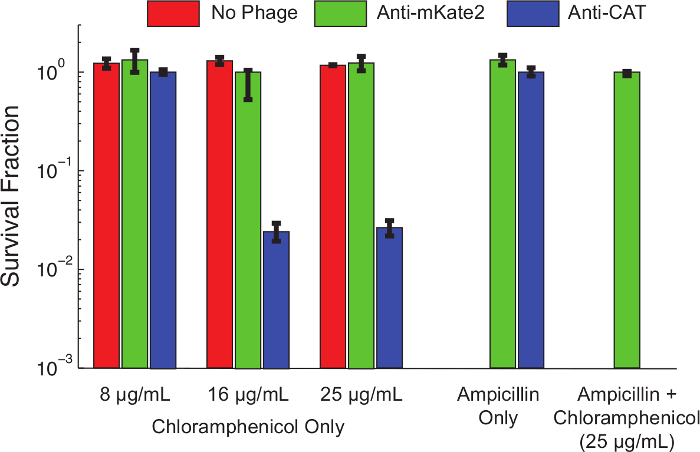
Figura 4: Derribo de CAT restaura la sensibilidad de cloranfenicol a una población genéticamente resistente E.. coli K12 MG1655 que expresa un gen CAT cromosómicamente integrado se dejaron sin tratar (barras rojas), se trata con un fagémido control de la orientación mKate2 (barras verdes), o se trata con un fagómido que expresa un anti-CAT Srna (barras azules). Después de 1 hora de infección, la viabilidad de la hormiga se indicaibiotics se evaluó mediante diluciones seriadas y de las planchas. Las cepas tratadas con fagémido anti-CAT murieron significativamente (> 90%) por cloranfenicol a concentraciones más altas, mientras que los tratamientos de control no se vieron afectados. La adición de ampicilina a las placas de cultivo selecciona positivamente para la infección fagémido y elimina las células no infectadas. En estas condiciones, no se observaron colonias resistentes a cloranfenicol después del tratamiento anti-CAT. Esto indica que la mayoría de los supervivientes representan un fracaso de la infección, en lugar de un fracaso de silenciamiento. Las barras de error representan la desviación estándar de 3 repeticiones. Por favor, haga clic aquí para ver una versión más grande de esta figura.
| secuencia diana CAT | 5 '- ATGGAGAAAAAAATCACTGGATAT - 3' |
| GUÍA secuencia de CAT | 5 '- ATATCCAGTGATTTTTTTCTCCAT - 3' |
Tabla 1:. Un ejemplo DIANA y GUÍA secuencia para el gen CAT Tenga en cuenta la relación inversa complemento.
| promotor pR | TAACACCGTGCGTGTTGACTATTTTACCTCTGGCGGTGATAATGGTTGC | ||||
| secuencia GUÍA | ATATCCAGTGATTTTTTTCTCCAT | ||||
| dominio de unión hfq | TTTCTGTTGGGCCATTGCATTGCCACTGATTTTCCAACATATAAAAAGACAAGCCCGAACAGTCGTCCGGGCTTTTTT TCTCGAG | ||||
| T1 / terminador TE | CTCGAGCCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTTTTGTCGGTGAA CGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA | ||||
Cuadro 2:. componentes de secuencia de la sRNA casete Cada secuencia está escrito 5'-3 '. El casete completa es la concatenación de estos 4 elementos en orden y comprende 292 pb.
| cebador | 5 '- CTGGCGGTGATAATGGTTGC [GUÍA] TTTCTGTTGGGCCATTGC - 3' |
| cebador inverso | 5 '- GCAATGGCCCAACAGAAA [TARGET] GCAACCATTATCACCGCCAG - 3' |
Tabla 3: Primer diseño para alterar elementos de guía existente, el cebador directo incluye los últimos 20 pb del promotor pR, la nueva secuencia GUÍA, y la primera de 18 pb del dominio de unión Hfq.. La secuencia diana es el complemento inverso exacto de la secuencia GUÍA. El cebador inverso es el complemento inverso exacto del cebador directo.
Tabla 4: Condiciones sugerido para el Single-cebador mutagénico de PCR.
| Paso | Temperatura | Hora |
| La desnaturalización inicial | 98 ° C | 30 segundos |
| 30 ciclos | 98 ° C | 10 sec |
| 55 ° C | 30 segundos | |
| 72 ° C | 120 sec | |
| extensión final | 72 ° C | 300 sec |
| Almacenamiento | 10 ° C |
Tabla 5: Protocolo sugerido termociclador para el Single-cebador mutagénico de PCR.
| Componente | Volumen |
| El ADN molde | 1 l |
| 10 mM Cebador | 0,5 l |
| 10 M cebador inverso | 0,5 l |
| Taq Mix 2X Maestro | 25 l |
| finales libre de nucleasar | 23 l |
| Volumen total | 50 l |
Tabla 6: Condiciones sugerido para la verificación de la secuencia de PCR.
| Paso | Temperatura | Hora |
| La desnaturalización inicial | 95 ° C | 30 segundos |
| 30 ciclos | 95 ° C | 30 segundos |
| 55 ° C | 30 segundos | |
| 68 ° C | 30 segundos | |
| extensión final | 68 ° C | 300 sec |
| Almacenamiento | 10 ° C |
Tabla 7: Sugerido termociclador ProProtocolo para la verificación de la secuencia de PCR.
El presente procedimiento logra la reducción de 80% en los niveles de fluorescencia mKate comparación con los controles no orientados. Esto está en línea con otros métodos desmontables ARN, donde no se observa silenciamiento completo y la eficiencia del 50-90% es típico 16,17. A nivel fenotípico, caídas CAT orientados fueron capaces de atenuar de manera significativa la resistencia al cloranfenicol, y eliminarlo bajo algunas condiciones.
El desmontables fenotipo detectable a nivel de la población después de sólo unas pocas horas después de la infección (Figura 3B). Esto ilustra una característica importante de suministro basado en el fago: a alta frecuencia desmontables se puede obtener directamente en cultivo discontinuo sin modificación genética previa. A diferencia de las modificaciones genéticas convencionales utilizando la transformación plásmido o integración genómica, la infección por fagos no requiere que una población se re-cultiva a partir de una sola colonia aislada. Esto permite que los efectos de la infección del fago para ser explored en poblaciones con dinámicas espaciales complejas 11, con estructuras espaciales pre-existentes, como biopelículas 18, o en las poblaciones naturales mezclados genéticamente 19.
Un paso crítico en este método es la producción de fagémido empaquetados en alto título. La carga metabólica asociada con la producción de partículas de fago puede dar lugar a altas tasas de mutación o pérdida del plásmido en la cepa de producción fagémido. Se recomienda que la cepa de producción de fagémido se cultivó directamente de una sola colonia cotransformed y no refrigerado, congelado o sub-cultivadas antes de la cosecha del fago. A baja eficiencia de co-transformación también se puede observar cuando se introduce el fagémido y ayudante plásmido de E. coli simultáneamente. En este caso, las eficiencias más altas se pueden obtener mediante la transformación del plásmido auxiliar primero, a continuación, la preparación de células competentes que llevan el plásmido auxiliar para la transformación posterior con el fagémido.
phagemid infección o expresión sRNA también impone una carga metabólica detectable en las células diana, y pueden dar lugar a alguna perturbación fenotípica. Por ejemplo, se observó una reducción en la fluorescencia mKate2 incluso cuando las células se infectaron con un fagémido orientación CAT (Figura 3). La infección con M13 no se cree que desencadenan respuestas de estrés sistémicos en E. coli 20, pero puede alterar patrones de transcripción indirectamente. Alternativamente, los marcadores GFP o resistencia a la ampicilina incluidos en el fagémido pueden competir por los recursos celulares, la reducción de la expresión y el crecimiento 9 mKate2. Por último, el propio casete sRNA puede alterar globalmente perfiles de expresión génica mediante titulación de la proteína Hfq, o a través de fuera del objetivo silenciamiento de ARNm. Efectos de destino situados son comunes en la orientación in vivo ARNi 21-23, pero que aún no se han investigado sistemáticamente para este sistema.
Una limitación de este método es que la infección efieficiencia es menor que 100%, lo que permite algunas bacterias no infectadas a persisten en la población. Los resultados de este trabajo y el trabajo anterior 10 sugieren que las células no infectadas representan 1-10% de la población final, y son responsables de la mayor parte de los fenotipos observados nonsilenced. Se conoce una variedad de rutas a M13-resistencia, con el ser la pérdida de mutaciones más común de la expresión de pilus 24. A la luz de estas limitaciones, los controles deben ser usados para confirmar altas tasas de infección y la eficiencia desmontables.
Otra limitación potencial para algunas aplicaciones es la transferencia ocasional de contaminar el fago auxiliar. Aunque M13K07 contiene una señal de empaquetamiento mutada, puede ser empaquetado en la cápside del fago a baja frecuencia y se transfiere a las poblaciones infectadas, lo que resulta en células competentes para la producción de fago y la propagación continua de fago más allá del evento infección inicial 25. Modificaciones al fago auxiliar han demostrado ser eficacesen la reducción de los envases no específica, aunque a veces a costa de la reducción de la producción de fagos 26.
Bacteriófago ingeniería se han convertido en una herramienta indispensable para E. la biología sintética coli, lo que permite la entrega rápida de nuevos genes de una población en crecimiento. Trabajos recientes han producido circuitos de comunicación intercelular 11 o expresado factores de transcripción para suprimir la resistencia a los antibióticos vías 27. El protocolo que aquí se presenta se suma a una creciente colección de herramientas que permiten el control de la fisiología bacteriana a través de los ARN programados. Nucleasas CRISPR-cas, cuando mutado para eliminar la actividad nucleasa, se ha demostrado que reprimir la transcripción en objetivos de genes de ARN de guiado 17,28. Por el contrario, sRNA silenciamiento funciona a nivel de traducción y no requiere expresión de la proteína exógena. La próxima generación de biotecnologías pueden unir control de la transcripción y de la traducción con la entrega mediada por fagos para programar fenotipo complejotipos en tiempo real.
The authors have nothing to disclose.
La financiación de este trabajo fue proporcionado por la Fundación Bettencourt Schueller en apoyo del equipo de París Bettencourt iGEM. Agradecemos a la unidad de investigación INSERM U1001 y Chantal Lotton para la asistencia técnica y asesoramiento. Fagémido Litmus28i_J23115-B0032-GFP fue proporcionado por Mónica Ortiz y Drew Endy, de Stanford.
| Name | Company | Catalog Number | Comments |
| Plasmid Miniprep Kit | Qiagen | 27104 | |
| DpnI Enzyme | NEB | R0176S | |
| Phusion High Fidelity Polymerase | NEB | M0530S | |
| Taq 2x Master Mix | NEB | M0270L | |
| M13KO7 Helper Phage | NEB | N0315S | |
| DH5α Competent Cells | Life Technologies | 18265-017 | |
| TOP10F' Cells | Life Technologies | C3030-03 | |
| LB Broth | Sigma | L3022-250G | |
| Ampicillin | Sigma | A9393-5G | |
| Kanamycin | Sigma | 60615-5G | |
| Chloramphenicol | Sigma | C0378-5G | |
| Tetracycline | Sigma | 87128-25G |
- Ohkumo, T., Masutani, C., Eki, T., Hanaoka, F. Use of RNAi in C. elegans. RNAi. , 129-137 (2008).
- Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., Mello, C. C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 391 (6669), 806-811 (1998).
- Iordanou, E., Chandran, R. R., Blackstone, N., Jiang, L. RNAi interference by dsRNA injection into Drosophila embryos. J Vis Exp. (50), e2477 (2011).
- Ramadan, N., Flockhart, I., Booker, M., Perrimon, N., Mathey-Prevot, B. Design and implementation of high-throughput RNAi screens in cultured Drosophila cells. Nat Protoc. 2 (9), 2245-2264 (2007).
- Tsong, T. Y. Electroporation of cell membranes. Biophys J. 60 (2), 297-306 (1991).
- Kim, W. J., Chang, C. -. W., Lee, M., Kim, S. W. Efficient siRNA delivery using water soluble lipopolymer for anti-angiogenic gene therapy. J Control Release. 118 (3), 357-363 (2007).
- Shi, Y. Mammalian RNAi for the masses. Trends Genet. 19 (1), 9-12 (2003).
- Na, D., Yoo, S. M., Chung, H., Park, H., Park, J. H., Lee, S. Y. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol. 31 (2), 170-174 (2013).
- Ceroni, F., Algar, R., Stan, G. -. B., Ellis, T. Quantifying cellular capacity identifies gene expression designs with reduced burden. Nat Methods. 12 (5), 415-418 (2015).
- Libis, V. K., Bernheim, A. G., et al. Silencing of Antibiotic Resistance in E. coli with Engineered Phage Bearing Small Regulatory RNAs. ACS Synth Biol. 3 (12), 1003-1006 (2014).
- Ortiz, M. E., Endy, D. Engineered cell-cell communication via DNA messaging. J Biol Eng. 6 (1), 16 (2012).
- Edelheit, O., Hanukoglu, A., Hanukoglu, I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9 (1), 61 (2009).
- Chung, C. T., Miller, R. H. Preparation and storage of competent Escherichia coli cells. Recombinant DNA Part I. , 621-627 (1993).
- Phornphisutthimas, S., Thamchaipenet, A., Panijpan, B. Conjugation in Escherichia coli. Biochem Mol Biol Educ. 35 (6), 440-445 (2007).
- Sezonov, G., Joseleau-Petit, D., D'Ari, R. Escherichia coli Physiology in Luria-Bertani Broth. J Bacteriol. 189 (23), 8746-8749 (2007).
- Mittal, V. Improving the efficiency of RNA interference in mammals. Nat Rev Genet. 5 (5), 355-365 (2004).
- Qi, L. S., Larson, M. H., et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 152 (5), 1173-1183 (2013).
- Lu, T. K., Collins, J. J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 104 (27), 11197-11202 (2007).
- Yosef, I., Manor, M., Kiro, R., Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci USA. 112 (23), 7267-7272 (2015).
- Karlsson, F., Malmborg-Hager, A. -. C., Albrekt, A. -. S., Borrebaeck, C. A. K. Genome-wide comparison of phage M13-infected vs. uninfected Escherichia coli. Can J Microbiol. 51 (1), 29-35 (2005).
- Senthil-Kumar, M., Mysore, K. S. Caveat of RNAi in plants: the off-target effect. Methods in molecular biology. 744, 13-25 (2011).
- Jackson, A. L., Linsley, P. S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 9 (1), 57-67 (2010).
- Cho, S. W., Kim, S., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Hagens, S., Blasi, U. Genetically modified filamentous phage as bactericidal agents: a pilot study. Lett Appl Microbiol. 37 (4), 318-323 (2003).
- Kasman, L. M., Kasman, A., Westwater, C., Dolan, J., Schmidt, M. G., Norris, J. S. Overcoming the phage replication threshold: a mathematical model with implications for phage therapy. J Virol. 76 (11), 5557-5564 (2002).
- Chasteen, L., Ayriss, J., Pavlik, P., Bradbury, A. R. M. Eliminating helper phage from phage display. Nucleic Acids Res. 34 (21), e145 (2006).
- Lu, T. K., Collins, J. J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci USA. 106 (12), 4629-4634 (2009).
- Bikard, D., Jiang, W., Samai, P., Hochschild, A., Zhang, F., Marraffini, L. A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41 (15), 7429-7437 (2013).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved