High-resolution Time-lapse Imaging and Automated Analysis of Microtubule Dynamics in Living Human Umbilical Vein Endothelial Cells
In This Article
Summary
Protocols for Human Umbilical Vein Endothelial Cell (HUVEC) culture, transient transfection of fluorescently-labeled markers of microtubule growth, live-cell imaging and automated analysis of interphase microtubule growth dynamics are detailed.
Abstract
The physiological process by which new vasculature forms from existing vasculature requires specific signaling events that trigger morphological changes within individual endothelial cells (ECs). These processes are critical for homeostatic maintenance such as wound healing, and are also crucial in promoting tumor growth and metastasis. EC morphology is defined by the organization of the cytoskeleton, a tightly regulated system of actin and microtubule (MT) dynamics that is known to control EC branching, polarity and directional migration, essential components of angiogenesis. To study MT dynamics, we used high-resolution fluorescence microscopy coupled with computational image analysis of fluorescently-labeled MT plus-ends to investigate MT growth dynamics and the regulation of EC branching morphology and directional migration. Time-lapse imaging of living Human Umbilical Vein Endothelial Cells (HUVECs) was performed following transfection with fluorescently-labeled MT End Binding protein 3 (EB3) and Mitotic Centromere Associated Kinesin (MCAK)-specific cDNA constructs to evaluate effects on MT dynamics. PlusTipTracker software was used to track EB3-labeled MT plus ends in order to measure MT growth speeds and MT growth lifetimes in time-lapse images. This methodology allows for the study of MT dynamics and the identification of how localized regulation of MT dynamics within sub-cellular regions contributes to the angiogenic processes of EC branching and migration.
Introduction
Angiogenesis is triggered by extracellular signaling cues that promote endothelial cells (ECs) to polarize, migrate with directional specificity, and form new vasculature in tissues that require blood supply. Contributing factors thought to drive angiogenesis are signals from the extracellular matrix (ECM). In response to physical cues, ECs extend new branches with directional specificity to guide directional movement in formation of the vascular network1,2. The architecture of EC morphology and branching is defined by localized regulation of the microtubule (MT) cytoskeleton in a way that is coordinated with the regulation of acto-myosin contractility3. In order for EC branches to form, myosin-II must be locally downregulated, resulting in localized membrane protrusions4. At the same time and in the same location within the cell, MT growth dynamics become modified resulting in slow and persistent MT growth to promote branch elongation and stability5. This coordinated cytoskeletal regulation allows ECs to polarize their morphology to facilitate directional migration.
The development of a polarized cell morphology requires polarized MT assembly6. In migrating epithelial cells, MTs grow slowly and persistently specifically at the leading edge of the cell7-9; while in neurons, the initiation and elongation of axons or dendrites requires that MTs are not only present, but that they are dynamically undergoing the processes of assembly and disassembly10-15. In this way, localized control of MT growth dynamics determines the architecture of the cell and allows for rapid remodeling of cell morphology as ECs navigate through their ECM.
MT growth dynamics are regulated by families of molecular motor proteins and non-motor proteins, termed Microtubule Associated Proteins or Microtubule Interacting Proteins (henceforth referred to as MAPs). MAPs can be locally activated or inactivated, typically through signaling cascades initiated at the cell-ECM interface16,17. Once active, MAPs function by binding to the MT and altering the kinetics of MT assembly and disassembly; thereby functioning to locally regulate MT dynamics in order to promote cell shape and polarity18-20. Mitotic Centromere Associated Kinesin (MCAK) is a Kinesin-13 family MAP that binds to MT ends and couples ATP hydrolysis to MT disassembly21-23. This functional role of MCAK is known to be critical within the mitotic spindle24-28, as well as during interphase29,30. Within interphase ECs, MCAK-mediated MT disassembly is regulated by localized Aurora-A induced phosphorylation of MCAK in response to wound-edge induced activation of the Rac1 small GTPase. The outcome of this signaling cascade is the inhibition of MCAK at the leading edge of ECs, resulting in polarized MT growth toward the leading edge and MCAK-induced MT disassembly at the trailing edge of migrating ECs31.
Traditional methodologies for measuring MT growth dynamics have typically used hand tracking of either fluorescently-labeled tubulin or, more recently, fluorescently-labeled MT End Binding protein 1 or 3 (EB1 or EB3, respectively). The fluorescent tubulin approach has the advantage of labeling the entire MT array. However, because the majority of mitotic cell types maintain a radial array of MTs during interphase8,32,33, MTs near the centrosome are very dense, and as a result, tracking MT growth and disassembly with fluorescent tubulin is usually limited to the peripheral regions of the cell. Because of these limitations, utilization of fluorescently-labeled EB proteins (EB1 or EB3) has been the more common approach to measuring MT dynamics. Because EBs only label the growing plus-ends of MTs, they appear as small (1-3 μm), brightly fluorescent comets, thus providing an easily discernable marker of MT polymerization with very limited background fluorescence34-37. While the fact that EBs label only a subset of the MT array represents a major strength of the EB approach, it also highlights one important limitation, that non-growing MTs are not labeled by the EB3 approach. Nevertheless, the ability to measure and track EB3 comets over time and to visualize MT growth within sub-cellular regions make this approach ideal for applications that require regional evaluation of MT organization.
Another critical limitation of traditional methodologies for measuring MT growth dynamics has been very large data sets and the time required for data analysis. This limitation stems from the need for hand tracking of hundreds of EB comets at high time resolution. Moreover, these measurements need to be done in a statistically significant number of cells and also performed under a variety of control and experimental conditions. Recently, these constraints have been overcome through computational analysis techniques specifically directed at tracking EB3 comets using time-lapse microscopy in living cells35. When used appropriately, this computational approach serves to both restrict the potential for human error associated with hand-tracking in addition to providing for a high-throughput method of measuring MT polymerization. Computational analysis of MT dynamics using the MatLab-based software package, plusTipTracker, allows for measurements of MT growth by tracking fluorescently-labeled EB3 comets5,31,35,38. Additionally, plusTipTracker software allows for calculations of MT catastrophe events (a.k.a. disassembly) based on the disappearance of EB3 comets within a growing MT track, and allows for sub-cellular (a.k.a. regional) analysis of MT growth dynamics within user-defined regions of interest. For our studies, MT growth dynamics were compared within branched regions versus non-branched regions, or within leading versus trailing cell edges. Data output from plusTipTracker software can also be used to generate color-coded track overlays for individual cells and sub-cellular regions.
Here we describe the methodology used to investigate the role of MCAK in modulating MT growth dynamics and the contribution of this MT depolymerase to the regulation of EC branching morphology and directional migration. Our approach used a primary human cell line, Human Umbilical Vein Endothelial Cells (HUVECs), which are easily cultured and highly amenable to electroporation-induced, transient transfection of plasmid cDNA. We then used high-resolution, time-lapse fluorescence microscopy of HUVECs transfected with MT End Binding protein 3 (EB3) and Mitotic Centromere Associated Kinesin (MCAK)-specific cDNA constructs to evaluate effects on MT dynamics transfected HUVECs. PlusTipTracker-based computational image analysis of EB3-labeled MT plus ends was to measure MT growth speeds and MT growth lifetimes in time-lapse images, to measure and compare effects of experimental manipulations on MT growth dynamics. This methodology allows for the study of MT dynamics and the identification of how localized regulation of MT dynamics within sub-cellular regions contributes to the angiogenic processes of EC branching and migration. These techniques are applicable to investigations of any MAP that can be fluorescently labeled and expressed in live cells.
Protocol
1. HUVEC Culture and Maintenance
- Prepare complete endothelial basal medium by adding the entire contents of the endothelial cell growth medium supplement package and 5% penicillin/streptomycin antibiotic mixture to phenol red free endothelial basal media (see Materials/Equipment list for details). Warm complete endothelial basal medium to 37 °C in a water bath.
- Remove one HUVEC cryovial from liquid nitrogen storage and thaw rapidly in a 37 °C water bath for 2 min. When ~ 80-90% thawed, add 500 μl of warmed complete endothelial basal medium to the cryovial, mix by pipetting, and dispense cells evenly into a 100 mm plastic tissue culture dish containing 15 ml of warmed complete endothelial basal medium.
- Place cells into a 37 °C incubator with 5% CO2. Allow cells to adhere overnight. Do not remove or change the cell culture medium.
- When the 100 mm dish is > 90% confluent, transfer HUVECs into a 75 cm2 tissue culture treated flask and maintain at >60% confluence at all times (see step 1.5).
Note: When maintained at a density >60%, HUVEC doubling time is consistently 18-20 hr.- For situations where passaging the cells is not necessary (i.e. the density is <90%), aspirate the medium and replace with fresh complete endothelial basal medium every 48 hr.
- When HUVECs reach >90% confluence, rinse 2 times with 10 ml of 1x PBS, and then remove the cells from the tissue culture dish by treating them 1.5 ml of 0.25% trypsin for 1-2 min at 37 °C. Note: Viability of HUVECs is greatly reduced by extending the length of trypsin exposure.
- Rescue cells by adding 8.5 ml complete endothelial basal medium to the trypsin/HUVEC mixture at a ratio of 4:1 endothelial basal media complete: trypsin (minimal ratio), and collect in a 15 ml conical tube.
- Centrifuge at 1,400 x g for 4 min. Aspirate the supernatant.
- Resuspend the pellet in 2 ml of complete endothelial basal medium and add the cells to a new 225 cm2 tissue culture flask containing 25 ml of complete endothelial basal medium.
2. Coverslip Preparation Prior to HUVEC Culture
- Coating Coverslips with Fibronectin Substrates.
- Prepare squeaky-clean 22 mm No. 1.5 coverslips39 and store at room temperature (RT) in 100% ethanol.
- Use a Bunsen burner to flame a coverslip and place it into a 35 mm petri dish using tweezers.
- Prepare a fibronectin/PBS mixture by mixing 10 μl of fibronectin stock (1 mg/ml) with 990 μl of PBS.
- Pipette 250 μl of the fibronectin/PBS mixture onto the coverslip in the 35 mm dish. Spread evenly to cover the surface of the coverslip.
- Incubate the dish for a minimum of 3 hr at 37 °C and rinse 3 times with 2 ml of 1x PBS before use.
- Coating Coverslips with Polyacrylamide (PA) Substrates.
- Coverslip Activation
- Place a 22 mm No. 1.5 coverslip in 50% 3-aminopropyltrimethyloxysilane for 10 min on a stir plate. After incubation, wash 10 times for 2 min each in double-distilled H2O (ddH2O).
- Incubate the coverslip in 0.5% glutaraldehyde for 30 min on a stir plate. After incubation, wash 10 times for 2 min in ddH2O.
- Polyacrylamide Preparation
- Prepare a PA gel with a compliance of 0.7 kilopascal (kPa) by adding 3.75 ml of 40% acrylamide, 0.3 ml of 2% bis-acrylamide, and 0.95 ml of ddH2O.
- Prepare a 10% working solution of ammonium persulfate (APS) and store on ice until use.
- To prepare the PA solution for 1 coverslip (total volume = 166.7 μl), add 0.83 μl of ddH2O, 41.7 μl of the bis-acrylamide solution (made in step 2.2.2.1), 124 μl of 10% APS, and 0.25 μl of TEMED.
- Immediately pipet 15 μl of PA solution onto a hydrophobic glass slide (see Reagents List) and cover with an activated 22 mm No. 1.5 coverslip. Allow the PA to polymerize for 20 min at RT.
- Remove the coverslip and transfer to a 35 mm dish. Wash 3 times with 2 ml of 1x PBS. Store for < 2 weeks at 4 °C.
- To bind fibronectin to the polyacrylamide gel, expose the polyacrylamide-coated coverslips to 2 mM sulfo-SANPAH with 7,500 J of UV light (254nm). Rinse once with ddH2O.
- Prepare a fibronectin/PBS mixture by mixing 10 μl of fibronectin stock (1 mg/ml) with 990 μl of PBS.
- Pipette 250 μl of the fibronectin/PBS mixture onto the coverslip(s) in a 35 mm petri dish. Spread evenly to cover the surface of the coverslip.
- Incubate the dish for a minimum of 3 hr at 37 °C and rinse 3 times with 2 ml of 1x PBS before use.
- Coverslip Activation
3. cDNA Transfection and HUVEC Culture on Coverslip
- Complete steps 1.5-1.6.
- Using a hemocytometer, count the cell number and collect a volume containing 100,000 cells/coverslip (for non-migration experiments) or 500,000 cells/coverslip (for migration experiments). Pellet the cells via centrifugation at 1,400 x g for 4 min.
Note: Migration protocol is described in section 9, below. - Resuspend cells to be transfected with 100 μl electroporation buffer. Combine with 1 μg cDNA and place into electroporation cuvette. Electroporate cells: DNA mixture using the program designated for HUVEC electroporation (e.g. program #: A-034).
- Rescue transfected cells with 500 μl complete endothelial basal medium and plate cells onto a fibronectin-coated 22 mm No. 1.5 coverslip (see Protocol 2.1, above). Incubate at 37 °C for 3-4 hr to allow time for expression of cDNA constructs.
4. Preparation of Specimen for Imaging
- Cut double-sided tape into 2.5 cm x 0.65 cm strips.
- Obtain a clean glass slide. Take two strips of double-sided tape and secure them horizontally along the top and bottom edge of the slide. Note: It is important to allow a small amount of space between the tape and the slide edge for sealing the chamber as described in step 4.5).
- Mount coverslip (from Step 3.4; cell-side down) such that 2 edges of the coverslip rest on the tape. Use forceps to press down gently to secure the coverslip.
- Using a micropipette, add 40 μl of imaging medium (complete endothelial basal medium supplemented with 25 μM HEPES) between the coverslip and slide. Ensure that the space between the coverslip and slide is completely filled with imaging medium. Aspirate excess imaging medium (if necessary).
- Seal all edges with warm VALAP (1:1:1 petroleum jelly: lanolin: paraffin). Check for leakages by pushing gently on the glass coverslip.
- Place mounted and sealed coverslip onto pre-warmed (37 °C) microscope stage.
5. Microscopy
- Using a spinning disk confocal microscope equipped with a 60X, 1.40 Numerical Aperture (NA) Differential Interference Contrast (DIC) oil immersion objective lens, an automated focus monitoring system (PFS), electronic shutter for transmitted illumination, an X-and-Y motored stage, band pass filters housed in an electronic filter wheel, and a monolithic laser combiner module, focus the microscope at the interface of the cell/coverslip and use the microscope stage joystick to locate a single cell.
- Click on the "Camera Settings" panel in the image software program. Within the "exposure time" dropdown window of the Camera Settings panel, select 400 msec.
Note: Exposure times may vary and should be determined empirically. - Click on the "ND Acquisition" panel in the image software program. Within the lambda ( λ) tab of the ND Acquisition panel, click on the check boxes to select the 488 and 561 nm lasers. Within the "time" tab of the ND Acquisition panel, set the acquisition time to 2 sec and the total time to 2 min.
- Click the "Run Now" button within the ND Acquisition panel to acquire a 2 min time-lapse image sequence at 2 sec acquisition intervals using a 400 msec exposure time for both the 488- and 561 nm illumination.
- In the "Optical Config" panel of the image software program and using the camera settings described in step 5.2, click on the 405 nm laser button and then click "Acquire" to capture a single image of blue fluorescent protein (BFP)-labeled proteins.
Note: It is generally advisable to limit cell exposure (frequency of image capture and/or exposure time) to 405 nm laser illumination as this wavelength can damage the cells over time.- For MCAK-shRNA studies, acquire a single 405 nm image to verify the expression of the BFP-labeled shRNA.
- In the "Optical Config" panel of the image software program and using the camera settings described in step 5.2, click on the DIC button and then click "Acquire" to capture a single DIC image of the cell for branching morphology analysis (see section 10, below).
- Following image acquisition, use the "brightness/contrast" function in the image software program to digitally increase the image signal.
- Export the brightness and contrast-adjusted images. Click "File" then click "Save as," then click to select ".tif" for the image file type.
Note: This typically generates 61 .tif image files from a single 2 min time-lapse acquisition when captured as described in Step 5.4.
6. MT Dynamics Analysis
- For EB3 tracking, use plusTipTracker software35, an open-source, MatLab-based computational software package designed for automatic detection, tracking, analysis and visualization of fluorescently labeled EBs.
- plusTipGetTracks Graphical User Interface (GUI)
- To access the plusTipGetTracks GUI, type plusTipGetTracks.
- For EB3 comet detection, use the GUI to browse and select the parent directory that contains the .tif images.
- Within the tracking parameters panel of the plusTipGetTracks GUI, enter the following values: Search Range Radius = 5 - 10; Minimum Sub-Track Length = 3; Max Gap Length (Frames) = 12; Max Shrinkage Factor = 1.0; Maximum Angle Forward = 25; Maximum Angle Backward = 10; Fluctuation Radius (Pixels) = 2; Frame Rate (sec) = 2; Pixel Size (µm) = 110.
Note: First check for the optimal tracking parameters using the plusTipParamSweepGUI on a representative set of .tif image files. The value entered for pixel size is specific to the objective lens used to acquire the time-lapse images (see Steps 5.1-5.2, above). - For Region of Interest (ROI) selection, create binary masks of the whole cell by manually tracing the cell edge using the DIC image of the cell to form a polygon.
- For Sub-Region of Interest (sub-ROI) selection, create leading and trailing edge masks by manually drawing a 2 point thick line across the wound-edge cell of interest using a modified plusTipTracker sub-ROI selection tool.
Note: The line should be drawn parallel to the wound edge at the position where the cell of interest extends beyond neighboring wound-edge cells31.
- plusTipSeeTracks GUI
- Verify and visually inspect the track overlays of the .tif images stored in the "ROI" folder by clicking the "Plot Tracks" button in the Track Overlays column. Repeat for all track overlays.
- Perform data grouping by clicking the "Create Groups" button in the Optional Settings column. Select the experimental data that belong to the same dataset by clicking on the data files. Save the selection by giving the "group" a name that can be easily identified (for example, "control group").
- For Sub-ROI MT growth dynamics measurements, enter the minimum number of image frames that one EB3 comet track must be detected within the sub-ROI in order to qualify as a "track" and extract MT growth excursions within the leading and trailing edge ROI by clicking on "manual selection" within the "Sub-ROIs" panel of the GUI.
Note: For example, use 3 frames (6 sec) as the minimum detection length to be extracted as a "track." Extracted tracks from sub-ROIs are stored within a sub-project folder within the original ROI folder for each cell. - MT growth sub-track and subpopulation analysis
- Calculate the mean MT growth speed and mean MT growth lifetime for "group" datasets (see step 6.3.2), for each ROI (see step 6.2.4), and for each sub-ROI (see step 6.2.5).
- Within the Quadrant Scatter Plots column of the plusTipSeeTracks GUI, click on "Select Groups" and click on the groups (created in Step 6.3.2) to be compared.
- Select "Growth speed" for the x-axis option and type the mean value for growth speed (from Step 6.3.4) into the box. Select "Growth lifetime" for the y-axis option and type the mean value for growth lifetime (from Step 6.3.4) into the box.
- Check the box next to "Remove tracks at start/end" and next to "Batch process on Groups." Click on "Make Plots" to generate a distribution profile for MT growth tracks for the whole cell, leading edges, and trailing edges sub-ROIs.
Note: For studies described here, the MT growth tracks are distributed into four subpopulations: slow and short-lived, slow and long-lived, fast and short-lived, and fast and long-lived. - Copy the mean values from step 6.3.4 into a spreadsheet software program and save the file with extension .xls.
- Click on "Data," then click on "Data Analysis" from the Data dropdown window. Select "t-test: two sample assuming equal variances" to compare mean MT growth speeds and MT growth lifetimes. Highlight data cells within the spreadsheet where t-test comparisons result in a p-value < 0.001.
7. Colocalization Analysis
- For background subtraction, using the “line tool” of the imaging software program, draw a Region of Interest (ROI) by clicking on the screen to create a square shape that is 10 µm2 on the green channel image (488 nm excitation) at a position where there is no cell fluorescence (the background). Repeat this for the red channel image (561 nm excitation) using the images from Step 5.8.
- Using the imaging software program, right click on the ROI from step 7.1 and select “set as background ROI.” Click on “Image,” then click on “subtract background” from the dropdown window to subtract the average background values (from Step 7.1) from the entire image. Perform background subtraction for cells co-expressing Em-Aurora A and either mCherry-MCAK, mApple-EB3, or mApple-tubulin.
- In the background subtracted red channel image only, click on the Binary ribbon and select "Define Threshold" and then select the "per channel" function to exclude the designated fluorescent marker.
Note: The thresholding step was performed on either the mCherry-MCAK, mApple-EB3, or mApple-tubulin image to allow drawing lines that specifically marked the thresholded object for co-localization analysis (see step 7.4). - Using the line tool in the imaging software, draw a 2-point thick line over the excluded objects within the thresholded image.
- Select the line objects drawn in step 7.4. Copy and paste the line objects from the red channel onto its corresponding green channel (Em-Aurora A) image.
- Measure the grayscale pixel intensity values under the line objects to calculate the co-localization and total Em-Aurora-A values for each individual cell by selecting “Measure,” then “Intensity Profile” from the dropdown window within the imaging software program.
- Organize the data by experimental treatment and sub-cellular region by exporting to a spreadsheet software program and saving the file as type .xls (for example, “control_grayscale_intensity.xls”).
Note: Data presented in this way represents the co-localization fraction of total measured Em-Aurora-A per sub-cellular region.
- Organize the data by experimental treatment and sub-cellular region by exporting to a spreadsheet software program and saving the file as type .xls (for example, “control_grayscale_intensity.xls”).
- Using the measured values from step 7.6, repeat step 6.3.7 to calculate the statistical significance using p <0.05 as the cutoff for significance.
8. Immunofluorescence
- Fix cells (see step 3.4) with paraformaldehyde/glutaraldehyde co-extraction/fixation buffer (PGF-PHEM; 4% paraformaldehyde, 0.15% glutaraldehyde, 0.2% detergent in 60 mM PIPES, 27.3 mM HEPES, 10 mM EGTA, and 8.2 mM MgSO4, pH 7.0) for 10 min at RT.
- Rinse 3 times for 5 min with 2 ml of 1x PBS.
- Treat 2 times for 15 min with 0.01 g/ml NaBH4 in 1x PBS to quench reactive aldehydes.
Note: NaBH4 forms bubbles that can cause the coverslips to float. It is critical that the coverslips remain submerged during these incubations. If the coverslips begin to float, use forceps to elevate one edge of the coverslip in order to allow the bubbles to escape. - Rinse 2 times with 1x PBS before blocking with 5% fat-free milk in 1x PBS on a rocking platform for 1 hr at RT.
- Incubate cells in primary antibodies (rabbit anti-pT288-Aurora A (1:1,000)) and rat anti-α-tubulin (1:500) prepared in 1x PBS solution. Incubate at 4 °C on a rocking platform overnight.
- Remove primary antibodies and rinse 3 times for 5 min in 1x PBS before incubating with secondary antibodies (donkey anti-rabbit (1:1,000) and goat anti-rat (1:1,000)) prepared in 5% fat-free milk for 2 hr at 37 °C.
- Mount coverslip on a glass slide in fluorescence-optimized mounting medium. Seal the coverslip edges to the slide using clear nail polish, and store in the dark at 4 °C.
9. Cell Migration Assay
- Perform HUVEC culture for migration assays as described above (Protocol 3: cDNA Transfection and HUVEC Culture on Coverslip, Steps 3.2-3.3).
- Following cell transfection, rescue transfected cells with 80 μl complete endothelial basal medium. Pipette the 80 μl sample as a single drop onto the center of a dried fibronectin-coated 22-mm round No. 1.5 coverslip. Do not spread the 80 μl drop. Incubate at 37 °C for 3-4 hr to allow time for cell adherence into a confluent monolayer.
Note: Drying the coverslip is necessary to prevent the 80 μl drop from spreading upon contact of the coverslip. This approach allows for HUVECs to be concentrated within a small area of the coverslip and thereby promotes the rapid formation of a confluent monolayer of cells. - Rinse 2 times in complete endothelial basal medium to remove un-attached cells.
- To create a "wound edge," scrape the coverslip with a sterilized razor blade by gently dragging a clean razor blade across the HUVEC monolayer (from Step 9.2). Hold the coverslip gently in place with forceps to prevent slippage of the razor blade during scraping.
- Allow the cells to recover in a 37 °C incubator for 3 hr. Assemble and mount coverslip(s) for imaging (see Protocol 4).
- Acquire phase-contrast and 488 nm images at 10 min intervals for 12 hr on the microscope using a 10x 0.45 NA phase objective and a 0.52 NA LWD condenser using the "ND Acquisition" panel of the image acquisition software program (described in Step 5.3).
10. Quantification of HUVEC Branching and Migration
- For branching analysis and to allow for even cell illumination and unbiased quantification, acquire a microscopic image of a HUVEC transfected (see Step 3.3) and expressing the mApple-C1 cDNA construct, a cell volume marker.
- Using the imaging software program, open the mApple-C1 image and label the position on both sides of the branch where the membrane displays the greatest angle of curvature. Connect those two points with a straight line. This line is the "branch origin."
- Using the line tool of the imaging software, visually identify branches that measure >10 μm in length from the "branch origin" determined in Step 10.2. Calculate the branch length by measuring the distance from the "branch origin" to the most distal point of the branch tip (refer to Figure 4).
- Count the number of branched protrusions that measure >10 μm in length to obtain "cell branch number."
- For migration analysis, using the images collected in Step 9.6, visually identify a wound-edge HUVEC based on physical location (i.e. cells physically at the edge of the wound) and expression profile (i.e., select a green wound edge cell if the experiment involves expression of GFP).
- Using the phase-contrast images of the cell, visually identify the nucleus (the semi-transparent spherical structure near the center of the cell), and then visually identify the nucleolus (a small, dark-colored, spherical structure within the nucleus) at the first time point of the time-lapse images. Click on the position of the nucleolus to label the x and y coordinates of the nucleolus.
- Using imaging software, hand-track the nucleolus in successive time-lapse images by clicking on the position of the nucleolus to label the x and y coordinates of the nucleolus in each frame of the time-lapse movie (Figure 4).
- Using the imaging software, click on “File” then click on Save As” then click to select the image file type “.xls.”
- Open the hand-tracking data file (from step 10.8) using a spreadsheet software program. Calculate the mean migration velocity and mean migration distance to origin.
- Using a spreadsheet software program, click on "Data," then click on "Data Analysis" from the Data dropdown window. Select Bonferroni-corrected, one-way analysis of variance test with >95% confidence as the cutoff for statistical significance.
Representative Results
HUVEC culture for live-cell microscopy
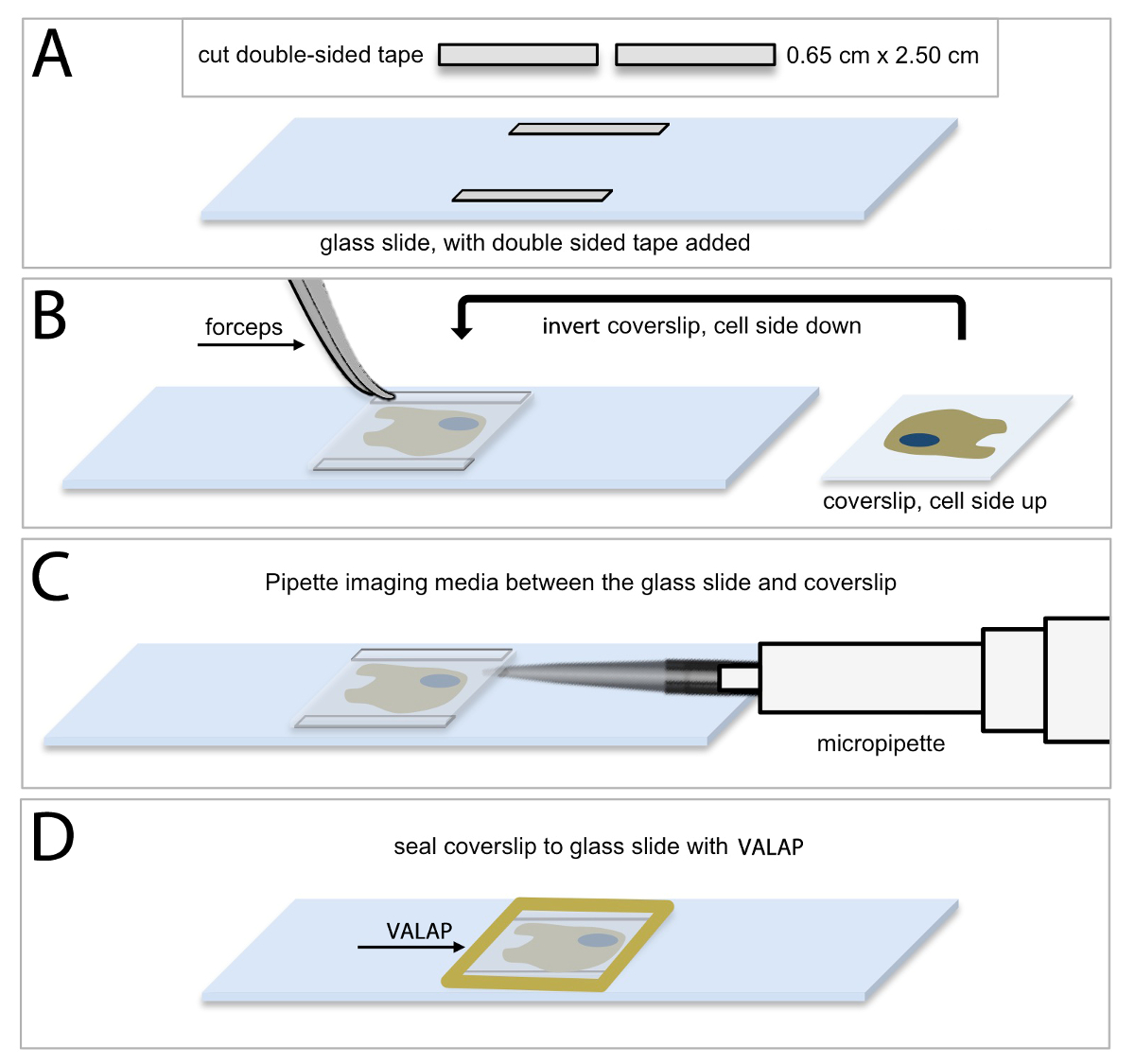
Live-cell imaging of HUVECs expressing fluorescent proteins requires that HUVECs are maintained in an environment that is suitable for normal cell growth and maintenance, as well as normal protein expression and function. Figure 1 shows a schematic depiction of the protocol used to prepare a buffered and closed system that maintains optimized optical characteristics for collecting time-lapse images of live cells. Thin strips of double-sided tape are placed onto a glass slide (Figure 1A) and then the coverslip containing the cultured HUVECs is inverted on top of the tape such that the cells are facing the slide (Figure 1B). In this way, the tape provides a small gap between the two glass surfaces, and creates a space in which the cells can be bathed in appropriate cell culture media (Figure 1C; see Protocol 4, above). In a final step, the glass coverslip is sealed to the glass slide using a petroleum jelly: lanolin: paraffin wax (1:1:1 mixture, VALAP; Figure 1D) to establish a closed system. This cell culture approach was used for both the shorter time sequence imaging of EB3 comets and for the longer time sequence branching and migration experiments. Additionally, the wax-like consistency of VALAP allows for easy removal from the glass slide and coverslip at the end of an imaging experiment such that supplemental approaches including fixation and immunolabelling (see Protocol 8), can be performed on the same coverslip and cell sample that was visualized using the live-cell imaging approach.

Figure 1: Methodology of HUVEC culture for live-cell microscopic imaging. (A) Cut 2 pieces of double-sided tape into 0.65 cm x 2.50 cm rectangular strips and secure one piece of double-sided tape horizontally along the top edge of the slide, and the other piece, along the bottom edge of the slide. It is important to allow a small amount of space between the tape and the slide edge for sealing the chamber as described in (D). (B) Using forceps remove the coverslip containing HUVECs and invert the coverslip (cell side down) on top of the pieces of tape. (C) Using a micropipette, add 40 μl of imaging media (complete endothelial basal medium supplemented with 25 μM HEPES) between the coverslip and the glass slide. Ensure that the space between the coverslip and slide is completely filled with imaging media and avoid bubbles. Use an aspirator along the edge of the coverslip to remove excess media, if necessary. (D) Use warm VALAP (1:1:1 vaseline : lanolin : paraffin) to seal around the edges of the coverslip. Check for imaging media leakages by pushing around gently on the coverslip. Use additional VALAP to remedy. Please click here to view a larger version of this figure.
{kind=link}
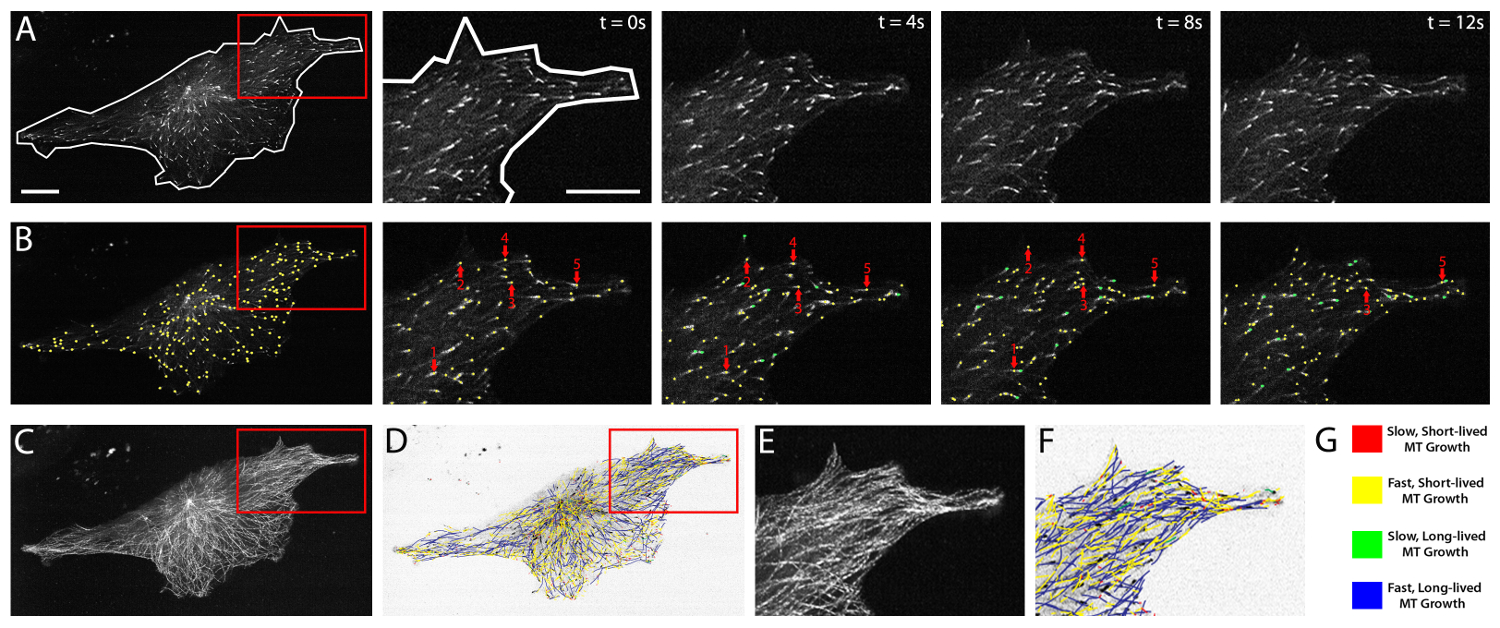
Automated tracking of fluorescently-labeled EB3 allows for comprehensive analysis of whole cell and regional MT growth dynamics. The collection of clear, distinct, and highly time and space-resolved microscopic images of the EB3 protein is essential to the analysis of EB3 movements within the cell. HUVEC expression of fluorescently-labeled EB3 varies widely from cell to cell and fluorescence intensity typically increases within the same cell over a period of hr. Thus, it is important to consider and monitor these changes in expression profile in order to identify potential effects of increased EB expression on MT growth dynamics. This is best accomplished by evaluating grey-scale fluorescence intensity profiles for each imaged cell, and then restricting data analysis to those cells that display a defined range of grey-scale expression profiles. Initial evaluation of expression profile data sets is critical, and should be compared with MT growth dynamics data that is obtained by plusTipTracker software for each cell. Expression profiles can be plotted against MT growth speed and MT growth lifetime for each cell within an experimental group, in order to determine the desired range of grey-scale expression and to identify potential outliers.
Figure 2 highlights a HUVEC cultured on a fibronectin substrate (see Protocol 2.1, above) and expressing near optimal levels of mApple-labeled EB3. Fluorescent EB3 comets are shown in a time-lapse series of images over a period of 12 sec (Figure 2A). Analysis of EB3 comets using plusTipTracker software involves an automated, frame-by-frame EB3 comet detection (green and yellow dots, Figure 2B). Comets detected within each frame are then linked, dependent upon their change in position from frame-to-frame, to generate EB3 tracks and a visual EB3 track overlay (Figure 2C-F). Track overlays are color-coded and color distinctions can be determined by parameters entered by the user. Commonly, these distinctions use the global mean of MT growth speed and MT growth lifetime5,31 and thereby split the data into four groups: slow and short-lived (red), slow and long-lived (yellow), fast and short-lived (green), and fast and long-lived MT growth (blue; Figure 2G). PlusTipTracker-mediated tracking of EB3-labeled MT growth dynamics also enables user-defined Regions of Interest (ROIs). PlusTipTracker extracts the MT growth track data from within the user-defined ROI (Figure 2A, B and F), thereby allowing for comparisons of MT growth dynamics within different regions of the same cell.

Figure 2: EB3 comet detection, tracking, and plusTipTracker data output. (A) time-lapse movie of a HUVEC expressing mApple EB3. Left panel shows a whole cell view and whole cell mask (white line) demarking the cell boundaries. Red box represents the zoomed region shown in the time-lapse images (right panels). Right panels show raw grey-scale time-lapse images of EB3 comets in successive imaging frames (t = 0 sec - t = 12 sec). (B) plusTipTracker detection of EB3 comets from the images shown in (A). Left panel shows a whole cell view of detected comets (yellow dots). Red box represents the zoomed region shown in the time-lapse images (right panels). Right panels highlight five detected EB3 comets (red arrows) and trace those five comets over the 12 sec imaging period. Green dots represent detected EB3 comets that did not exist (or were not detected) in the previous time interval. (C) Maximum intensity projection of 61 frames (2 min movie) of the EB3 comets shown in (A). (D) plusTipTracker MT track overlay of the same 61 frames (2 min movie) shown in (C). (E) Zoom of maximum intensity projection (red box in C). (F) Zoom of plusTipTracker MT track overlay of maximum intensity projection (red box in D). (G) Color-coded legend for track overlays shown in (D) and (F). Designation as slow, fast, short-lived or long-lived is based on the mean value for all growth tracks measured in a group of ten mApple-EB3 expressing HUVECs. For easier visualization, track overlays in (D) and (F) are generated using inverted pixel intensities of the cell image shown in (A). Scale bars = 20 μm. Please click here to view a larger version of this figure.
{kind=link}
HUVEC branching and migration patterns are sensitive to mechano-physical signals from the ECM.
HUVECs are well known to respond to various types of signaling cues and, in doing so, to undergo morphological alterations that dictate an appropriate cellular response to the particular signaling cue(s)36,40-42. HUVECs cultured on fibronectin coated glass substrates (Figure 3A), or on stiff 2-dimensional polyacrylamide ECMs (55 kPa) typically display a round or elongated morphology with few distinct branched protrusions (Figure 3B). However, when cultured on very soft 2-dimensional polyacrylamide ECMs (0.7 kPa), HUVECs display a highly branched morphology that is known to result from compliance-induced downregulation of myosin-II contractility4,5,31 (Figure 3C). Thus, HUVECs regulate their cellular morphology through ECM mechanosensing, and this is accomplished through localized modulation of MT dynamics and acto-myosin contractility.
Because HUVECs display a wide variety of morphological attributes, in order to measure HUVEC branching it is critical to first define what a branched protrusion is, and then to define how a branched protrusion will be measured objectively. One technique uses a four-step approach by which a cell boundary or "mask" is generated by hand tracing the edges of the cell using an image of a cell expressing a fluorescent volume marker to define the cell edges (Figure 3D). Following mask generation, branch origins are defined by locating greatest angle of curvature between the branched protrusion and the cell body. A line is drawn to demark the "branch origin" (purple angled lines; Figure 3E). Branch number is measured simply by counting the number of branches with a defined branch origin (Figure 3F). Branch length is measured as the distance from the branch origin to the distal branch tip (Figure 3G). To avoid inclusion of small lamellipodial projections from data analysis, additional criterion require that branches must be longer ("branch length") than they are wide (at the "branch origin"), and that branches must be at least ten microns in length5,31.

Figure 3: Analysis of HUVEC branching morphology. (A-C) DIC image examples of HUVECs cultured on fibronectin coated glass ECMs (A) stiff (55 kPa) polyacrylamide ECMs (B) and soft (0.7 kPa) polyacrylamide ECMs (C). Branching is dramatically increased in HUVECs cultured on 0.7 kPa polyacrylamide ECMs (C) as compared to stiffer ECMs (A, B). (D). DIC microscopic image (left image) of a transfected HUVEC expressing a mApple-C1 cDNA construct (a cell volume marker; right image). (E) Define the branch origin by locating the greatest angle of curvature (i.e. the position where the membrane displays the greatest curvature; purple-colored lines) on either side of the branch and then connect the angles with a straight line (blue lines). (F) Identify and selecting branched protrusions that extend >10 μm from the "branch origin" (defined in E). Count the number of branched protrusions to obtain the "branch number." (G) Measure the distance from the center of the "branch origin" to the most distal point of the branch using the line tool in the annotations and measurements application of NIS Elements Software to obtain the "branch length." Scale bars = 20 μm. Please click here to view a larger version of this figure.
{kind=link}
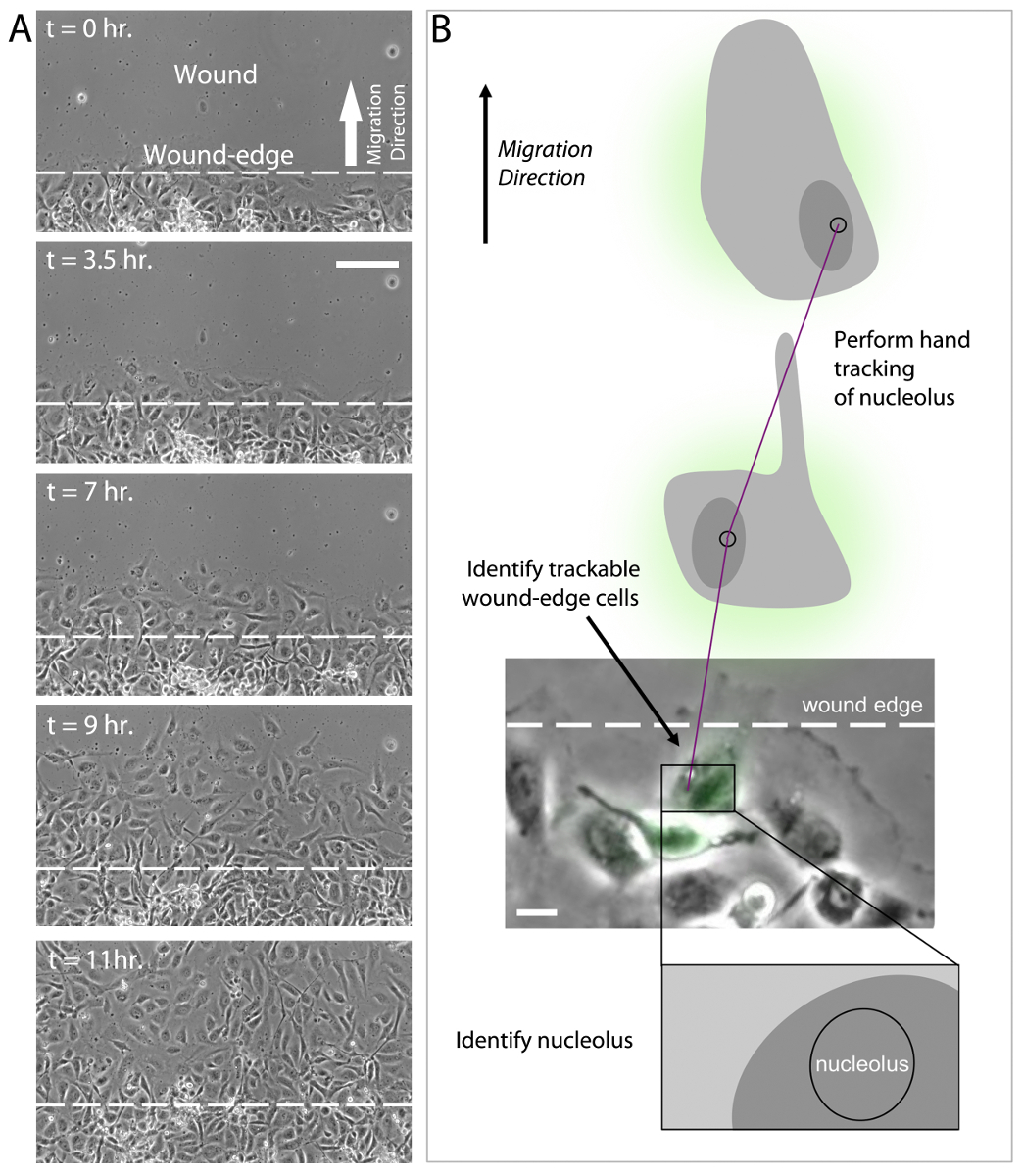
In addition to sensing ECM compliance, HUVECs also sense and respond to the elimination of adjacent cellular contacts as induced by scratch wounding. In the migration assay, HUVECs cultured in a monolayer are subjected to a scratch wound and migration characteristics are measured (see Protocol 9, above). After induction of the wound by scratching the monolayer, wound-edge HUVECs polarize by developing a protrusive leading edge that extends into the wound (Figure 4A, t = 0 hr). Over a time course of 10-12 hr, polarized wound-edge HUVECs migrate into the wound area and fill the wound, establishing a new confluent monolayer (Figure 4A, t = 11 hr). As with HUVEC branching, current evidence suggests that polarization and migration is critically dependent upon the coordinated re-organization of the MT and actin cytoskeletons43-49. This is achieved, in part, through localized inhibition of MCAK-induced MT disassembly at the leading edge, but not at the trailing edge of wound-edge HUVECs31. The sensitivity of MT dynamics to wound-induced polarization is initiated by the Rac1 GTPase, which targets numerous downstream regulator proteins, including Aurora-A kinase that phosphorylates and inhibits MCAK at the leading edge, and other kinases that are capable of inhibiting or activating MAPs to locally modulate MT growth and shrinkage18,31,36,48,50-53. Extensive experimental evidence supports the notion that coordination of leading edge protrusion and trailing edge contraction is governed through cycles of Rac1 activation to promote MT assembly at the leading edge and RhoA activation to acto-myosin contractility at the trailing edge, thereby driving directed motility9,41,51,52,54-58.
To date, there is not an established methodology for performing automated tracking of cell migration. This is a result of difficulty in computationally analyzing cell-ECM boundaries that are consistently changing as wound-edge cells polarize and migrate. While many software programs are capable of tracking fluorescently labeled cells59-64, the population of fluorescently labeled HUVECs in wound-edge migration experiments is approximately 30-40% of the total HUVEC population, and therefore the majority of the wound-edge cells must be visualized by phase-contrast or DIC imaging. Additionally, it is important when measuring wound-edge migration to evaluate fluorescent and non-fluorescent cells within the same wound in order to identify specific effects of expression constructs both within experimental study groups and between experimental study groups. Currently, the best approach to measure HUVEC migration is to use a hand tracking approach in which a wound-edge cell is first identified and then the nucleus and nucleolus are identified (Figure 4B). The nucleolus is used as the position determinant for cell tracking due to its high light scattering density and relatively small size. These two features of the nucleolus make it easier to identify and reduce measurement error by limiting the area in which the user can place individual track positions. HUVEC migration tracks are then generated by clicking on the nucleolus in successive time-lapse image frames. Finally, migration velocity and migration distance to origin are measured and analyzed for control and experimental cell groups (Figure 4B).

Figure 4: Analysis of wound-edge HUVECs and migration tracking. (A) 20X phase-contrast images of an 11 hr time-lapse imaging series of scratch wound-edge HUVECs (a.k.a. "Migration Assay"). The wound and direction of migration are indicated in panel t = 0 hr. HUVECs are shown to cross the wound-edge and advance into the wound until a monolayer of cells is formed (t = 3.5 hr - t = 11 hr). (B) Analysis of migration assay time lapse imaging (as shown in A) requires first the identification of a trackable wound-edge HUVEC based on cell location (i.e. cells physically at the edge of the wound) and expression profile (i.e., select a green wound edge cell if the experiment involves expression of GFP). Using the phase contrast image, identify the nucleus and the nucleolus. Hand track the nucleolus in successive time-lapse images using the annotations and measurements application in NIS Elements software to determine migration velocity and distance to origin. Scale bars = 100 μm (A), 20 μm (B). Please click here to view a larger version of this figure.
{kind=link}
Discussion
Using HUVECs in Morphology and Migration Experiments.
Live-cell imaging of EB3-labeled MT growth dynamics within HUVECs requires appropriate and reproducible cell culture conditions in order to measure and evaluate changes in MT growth and HUVEC morphology, such that they can then be assigned to an ECM signaling cue. HUVECs represent an excellent model for studying cell shape and motility. They are highly amenable to transfection with fluorescently-labeled cDNAs, they exhibit dramatic morphologies in response to both soluble and physical signaling cues, and they maintain the ability to organize themselves into vascular tubes when provided appropriate cell culture conditions65-70. Primary cell cultures, such as HUVECs, require careful handling and monitoring of cell passage number in order to ensure the cells are healthy and that they will behave normally during experimentation. For example, when conducting experiments investigating the cytoskeleton and/or cell morphology, HUVECs should not be used for experiments beyond the tenth cell culture passage, as HUVECs typically begin to display non-uniform morphologies around passage twelve to fifteen.
Cell density is also a critical regulator of HUVEC health and proliferation. HUVEC branching investigations (see Protocol 10.1-10.4, above) use relatively low cell density cultures in order to provide the cells physical space to interact with their ECM and to extend branched protrusions. In contrast, in wound-edge migration studies (see Protocol 10.5-10.8), HUVECs are culture in a monolayer prior to wounding. As such, wound-edge HUVECs display a relatively un-branched morphology, extend leading edge protrusions, and exhibit cell-cell interactions with their immediate neighbors as they migrate into the wound.
Caveats and Advantages of Tracking MT Dynamics in Living HUVECs.
Spinning disk microscopy is an excellent approach to test experimental hypotheses that require confocal image clarity with high time-resolution. With the appropriate camera and laser module, this microscopy approach is sensitive to low emission fluorescence and can aid in reduced photobleaching compared to other types of fluorescence microscopy. Importantly, this approach is also well suited for moderate to high time-resolution imaging as is required for generating comprehensive measurements of MT growth dynamics using the EB3 labeling approach, in a variety of different cell types.
While the EB3 methodology of labeling growing MT plus-ends has distinct advantages over other methods of fluorescently labeling MTs, it also has unique disadvantages. For example, there is a proportion of the MT array that is not actively growing at any given time, including both the population of disassembling MTs and the population of stabile, non-dynamic MTs71-74. EB3 would not label these two populations of MTs and therefore the contribution of these two populations would not be included in experimental data analysis. Alternative methodologies for evaluating the entire MT array have focused on expression of fluorescently-labeled tubulin, which incorporates into all populations of MTs and allows for identification of growing, shrinking, and stabile MTs. However, because of the relatively high density of the MT array near the center of the cell and adjacent to the microtubule organizing center (MTOC), labeling the entire MT array creates a disadvantage in performing image analysis of MT ends that are not near the cell periphery75-77. Experiments using two-color co-labeling of MTs with fluorescent tubulin and fluorescent EB3 in live cells have revealed, qualitatively, that the vast majority of tubulin-labeled MTs are also EB3-labeled78. Additionally, there has not been an efficient method of automated tracking of MTs labeled with fluorescent tubulin. This means that data collection and measurements of MT dynamics in cells expressing fluorescent tubulin require hand tracking of individual MTs, a process that is error prone and tedious. As a result, automated computational analysis of EB3 labeled MTs has become the preferred methodology for measuring MT dynamics as it can be applied to MT dynamics experiments across numerous cell types and within a variety of experimental regimes35,79.
Linking MT Growth Dynamics to HUVEC Morphology and Migration.
From the perspective of a cytoskeletal investigator, one extremely useful adaptation of the plusTipTracker automated tracking analysis is the ability to measure and evaluate both whole cell and regional changes in MT growth dynamics. The requirement for a cell to become polarized is an essential pre-requisite for directional cell migration. As such, polarized signaling cues have long been known as key driving factors for directional cell migration30,31,51,54,56,80,81. For example, soluble signaling molecules such as VEGF induce actin polymerization and MT growth toward the VEGF cue in endothelial cell types2,82-87. Additionally, in response to physical interactions with the ECM (for example, compliance and dimensionality mechanosensing), HUVECs modulate their MT dynamics by locally regulating MAPs to promote MT growth within elongating branched protrusions, and this serves to guide HUVEC migration in the direction of the cue5,31,88. Thus, polarization in response to extracellular signaling cues highlights the need for experimental evaluation of localized changes in cytoskeletal dynamics.
Simultaneous live-cell imaging of MT growth dynamics (using the EB3 approach) and HUVEC cell migration is extremely difficult to perform because MTs grow on a time scale of seconds, while changes in cell morphology and directional migration occur on a time scale of hours. Yet both processes are intimately linked. Moreover, continuous, rapid imaging of fluorescently-labeled EB3 on the second time scale leads to photobleaching of the EB3 signal. Attempts to overcome this issue by conducting short imaging series of EB3 (1-2 min acquisition series) every 30 minutes have been successful, but could only be accomplished in a small number of ECs that had optimal expression of EB3 and that did not display adverse effects to repeated laser illumination5.
An alternative approach that allows for interpretations of how MT growth dynamics contribute to changes in cell morphology and migration is the plusTipTracker Region of Interest (ROI) tool35 (see Protocol 6.6). This methodology allows for whole cell MT growth tracks to be sub-divided into user-defined regions, from which MT growth tracks can then be extracted and analyzed. For example, comparisons of "branched" versus "non-branched" regions of the cell have revealed that MTs grow more slowly and more persistently within branched regions as compared to the non-branched cell periphery5. In polarized wound-edge HUVECs, comparisons of the leading edge and trailing edge MT growth dynamics revealed that MCAK-induced MT disassembly is inhibited specifically within the leading edge, but not the trailing edge. Regional evaluation of leading and trailing edge MT growth dynamics in HUVECs expressing Rac1 and/or phosphorylation mutants of MCAK further revealed that Rac1-induced activation of Aurora-A kinase specifically and locally inhibits MCAK activity within the leading edge to drive HUVEC polarization and directional migration31. Thus, the combination of automated tracking of MT growth dynamics and regional evaluation of MT growth dynamics within the branched protrusions and/or the leading edge of wound-edge HUVECs has allowed us to link MT growth dynamics to HUVEC morphology and migration.
The protocols described are designed to provide a generally straightforward and highly repeatable methodology for HUVEC culture and experimental investigation. Nevertheless, many of the protocol details are complex and time consuming, which, in turn, provides opportunity for errors or difficulties in conducting the experimental methodology. While it was attempted to address potential problems throughout the protocol, here is provided additional, detailed troubleshooting of potential problems that are less common or otherwise abbreviated as notes within the methodology protocols.
Related to coating coverslips with polyacrylamide substrates (Protocol 2.2), a common problem that arises is the complex nature of activating glass coverslips, coupling polyacrylamide to the glass, activating polyacrylamide, and then coupling ECM to the polyacrylamide. One particularly difficult and potentially problematic step is culturing HUVECs onto the fibronectin/polyacrylamide gel following exposure of the polyacrylamide-coated coverslips to 2 mM sulfo-SANPAH (step 2.2.2.6, above). It is critical to the success of culturing HUVECs on these substrates that sulfo-SANPAH be completely removed from the experimental system following ECM conjugation. This can be best accomplished by washing with ddH2O and incubating the coverslips in ddH2O on a shaker overnight. If cells cultured on the polyacrylamide ECMs do not survive, or if viability is less than expected, increased and repeated washing of sulfo-SANPAH after conjugation to fibronectin (step 2.2.2.9) is recommended to potentially alleviate this problem.
Related to Protocol 3 (cDNA transfection and HUVEC culture on a coverslip), a major influence to the success of the electroporation procedure is the handling of the HUVECs both during trypsinization of the cells to remove them from the plastic flask, and following the conclusion of the electroporation (steps 3.3-3.4, above). It is critical to carefully monitor the HUVECs while in trypsin, and not to exceed the time needed for the cells to "round-up" on the surface of the flask (typically 1-2 min). Excessive time in trypsin is a major cause of HUVEC death, which is typically not realized until the cells are plated onto fibronectin-coated coverslips (step 3.4) and then fail to attach to the substrate.
Of similar importance, HUVECs (and most primary cell types) do not fare well when suspended for long times in the electroporation buffer. During larger-scale experiments, for example, where the user has five or more conditions that require electroporation, it is better to keep the cells pelleted in individual tubes (1 tube per transfection) and to mix them, one-at-a-time, in the electroporation buffer immediately before electroporation. This approach minimizes incubation time in electroporation buffer and maximized HUVEC survival. Additionally, immediate rescue in complete culture media is also critical following electroporation. Rescue delays of one minute or more following electroporation can result in near complete HUVEC death and thus should be avoided.
Related cell migration assay (Protocol 9), the technique of culturing HUVECs at extremely high density depends on a critical modification (described in step 9.2) of the traditional HUVEC cell culture methodology (described in Protocol 3) that requires drying of the fibronectin coated coverslip in order to enable HUVEC culture into a very small area of the coverslip. Often times, if the coverslip is not completely dried or if the prior coating of the coverslip with fibronectin (steps 2.1 or 2.2) was not efficient, the media-containing cells that is placed on the coverslip will lose its surface tension and expand to the edges of the coverslip, thereby reducing the density of cells on the coverslip. One method to troubleshoot this potential problem is to aspirate the media from the coverslip and then place the coverslip (still in its 35mm dish) into the back of the biosafety cabinet for 5-10 min. This adaptation can help to allow the flow of air within the cabinet to assist in drying the dish. If any visible liquid remains on the coverslip after air drying, aspirate the liquid spots and again allow to air dry for 5-10 additional min in the biosafety cabinet. It is important to note that there is no way to completely avoid this potential problem, but it does become less of an issue with repeated practice of the protocol. It is also a troubleshooting recommendation that when preparing coverslips for the cell migration assay (or any assay, in general), to prepare extra coverslips and have extra HUVECs ready-to-go in case of problems along the way.
With these troubleshooting considerations in mind, it is also important to highlight the utility of the protocols discussed and their implications in future cell biology research. The applicability to the polyacrylamide approach is wide-ranging, and includes not only two-dimensional studies of cell-ECM engagement (described above), but also three-dimensional investigations5. This application is critical to increasing our understanding of how HUVECs regulate cell shape and migration in physiological environments and should provide investigators a basic understanding of how morphological changes are coordinated with cytoskeletal changes. While the protocols described here are focused on measurements of MT growth dynamics, the majority of the protocols can and should be adapted to measure any number of other microscopically measureable aspects of cell-ECM interactions. With respect to MT dynamics, there are numerous MAPs, including at least 14 families of kinesins89, each with a potential role in contributing to HUVEC polarity and directed migration, and all of which can be investigated using the protocols presented.
Future investigations should also be targeted at cell-ECM engagement in normal versus diseased states. HUVEC protocols presented here are applicable to investigations of normal vascular homeostatic processes, as well as to disease states including heart disease, retinopathy, tumor growth and metastasis, to name a few. Conjugating visibly transparent ECMs and polyacrylamide substrates of amenable compliance with microscopic studies will allow for cell-ECM engagement studies such that endothelial cell vascular angiogenesis can be monitored with high spatial and temporal resolution. These types of experimental approaches have already begun to reveal important differences in endothelial cell shape, polarity, migration, and the effects of proteins that regulate these processes5,31,90. Future efforts should aim to identify how the combination of ECM compliance and dimensionality mechanosensing may serve to locally control proteins that target and regulate cytoskeletal dynamics.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
Special thanks to Dr. Clare M. Waterman for mentorship and discussions, Michelle Baird and Mike Davidson for providing many of the fluorescent cDNA constructs used in these studies, and Kathryn Applegate and Gaudenz Danuser for developing plusTipTracker MT analysis software. This work was supported, in part, by a grant to K.A.M from the National Heart Lung and Blood Institute (NHLBI) and the National Institutes of Health (NIH). (Grant: 4K22HL113069).
Materials
| Name | Company | Catalog Number | Comments |
| Aurora A Inhibitor I | Selleck Chemicals | S1451 | |
| 2% Bis solution | Bio-Rad | 1610142 | |
| 3-aminopropyltrimethyloxysilane | Sigma-Aldrich | A3648 | |
| 40% acrylamide | Bio-Rad | 1610140 | |
| 405, 488, 561, 640 nm high power MLC400 laser combiner module | Agilent Technologies | 99462 | |
| Ammonium persulfate | Sigma-Aldrich | A3678 | |
| anti–mouse IgG immunoprecipitation beads | TrueBlot Reagents; Rockland Inc. | 88-1688-31 | |
| anti–rabbit IgG immunoprecipitation beads | TrueBlot Reagents; Rockland Inc. | 88-7788-31 | |
| ET455/50m, DAPI-ET Emission Filter | Chroma Technology Corp. | ET455/50m | |
| ET525/36m Emission Filter | Chroma Technology Corp. | ET525/36m | |
| ET605/70m Emission Filter | Chroma Technology Corp. | ET605/70m | |
| ET700/75m Emission Filter | Chroma Technology Corp. | ET700/75m | |
| Bovine Serum Albumin Fraction V | RPI Products | A30075-100.0 | |
| Clara cooled charge-coupled device camera | Andor Technology | 77026010 | |
| Cy3 donkey anti-rabbit | Jackson ImmunoResearch | 711-165-152 | |
| DPBS | Thermo Fisher Scientific | 14190250 | |
| Dyelight 649 goat anti-rat | Jackson ImmunoResearch | 112-496-068 | |
| EGM-MV SingleQuot Supply & Growth Factors (contents shown below) | Lonza | CC-4143 | |
| Fetal bovine serum 25 mL | |||
| Bovine brain extract 2.0 mL | |||
| Gentamycin sulfate 0.5 mL | |||
| Epidermal growth factor 0.5 mL | |||
| Hydrocortisone 0.5 mL | |||
| Penicillin/Streptomycin (10,000 IU Pen/mL, 10,000 ug Strep/mL) 10.0 mL | |||
| EGTA | Sigma-Aldrich | E3884-100G | |
| Electronic shutter | Sutter instrument | 77016099 | |
| Fibronectin | EMD Millipore | FC010 | |
| Glutaraldehyde | Thermo Fisher Scientific | S80026 | |
| HALT Protease inhibitor | Thermo Fisher Scientific | PI-78425 | |
| HEPES | Sigma-Aldrich | H4034 | |
| HRP goat anti-mouse | Jackson ImmunoResearch | 115-036-062 | |
| HRP goat anti-rabbit | Jackson ImmunoResearch | 111-036-045 | |
| Immuno-Blot PVDF Membrane | Bio-Rad | 162-0174 | |
| Lanolin | Thermo Fisher Scientific | 8006-54-0 | |
| Magnesium sulfate | Sigma-Aldrich | M2643-500G | |
| Microscope | Nikon TiE | MEA53100 | |
| Motorized stage | ASI Technologies | MS-2000 | |
| Mounting medium | Dako | CS70330-2 | |
| Mouse anti-Aurora A | Abcam | ab130156 | |
| Mouse anti-GAPDH | Abcam | ab9484 | |
| Mouse anti-MCAK | Abcam | ab50778 | |
| Mouse anti-tubulin (DM1A) | Cell Signaling Technology | 3873S | |
| Nail Polish | Electron Microscopy Sciences | 72180 | |
| NIS Elements software | Nikon | MQS310000 | |
| Nucleofector Kit V | Lonza | VACA-1003 | Contains the electroporation buffer |
| Paraffin | Thermo Fisher Scientific | P31-500 | |
| Penicillin-Streptomycin (10,000IU Pen/mL,10,000ug Strep/mL) | MP Biomedicals | 91670249 | |
| Petroleum jelly | Dow Corning | 2021854-1113 | |
| Pipes | Sigma-Aldrich | P6757-100G | |
| plusTipTracker | |||
| Rabbit anti-GFP | Abcam | ab290 | |
| Rabbit anti–pT288–Aurora A | Abcam | ab18318 | |
| Rat anti–α-tubulin | AbD Serotec | MCA78G | |
| Sodium borohydride | Thermo Fisher Scientific | 678-10 | |
| Spinning disk | Andor Technology | Yokagawa CSU-X1 | |
| Sulfo-SANPAH | Thermo Fisher Scientific | PI-22589 | |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | 34080 | |
| Triton X-100 | Thermo Fisher Scientific | BP151-500 | |
| Trypsin | Corning | MT25053CI | |
| Tween-20 | Thermo Fisher Scientific | BP337 |
References
- Gerhardt, H., Betsholtz, C. How do endothelial cells orientate?. EXS. 94, 3-15 (2005).
- Gerhardt, H., et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 161, 1163-1177 (2003).
- Bayless, K. J., Johnson, G. A. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J Vasc Res. 48, 369-385 (2011).
- Fischer, R. S., Gardel, M., Ma, X., Adelstein, R. S., Waterman, C. M. Local cortical tension by myosin II guides 3D endothelial cell branching. Curr Biol. 19, 260-265 (2009).
- Myers, K. A., Applegate, K. T., Danuser, G., Fischer, R. S., Waterman, C. M. Distinct ECM mechanosensing pathways regulate microtubule dynamics to control endothelial cell branching morphogenesis. J Cell Biol. 192, 321-334 (2011).
- Rodriguez, O. C., et al. Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat Cell Biol. 5, 599-609 (2003).
- Waterman-Storer, C. M., Salmon, E. D. Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J Cell Biol. 139, 417-434 (1997).
- Waterman-Storer, C. M., Salmon, E. D. Microtubule dynamics: treadmilling comes around again. Curr Biol. 7, R369-R372 (1997).
- Wittmann, T., Bokoch, G. M., Waterman-Storer, C. M. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J Cell Biol. 161, 845-851 (2003).
- Ahmad, F. J., Pienkowski, T. P., Baas, P. W. Regional differences in microtubule dynamics in the axon. J Neurosci. 13, 856-866 (1993).
- Dent, E. W., Callaway, J. L., Szebenyi, G., Baas, P. W., Kalil, K. Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J Neurosci. 19, 8894-8908 (1999).
- Dent, E. W., Kalil, K. Axon branching requires interactions between dynamic microtubules and actin filaments. J Neurosci. 21, 9757-9769 (2001).
- Dehmelt, L., Nalbant, P., Steffen, W., Halpain, S. A microtubule-based, dynein-dependent force induces local cell protrusions: Implications for neurite initiation. Brain Cell Biol. 35, 39-56 (2006).
- van Veen, M. P., van Pelt, J. Dynamic mechanisms of neuronal outgrowth. Prog Brain Res. 102, 95-108 (1994).
- Myers, K. A., He, Y., Hasaka, T. P., Baas, P. W. Microtubule transport in the axon: Re-thinking a potential role for the actin cytoskeleton. Neuroscientist. 12, 107-118 (2006).
- Stehbens, S., Wittmann, T. Targeting and transport: how microtubules control focal adhesion dynamics. J Cell Biol. 198, 481-489 (2012).
- Stehbens, S. J., et al. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat Cell Biol. 16, 561-573 (2014).
- Wittmann, T., Waterman-Storer, C. M. Spatial regulation of CLASP affinity for microtubules by Rac1 and GSK3beta in migrating epithelial cells. J Cell Biol. 169, 929-939 (2005).
- Kumar, P., et al. GSK3beta phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J Cell Biol. 184, 895-908 (2009).
- Al-Bassam, J., Chang, F. Regulation of microtubule dynamics by TOG-domain proteins XMAP215/Dis1 and CLASP. Trends Cell Biol. 21, 604-614 (2011).
- Desai, A., Verma, S., Mitchison, T. J., Walczak, C. E. Kin I kinesins are microtubule-destabilizing enzymes. Cell. 96, 69-78 (1999).
- Hunter, A. W., et al. The kinesin-related protein MCAK is a microtubule depolymerase that forms an ATP-hydrolyzing complex at microtubule ends. Mol Cell. 11, 445-457 (2003).
- Lee, T., Langford, K. J., Askham, J. M., Bruning-Richardson, A., Morrison, E. E. MCAK associates with EB1. Oncogene. 27, 2494-2500 (2008).
- Walczak, C. E., Mitchison, T. J., Desai, A. XKCM1: a Xenopus kinesin-related protein that regulates microtubule dynamics during mitotic spindle assembly. Cell. 84, 37-47 (1996).
- Maney, T., Hunter, A. W., Wagenbach, M., Wordeman, L. Mitotic centromere-associated kinesin is important for anaphase chromosome segregation. J Cell Biol. 142, 787-801 (1998).
- Lan, W., et al. Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr Biol. 14, 273-286 (2004).
- Ganem, N. J., Upton, K., Compton, D. A. Efficient mitosis in human cells lacking poleward microtubule flux. Curr Biol. 15, 1827-1832 (2005).
- Wordeman, L., Wagenbach, M., von Dassow, G. MCAK facilitates chromosome movement by promoting kinetochore microtubule turnover. J Cell Biol. 179, 869-879 (2007).
- Kline-Smith, S. L., Walczak, C. E. The microtubule-destabilizing kinesin XKCM1 regulates microtubule dynamic instability in cells. Mol Biol Cell. 13, 2718-2731 (2002).
- Moore, A. T., et al. MCAK associates with the tips of polymerizing microtubules. J Cell Biol. 169, 391-397 (2005).
- Braun, A., et al. Rac1 and Aurora A regulate MCAK to polarize microtubule growth in migrating endothelial cells. J Cell Biol. 206, 97-112 (2014).
- Tanenbaum, M. E., Medema, R. H., Akhmanova, A. Regulation of localization and activity of the microtubule depolymerase MCAK. Bioarchitecture. 1, 80-87 (2011).
- Kushner, E. J., et al. Excess centrosomes disrupt endothelial cell migration via centrosome scattering. J Cell Biol. 206, 257-272 (2014).
- Siekmann, A. F., Affolter, M., Belting, H. G. The tip cell concept 10 years after: new players tune in for a common theme. Exp Cell Res. 319, 1255-1263 (2013).
- Applegate, K. T., et al. plusTipTracker: Quantitative image analysis software for the measurement of microtubule dynamics. J Struct Biol. 176, 168-184 (2011).
- Montenegro Gouveia, S., et al. In vitro reconstitution of the functional interplay between MCAK and EB3 at microtubule plus ends. Curr Biol. 20, 1717-1722 (2010).
- Honnappa, S., et al. An EB1-binding motif acts as a microtubule tip localization signal. Cell. 138, 366-376 (2009).
- Stout, A., D'Amico, S., Enzenbacher, T., Ebbert, P., Lowery, L. A. Using plusTipTracker software to measure microtubule dynamics in Xenopus laevis growth cones. J Vis Exp. , e52138 (2014).
- Waterman-Storer, C. M. Microtubule/organelle motility assays. Curr Protoc Cell Biol. Chapter 13, Unit 13 (2001).
- Kutys, M. L., Yamada, K. M. An extracellular-matrix-specific GEF-GAP interaction regulates Rho GTPase crosstalk for 3D collagen migration. Nat Cell Biol. 16, 909-917 (2014).
- Friedl, P., Wolf, K., Zegers, M. M. Rho-directed forces in collective migration. Nat Cell Biol. 16, 208-210 (2014).
- Hellstrom, M., et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 445, 776-780 (2007).
- Semina, E. V., et al. T-cadherin activates Rac1 and Cdc42 and changes endothelial permeability. Biochemistry (Mosc). 74, 362-370 (2009).
- Liang, Z. W., et al. Nestin-mediated cytoskeletal remodeling in endothelial cells: novel mechanistic insight into VEGF-induced cell migration in angiogenesis. Am J Physiol Cell Physiol. 308, C349-C358 (2015).
- Kamath, K., Smiyun, G., Wilson, L., Jordan, M. A. Mechanisms of inhibition of endothelial cell migration by taxanes. Cytoskeleton (Hoboken). 71, 46-60 (2014).
- Pasquier, E., et al. Antiangiogenic concentrations of paclitaxel induce an increase in microtubule dynamics in endothelial cells but not in cancer cells. Cancer Res. 65, 2433-2440 (2005).
- Sun, X., et al. Regulation of tumor angiogenesis by the microtubule-binding protein CLIP-170. Protein Cell. 4, 266-276 (2013).
- Lyle, K. S., Corleto, J. A., Wittmann, T. Microtubule dynamics regulation contributes to endothelial morphogenesis. Bioarchitecture. 2, 220-227 (2012).
- Ganguly, A., Yang, H., Zhang, H., Cabral, F., Patel, K. D. Microtubule dynamics control tail retraction in migrating vascular endothelial cells. Mol Cancer Ther. 12, 2837-2846 (2013).
- Wittmann, T., Bokoch, G. M., Waterman-Storer, C. M. Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J Biol Chem. 279, 6196-6203 (2004).
- Waterman-Storer, C. M., Worthylake, R. A., Liu, B. P., Burridge, K., Salmon, E. D. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol. 1, 45-50 (1999).
- Pasapera, A. M., et al. Rac1-dependent phosphorylation and focal adhesion recruitment of myosin IIA regulates migration and mechanosensing. Curr Biol. 25, 175-186 (2015).
- Cooper, J. R., Wagenbach, M., Asbury, C. L., Wordeman, L. Catalysis of the microtubule on-rate is the major parameter regulating the depolymerase activity of MCAK. Nat Struct Mol Biol. 17, 77-82 (2010).
- Hu, Y. L., et al. Roles of microtubule dynamics and small GTPase Rac in endothelial cell migration and lamellipodium formation under flow. J Vasc Res. 39, 465-476 (2002).
- Davis, G. E., Koh, W., Stratman, A. N. Mechanisms controlling human endothelial lumen formation and tube assembly in three-dimensional extracellular matrices. Birth Defects Res C Embryo Today. 81, 270-285 (2007).
- Spaargaren, M., Bos, J. L. Rab5 induces Rac-independent lamellipodia formation and cell migration. Mol Biol Cell. 10, 3239-3250 (1999).
- Petrie, R. J., Yamada, K. M. At the leading edge of three-dimensional cell migration. J Cell Sci. 125, 5917-5926 (2012).
- Westenskow, P. D., et al. Ras pathway inhibition prevents neovascularization by repressing endothelial cell sprouting. J Clin Invest. 123, 4900-4908 (2013).
- Chernomoretz, A., Bush, A., Yu, R., Gordon, A., Colman-Lerner, A. Using Cell-ID 1.4 with R for microscope-based cytometry. Curr Protoc Mol Biol. Chapter 14, (2008).
- Tarnawski, W., et al. A robust algorithm for segmenting and tracking clustered cells in time-lapse fluorescent microscopy. IEEE J Biomed Health Inform. 17, 862-869 (2013).
- Rambaldi, D., Pece, S., Di Fiore, P. P. flowFit: a Bioconductor package to estimate proliferation in cell-tracking dye studies. Bioinformatics. 30, 2060-2065 (2014).
- Cordelieres, F. P., et al. Automated cell tracking and analysis in phase-contrast videos (iTrack4U): development of Java software based on combined mean-shift processes. PLoS One. 8, e81266 (2013).
- Henry, K. M., et al. PhagoSight: an open-source MATLAB(R) package for the analysis of fluorescent neutrophil and macrophage migration in a zebrafish model. PLoS One. 8, e72636 (2013).
- Klein, J., et al. TLM-Tracker: software for cell segmentation, tracking and lineage analysis in time-lapse microscopy movies. Bioinformatics. 28, 2276-2277 (2012).
- Gerhardt, H., Betsholtz, C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 314, 15-23 (2003).
- Geudens, I., Gerhardt, H. Coordinating cell behaviour during blood vessel formation. Development. 138, 4569-4583 (2011).
- Davis, G. E., Stratman, A. N., Sacharidou, A., Koh, W. Molecular basis for endothelial lumen formation and tubulogenesis during vasculogenesis and angiogenic sprouting. Int Rev Cell Mol Biol. 288, 101-165 (2011).
- Tung, J. J., Tattersall, I. W., Kitajewski, J. Tips stalks, tubes: notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. Cold Spring Harb Perspect Med. 2, a006601 (2012).
- Davis, G. E., et al. Control of vascular tube morphogenesis and maturation in 3D extracellular matrices by endothelial cells and pericytes. Methods Mol Biol. 1066, 17-28 (2013).
- Kim, D. J., Martinez-Lemus, L. A., Davis, G. E. EB1, p150Glued, and Clasp1 control endothelial tubulogenesis through microtubule assembly, acetylation, and apical polarization. Blood. 121, 3521-3530 (2013).
- Desai, A., Mitchison, T. J. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 13, 83-117 (1997).
- Drechsel, D. N., Kirschner, M. W. The minimum GTP cap required to stabilize microtubules. Curr Biol. 4, 1053-1061 (1994).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104, 277-288 (1987).
- Kirschner, M., Mitchison, T. Beyond self-assembly: from microtubules to morphogenesis. Cell. 45, 329-342 (1986).
- Malikov, V., Cytrynbaum, E. N., Kashina, A., Mogilner, A., Rodionov, V. Centering of a radial microtubule array by translocation along microtubules spontaneously nucleated in the cytoplasm. Nat Cell Biol. 7, 1213-1218 (2005).
- Wieczorek, M., Bechstedt, S., Chaaban, S., Brouhard, G. J. Microtubule-associated proteins control the kinetics of microtubule nucleation. Nat Cell Biol. 17, 907-916 (2015).
- Wiese, C., Zheng, Y. Microtubule nucleation: gamma-tubulin and beyond. J Cell Sci. 119, 4143-4153 (2006).
- Shin, W. D., Fischer, R. S., Kanchawong, P., Kim, Y., Lim, J., Myers, K. A., Nishimura, Y., Plotnikov, S. V., Thievessen, I., Yarar, D., Sabass, B., Waterman, C. M. . Live cell imaging: A laboratory. , 119-138 (2010).
- Matov, A., et al. Analysis of microtubule dynamic instability using a plus-end growth marker. Nat Methods. 7, 761-768 (2010).
- Gundersen, G. G., Bulinski, J. C. Selective stabilization of microtubules oriented toward the direction of cell migration. Proc Natl Acad Sci U S A. 85, 5946-5950 (1988).
- Wittmann, T., Waterman-Storer, C. M. Cell motility: can Rho GTPases and microtubules point the way?. J Cell Sci. 114, 3795-3803 (2001).
- Jakobsson, L., Bentley, K., Gerhardt, H. VEGFRs and Notch: a dynamic collaboration in vascular patterning. Biochem Soc Trans. 37, 1233-1236 (2009).
- Blanco, R., Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb Perspect Med. 3, a006569 (2013).
- Gerhardt, H. VEGF and endothelial guidance in angiogenic sprouting. Organogenesis. 4, 241-246 (2008).
- Napione, L., et al. Unraveling the influence of endothelial cell density on VEGF-A signaling. Blood. 119, 5599-5607 (2012).
- Benedito, R., et al. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 484, 110-114 (2012).
- Czeisler, C., Mikawa, T. Microtubules coordinate VEGFR2 signaling and sorting. PLoS One. 8, e75833 (2013).
- Howes, S. C., Alushin, G. M., Shida, T., Nachury, M. V., Nogales, E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol Biol Cell. 25, 257-266 (2014).
- Hirokawa, N., Noda, Y., Tanaka, Y., Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 10, 682-696 (2009).
- Fischer, R. S., Myers, K. A., Gardel, M. L., Waterman, C. M. Stiffness-controlled three-dimensional extracellular matrices for high-resolution imaging of cell behavior. Nat Protoc. 7, 2056-2066 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved