Handling and Assessment of Human Primary Prostate Organoid Culture
In This Article
Summary
Here, we present a protocol to guide human primary prostate organoid handling then suggest endpoints to assess phenotype. Seeding, culture maintenance, recovery from matrix gel, morphologic quantification, embedding and sectioning, FFPE sectioning, whole-mount staining, and application of commercial assays are described.
Abstract
This paper describes a detailed protocol for three-dimensional (3D) culturing, handling, and evaluation of human primary prostate organoids. The process involves seeding of epithelial cells sparsely in a 3D matrix gel on a 96-well microplate with media changes to cultivate expansion into organoids. Morphology is then assessed by whole-well capturing of z-stack images. Compression of z-stacks creates a single in-focus image from which organoids are measured to quantify a variety of outputs, including circularity, roundness, and area.DNA, RNA, and protein can be collected from organoids recovered from the matrix gel. Cell populations of interest can be assessed by organoid dissociation and flow cytometry. Formalin-fixation-paraffin-embedding (FFPE) followed by sectioning is used for the histological assessment and antibody staining. Whole-mount immunofluorescent staining preserves organoid morphology and facilitates observation of protein localization in organoids in situ. Commercial assays that are traditionally used for 2D monolayer cells can be modified for 3D organoids. Used together, the techniques in this protocol provide a robust toolbox to quantify prostate organoid growth, morphologic characteristics, and expression of differentiation markers.
Introduction
Organoids are a valuable tool to study organogenesis and disease. They provide a less expensive alternative to animal models, and patient-derived organoids are evolving as a strategy for personalized medicine1,2,3. This three-dimensional (3D) culture system involves seeding stem or progenitor cells (harvested from tissue or induced pluripotent stem cells) into a gel of extracellular matrix components (matrix gel)4. The cells proliferate and differentiate, resulting in organotypic structures that recapitulate the cellular hierarchy and morphology of the organ of interest's functional unit. Organoids have been grown using cells originating from a variety of organs, including the salivary gland, stomach, intestine, liver, prostate, lung, and brain4. Although there are numerous protocols describing steps to establish prostate organoids5,6, it is challenging to find sufficiently detailed methods on how to achieve quantitative endpoints from organoids. This paper summarizes methods developed for human primary prostate cells and details a series of suggested endpoints to assess the organoid phenotypes. These techniques were optimized for prostate organoids and may be applicable to other 3D cell cultures.
Prostate organoid cultures have recently emerged as a valuable in vitro model that lacks the limitations of monolayer cultures using established immortalized cell lines. Benign prostate epithelium and prostate cancer is challenging to model in vitro using immortalized cell lines. The number of benign cell lines is limited, and all have undergone transformation with oncogenes7. Primary prostate epithelial cells in monolayers do not differentiate into luminal cells and lack androgen receptors8. The majority of prostate cancer cell lines do not have functional androgen receptors, a significant mediator of early disease state, and lack key genetic changes that have been found in patient tumors9. Prostate organoid cultures can be grown easily from benign epithelium and are valuable in studying stem cell properties, progenitor cells, differentiation, and effects of experimental changes in the microenvironment4,5. Prostate cancer organoids can be grown as part of a precision medicine approach to modeling patient disease and response to therapies10.
This protocol has been compiled from existing protocols that use various cell types, but it has been optimized here for use in human primary prostate cells. It compliments protocols outlined by Sawyers, Clevers, and Shen5,6 that describe growth, passaging, bright-field imaging, cryopreservation, and RNA and DNA isolation of prostate mouse and human organoids. The whole-mount protocol is modified from Mahé et al.11, who used gastrointestinal epithelial organoids and describes live imaging and frozen and paraffin embedding. Borten et al.12 described the analysis of breast, colon, and colorectal cancer organoid morphology from bright-field imaging. Additionally, Richards et al.13 used a method for the morphologic assessment of prostate organoids. Finally, Hu et al.14 described a method of adhering prostate organoids to a chamber slide overnight prior to the immunofluorescent staining to observe single cells and dispersion of spheres for flow cytometry analysis.
The goal of this protocol is to demonstrate in sufficient detail these technically challenging methods as one protocol, including cell seeding; media and matrix gel maintenance; collection of cells for flow cytometry; RNA, DNA, and protein extraction; morphologic assessment from bright-field z-stack analysis; embedding, processing and sectioning for histological staining; and whole-mounting for immunofluorescence or fluorescent probe assays. The biological relevance and interpretation of these various endpoints will vary among experimental design and antibodies used for analysis. Through utilization of this protocol, users should feel prepared to set up an experiment with a toolkit of endpoints.
Human primary prostate epithelial (PrE) cells were established in the lab by the protocol described by Peehl15,16 and maintained to a limited number of passages (up to four) as previously described17, but they are also available from commercial vendors. Hepatocyte serum-free media-based6, KSFM-based13, and R-spondin 1-conditioned5 media are all published as having successfully produced organoids. KSFM-based requires the fewest number of additives, so it is described here.
Protocol
The following protocol was optimized using human primary prostate epithelial cells harvested from patient tissue at the University of Illinois at Chicago Hospital. All human tissues used for these experiments were acquired via an Institutional Review Board-approved protocol and/or exemption at the University of Illinois at Chicago. While the culture conditions will vary depending on the cell type of interest, the endpoints can be applied to organoids of other tissues.
1. Seeding of Epithelial Cells into Matrix Gel and Changing Media
- Collect necessary materials: human primary prostate epithelial cells (PrE), matrix gel on ice, 96-well microplate, keratinocyte serum-free media (KSFM) on ice, charcoal stripped fetal bovine serum (FBS), and dihydrotestosterone (DHT).
- Prepare KSFM-based organoid media by combining 47.5 mL of KSFM with 2.5 mL of charcoal stripped FBS and dihydrotestosterone (DHT) to a final concentration of 10 nM DHT, then reserve on ice.
- Plate cells into a 3D matrix in a cell culture hood using sterile technique.
- Gently mix cold KSFM-based organoid media with ice-cold matrix gel to obtain a 50% matrix gel mixture by slowly pipetting up and down, avoiding bubbles.
NOTE: Matrix gel should be thawed overnight at 4 °C and kept on ice whenever possible. It solidifies quickly at room temperature (RT). - Pre-wet a pipette tip in cold media and transfer 40–50 μL of 50% matrix gel onto the bottom of each well to be used on a 96-well plate. Coat the bottom of the well to form a base layer.
NOTE: Do not fully dispense to the second stop on pipette, as this can create bubbles. - Place the 96-well plate into 37 °C incubator for 30–45 min to solidify the base layer.
NOTE: Do not plate epithelial cells onto the base layer until it has solidified, or cells may fall through the matrix onto the bottom of the plate and grow as a monolayer. - Mix epithelial cells suspended in KSFM-based organoid media with matrix gel to achieve a final concentration of 100–1,000 cells/100 μL and 33% matrix gel. Plate epithelial cells on top of base layers using 100 μL of 33% matrix gel/cell mixture per well.
NOTE: Previous experiments have shown that seeding epithelial cells too densely in 3D will restrict the size of organoid growth, while seeding too sparsely can result in very low yield13. It is important to optimize seeding conditions for the cell type of interest, based on the patient-specific organoid formation efficiency.
- Gently mix cold KSFM-based organoid media with ice-cold matrix gel to obtain a 50% matrix gel mixture by slowly pipetting up and down, avoiding bubbles.
- Place the 96-well plate in a 37 °C incubator for 30–45 min to allow the matrix gel to solidify, then gently add 100 μL of KSFM-based organoid media on top of cells by pipetting slowly against the edge of the well.
- Change the media every 2–3 days. Pipetting against the side of the well with space between the media and matrix gel, carefully remove 50 μL of media and replace with 50 μL of fresh media.
NOTE: For a long-term culture, the matrix gel needs to be changed every 2–3 weeks. If the matrix gel culture appears unstable and displays translucent wrinkles under the microscope, then the matrix gel has broken down and needs to be replaced.
2. Collection of Organoids from Matrix Gel
NOTE: Collection of organoids is necessary for matrix gel changes, passaging or endpoints of RNA extraction, DNA extraction, protein extraction, and flow cytometry.
- Warm neutral protease and Hanks' balanced salt solution (HBSS) in a 37 °C water bath.
- Carefully remove media from wells without disrupting the matrix gel.
- Add ~200 μL of neutral protease to each well at a ~1:2 ratio of matrix gel:neutral protease and incubate for 20 min at 37 °C.
- Mechanically dissociate the matrix gel:neutral protease mixture by pipetting up and down.
- Incubate for 20 min at 37 °C.
- Transfer the neutral protease-matrix gel-cell mixture to a microcentrifuge tube or conical tube, depending on the volume. Centrifuge at 300 x g for 3–5 min to pellet cells.
NOTE: If no cell pellet appears, the matrix gel may not be completely dissociated and can be visualized as a translucent phase at the bottom of the tube distinct from the pink supernatant. Remove the supernatant and add more neutral protease at a 1:2 ratio to matrix the gel pellet, incubate for another 20–40 min at 37 °C, and repeat centrifugation at 300 x g. - When an organoid pellet is acquired, remove the supernatant. Proceed with passaging (see step 2.7.1), organoid lysis for RNA, DNA, or protein extraction (see step 2.7.2), processing single cells by flow cytometry, and single-cell sequencing (see step 2.7.3).
- For long-term culture, gently resuspend intact organoids in 33% fresh matrix gel and plate by following steps described in steps 1.1–1.5. To dissociate organoids and replate as single cells, resuspend the pellet in 1 mL of warm cell-dissociation enzymes and incubate at 37 °C for 10–15 min, wash once with 5 mL of HBSS, and re-plate in the fresh matrix gel following steps 1.1–1.5.
- Resuspend the organoid pellet in an appropriate lysis buffer for RNA extraction, DNA extraction, or protein extraction as listed in the Table of Materials and follow the manufacturer’s protocol.
- Resuspend organoids in 1 mL of warm cell-dissociation enzymes and incubate at 37 °C for 10–15 min to dissociate the organoids into single cells. Wash once with 5 mL of HBSS and proceed with the protocol for flow cytometry5 or single-cell sequencing18.
NOTE: To dissociate into single cells at low concentrations of matrix gel, the neutral protease may not be needed, and cell-dissociation enzymes can be applied directly to the well after step 2.2.
3. Whole-well Image Acquisition and Analysis of Organoid Morphology
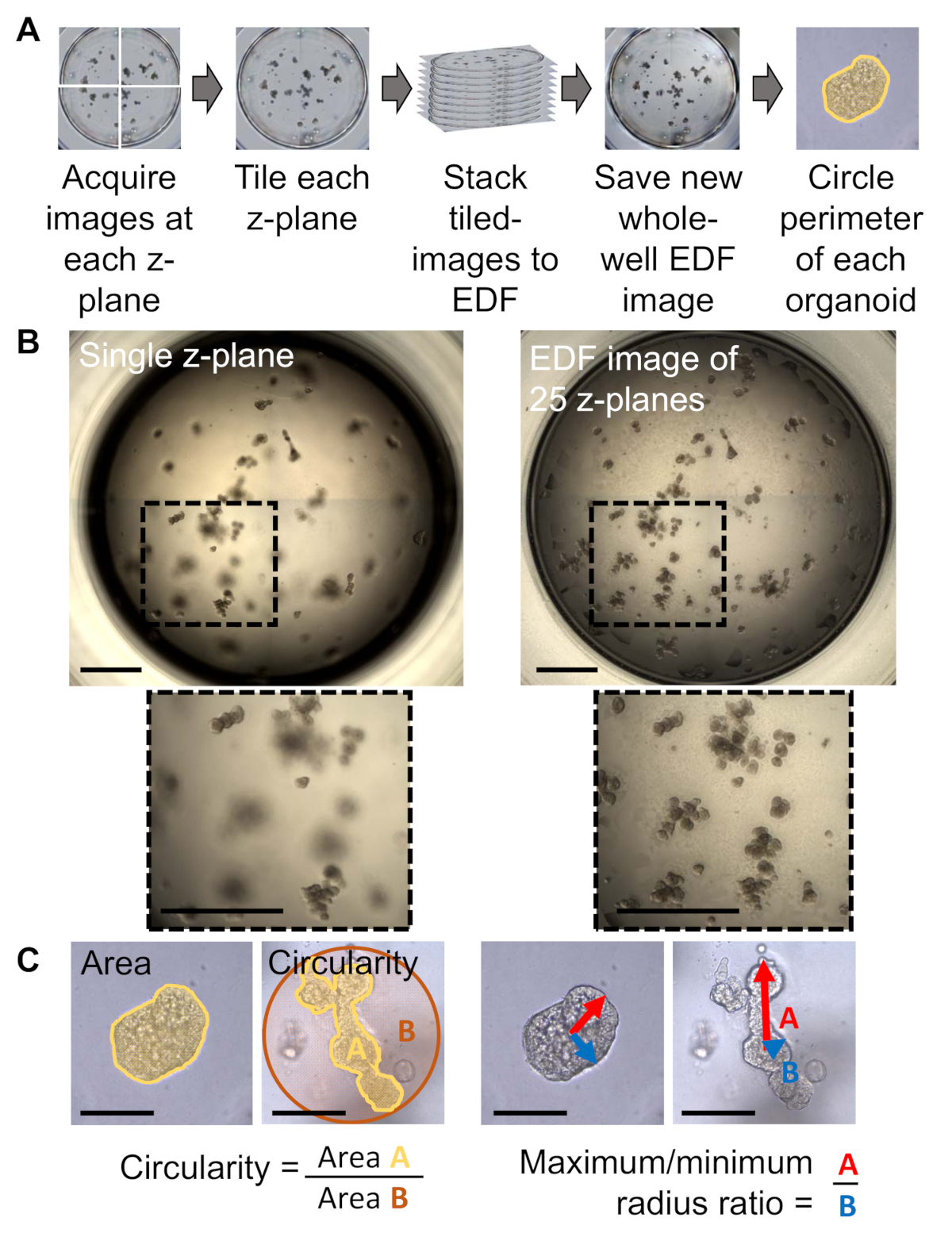
- Acquire whole-well z-stack images using a transmitted light inverted microscope with a motorized X/Y scanning stage (workflow illustrated in Figure 1A).
- With the focal position adjusted to the bottom of the well, begin focusing upward until the first organoid is encountered, which will be in the center of the well due to the meniscus from the base layer. Set the bottom z-plane at this position.
- Continue focusing upward until the last organoid is in focus, which will be in the periphery of the well. Set the top z-plane at this position.
NOTE: After setting the top and bottom of the z-stack, ensure that these positions capture the organoids within all remaining wells and adjust the top/bottom as needed. This allows for the z-range to be used for a single scan that includes every organoid within each well. - Capture 15–35 z-planes per well, using a larger number for greater resolution. Capture the entire well image, merge and collect quadrants or subsections if needed.
NOTE: It is recommended to take multiple z-planes as opposed to a single z-plane, as illustrated in Figure 1B in which organoids are out of focus when capturing only one z-plane. - Save z-stack images for downstream analysis.
- Analyze images using image software with freeform drawing capabilities.
- Open a single z-plane image set from step 3.1.4.
- If needed, tile the images taken at each individual z-plane into a single, whole-well image. Save the new image as a separate file. Repeat this process for each z-plane acquired.
- Using image software, open the whole-well image for every z-plane from a single well and create an extended depth of field (EDF) image from the stack of z-planes.
NOTE: This type of image will select the best focus region of each z-plane to create a single in-focus image of the well. - Set the pixel scale of the software to the scale bar of the image that is being measured.
- Using the freeform drawing tool of the image software, trace the perimeter of every organoid in the well.
- Obtain the measurement metrics for traced organoid perimeters from the image software.
NOTE: Recommended parameters are area, circularity, and maximum/minimum radius ratio. Representative results illustrating the application of these metrics are shown in Figure 1C. Area is calculated from the polygon defined by the organoid’s perimeter. Circularity is the ratio of the area of the organoid to the area of a circle with a diameter equal the organoid’s maximum ferret, where 0 is less circular and 1 is more circular. Maximum/minimum radius ratio is the ratio between the maximum radius and minimum radius of the traced organoid.
Circularity =
Radius ratio =
4. Formalin Fixation and Paraffin Embedding of Organoids for Histological Endpoints
- Prepare embedding supplies: HBSS, 2% agar solution, histological marking dye, histology gel, pipette tip mold, tape, plunger from a 1 cc insulin syringe, ice pack, cassettes, 10% neutral buffered formalin (NBF), pencil, and 75% histological grade ethanol (EtOH).
- Prepare two microcentrifuge tubes of 2% agar solution. Incubate in a water bath or on a heat block at 100–110 °C to maintain agar in liquid form. Add 1–2 drops of histologic marking dye to one of the 2% agar solutions, which will denote the top of the agar plug and assist in maintaining orientation during embedding. Mix well.
- Warm the histology gel in a water bath or heat block at 55–65 °C.
- Using a 1000 μL pipette tip, create molds by cutting out a small section of the pipette tip to make a cylinder that holds ~50 μL. Create a second, wider mold that fits around the first mold.
- Create a cold block by wrapping an ice pack with tape (adhesive side up) and adhering molds to the tape.
- Create a plunger by removing the plunger from a 1 cc insulin syringe.
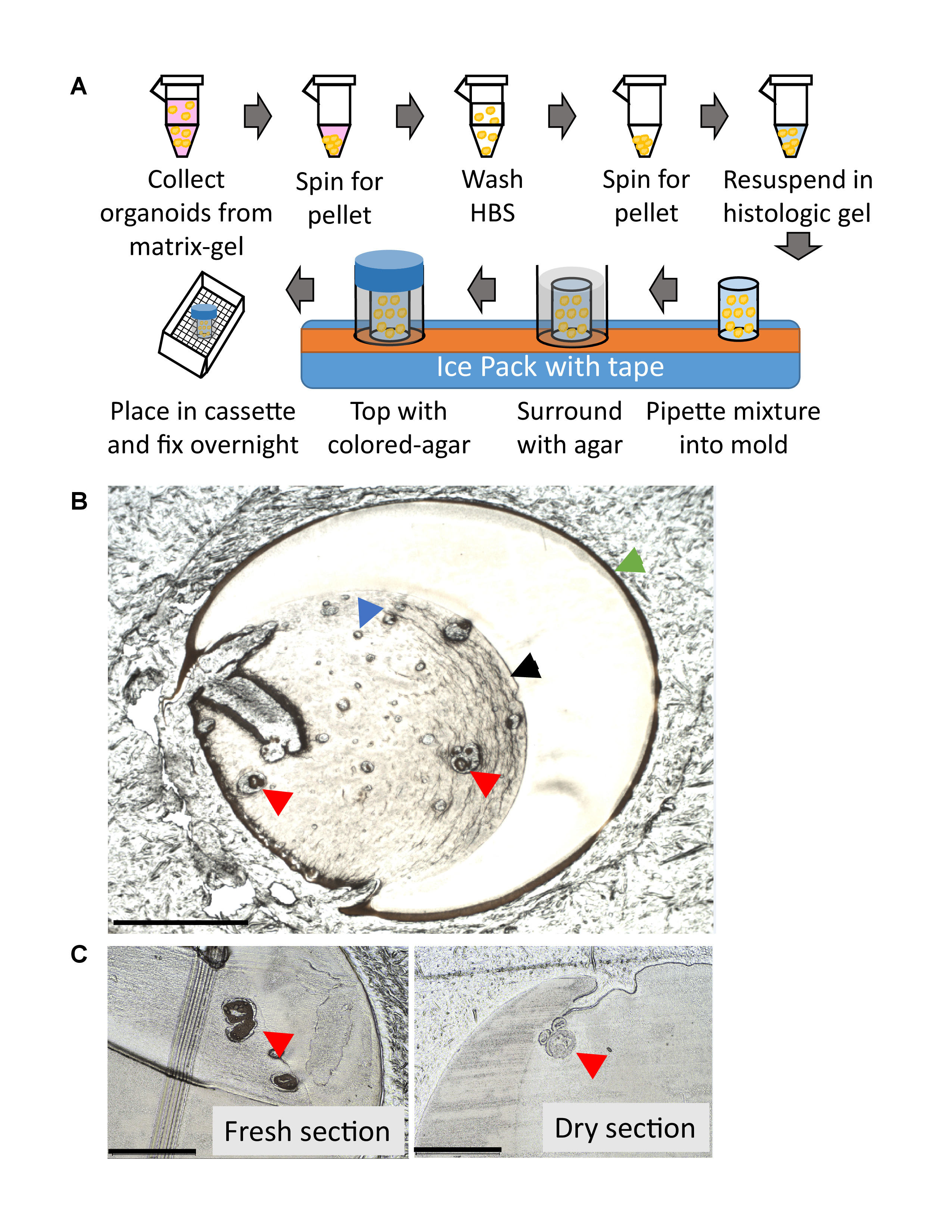
- Embed the organoids in a histology gel plug for processing, following the workflow depicted in Figure 2A.
- Dissociate the matrix gel and pellet organoids by following steps 2.1–2.7.
- Wash the organoid pellet once with 1 mL of HBSS and re-pellet by spinning for 3–5 min at 300 x g and removing the supernatant.
- Re-suspend the organoid pellet in 30–50 μL of liquid histology gel. Mix gently by pipetting up and down slowly. Transfer the histology gel/organoid mixture into a mold on the cold block and allow the specimen to cool and solidify into a plug.
- Use a plunger to push the plug out of the small mold and into the larger mold. Pipette clear 2% agar around the plug to fill the larger mold.
- Pipette histologic dye-tinted 2% agar on top of the plug to denote the top of the sample.
- Once cooled, use a plunger to transfer the plug into a histology cassette. Label the cassette using a pencil and place cassette into 10% NBF for fixation overnight.
- Transfer the cassette from 10% NBF to 70% histological grade EtOH.
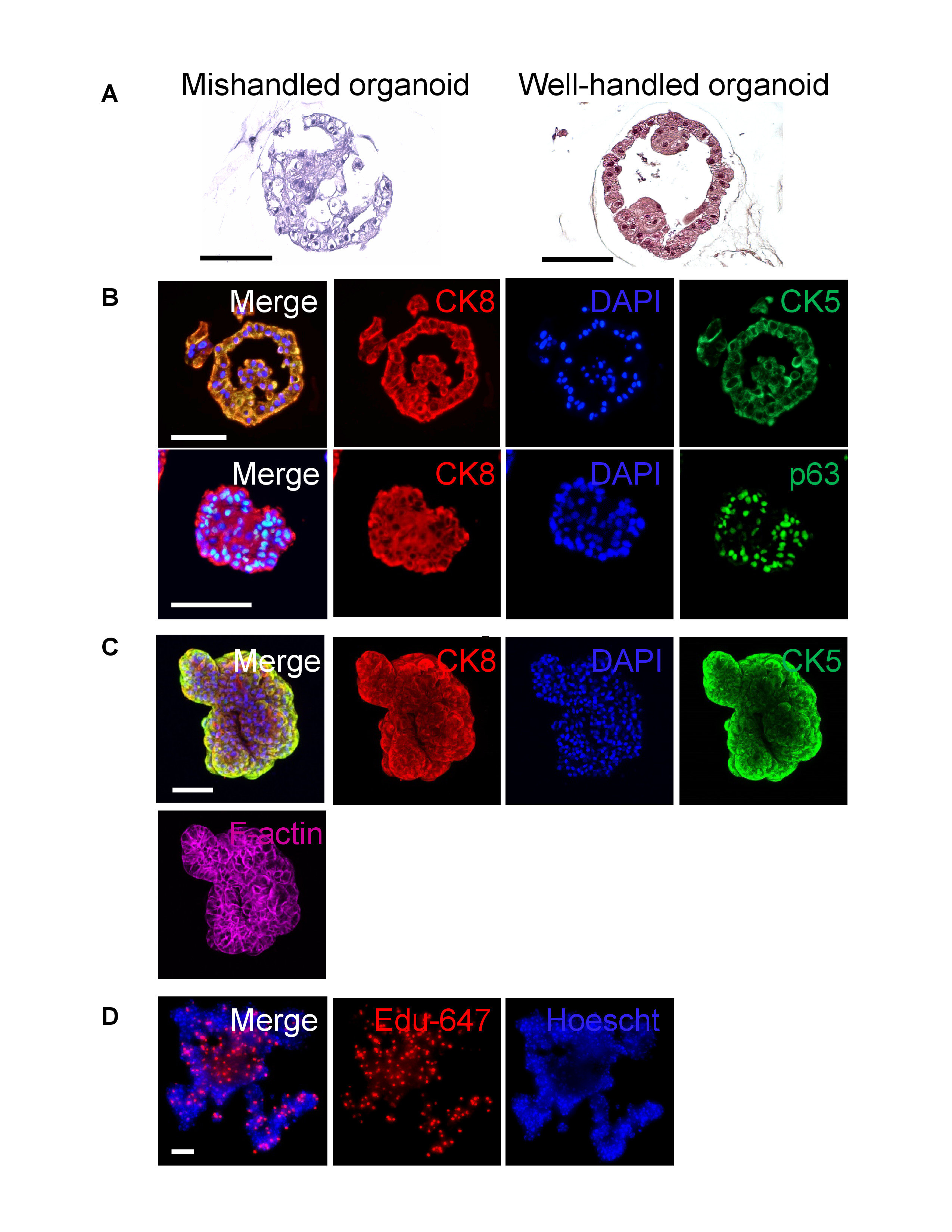
- Process the histology gel plug using a pre-set biopsy protocol on a processor, if available. If preset is not available, input the following sequence and lengths: 70% EtOH for 6 min, 70% EtOH for 10 min, 80% EtOH for 10 min, 95% EtOH 2 times for 10 min, 100% EtOH 3 times for 10 min, xylene 3 times for 15 min, and paraffin 3 times for 15 min. Deviating from the recommended processing sequence can result in ruptured organoids (Figure 3A).
- Embed the histology gel plug with the colored portion facing up, as this will allow the discolored portion containing the organoids to be sectioned into first.
- Section the FFPE histology gel blocks:
- Collect necessary supplies: FFPE histology gel blocks, histology pen, tissue float water bath, ice bucket with ice, microtome, microtome blades, embedding workstation, positively charged microscope slides, and laboratory oven.
- Fill water bath until very full and set to 40 °C, then pre-warm laboratory oven to 45–50 °C.
- Prepare an ice bucket, then fill with ice and add water to create an ice-water slurry. Chill the blocks on ice until they are very cold.
NOTE: Blocks can be set on ice for up to 1 day, but if left overnight, the blocks will become hydrated and will need to be reprocessed. - Insert a block into microtome cassette clamp, add the blade to the knife holder, and unlock any protective locking measures on the machine.
- Begin by trimming the face of the paraffin block (facing off) at a 10 µm thickness. Once a section emerges that contains the plug area, stop trimming, lock the microtome rotor, and transfer the block back onto the ice to chill. If several blocks are to be sectioned, face-off each block before moving to step 4.5.6.
- Move the blade to a new, unused portion and transfer the block back to the clamp. Unlock the machine and begin trimming at 5 μm per section.
NOTE: 5 μm is recommended for best results, but 3–10 μm is also acceptable. - Using tweezers, transfer the section to the 40 °C water bath and allow the section to spread out. Transfer the section onto a slide by dipping vertically into the water.
NOTE: Dipping at an angle can transfer bubbles from the bottom of the bath to the section and cause distortion of the sample. - Visualize the section under microscope (Figure 2B,C).
NOTE: If an organoid present, it will appear similar to the red arrow in Figure 2B. Bubbles (Figure 2B, blue arrow) can appear similar to organoids and are easier to discern on a fresh-slide as dry organoids appear translucent (Figure 2C). If no organoids are present, section further into the block.
- When slides containing organoids have been acquired, circle the area around the section on the back of slide with a histology pen and bake overnight at 45–50 °C.
- Collect slides and proceed to H&E (see step 4.9.1), immunohistochemical (see step 4.9.2), or immunofluorescent staining (see step 5).
NOTE: Representative results are shown in Figure 3B.- To perform H&E staining, obtain a kit from the materials list and follow the manufacturer’s specifications.
- To perform immunohistochemical staining, obtain a kit and primary antibody from the materials list and follow the manufacturer’s specifications.
5. Immunofluorescent Staining of FFPE and Sectioned Organoids
- Collect supplies: baked slide from step 4, xylene, 100% EtOH, 95% EtOH, 70% EtOH, deionized water (DI H2O), coplin jars, 1x antigen retrieval solution (sodium citrate buffer or EDTA-Tris buffer), phosphate-buffered saline (PBS), 5% normal horse serum (NHS) with 0.1% non-ionic detergent in PBS, 1% bovine serum albumin (BSA) with 0.3% non-ionic detergent in PBS, staining rack, staining dish, decloaking chamber, hydrophobic barrier pen, humidity chamber, 1° and 2° antibodies of interest, and counterstains of interest.
- Deparaffinize and rehydrate the slides by dipping into coplins filled with the following solutions: xylene (3 dips, 5 min each), 100% EtOH (2 dips, 3 min each), 95% EtOH (2 dips, 3 min each), 70% EtOH (2 dips, 3 min each), and DI H2O (2 dips, 3 min each).
- Fill the staining dish with antigen retrieval solution and pre-heat decloaking chamber to 100 °C.
- Perform antigen retrieval by immersing slides into the pre-heated staining dish and incubate for 5–10 min at 100°C. Once the cycle is complete, allow the decloaking chamber to sit for 20 min before opening the lid.
- Once the slide dish has cooled to RT, place the staining dish under running DI H2O to rinse the slides for 5 min.
- Remove the slides from the staining dish, draw a circle around the sample with a hydrophobic barrier pen, and lay the slide on the humidity chamber.
- Block any non-specific antibody staining by applying 5% NHS with 0.1% non-ionic detergent in PBS to the hydrophobic circle around the sample (about 30 μL is needed to cover the sample).
- Wash slides in coplins filled with PBS 3 times for 5 min each.
- Add the 1° antibodies (Table of Materials) diluted in 1% BSA with 0.3% non-ionic detergent in PBS to the hydrophobic circle around the sample (about 30 μL should cover the section), then incubate for 1 h at RT or overnight at 4 °C in humidity chamber.
- Wash the slides in coplins filled with PBS 3 times for 5 min each.
- Add the 2° antibody diluted in 1% BSA with 0.3% non-ionic detergent in PBS to the hydrophobic circle around the sample (about 30 μL will cover the area), then incubate for 1 h at RT in humidity chamber.
- Wash the slides in coplin filled with PBS 3 times for 5 min each.
- Add DAPI, diluted to the manufacturer’s specifications, in 1% BSA with 0.3% non-ionic detergent in PBS to the hydrophobic circle around the sample, then incubate for 2–5 min at RT in the dark (30 μL).
- Wash the slides in coplins filled with PBS 3 times for 5 min each.
- Mount the slides with antifade mounting media and coverslip, then visualize the results under a microscope.
6. Whole-mounting of Organoids for Immunofluorescent Staining
- Collect supplies: chamber slide or confocal dish, phosphate-buffered saline (PBS), fixative, 50 mM NH4Cl in PBS, 0.1% non-ionic detergent in PBS, 5% NHS with 0.1% non-ionic detergent in PBS, 1% BSA with 0.3% non-ionic detergent in PBS, 1° and 2° antibodies of interest, and counterstains of interest.
- Carefully remove as much media as possible without disrupting the organoid culture by pipetting against the side of well, leaving space between media and matrix gel.
- Using a trimmed, pre-wet with media or PBS, 1,000 μL pipette tip, draw up one drop (25–50 μL) of matrix gel that contains the organoid(s) of interest and dispense onto the middle of a chamber slide well or confocal dish.
NOTE: 33% matrix gel is not a solid. It is a gelatinous fluid that handles similar to a Jello that has not fully set. A trimmed pipette tip with a large opening will prevent organoids from breaking during transfers. - If organoids remain in the parent culture, replace media with warm KSFM-based media.
- With the naked eye or dissecting scope, aspirate the excess media and matrix gel from the chamber slide with a fine-tip pipette (10–200 μL).
NOTE: It is optional to pretreat the chamber slide with an adherence reagent. - Plate an equal volume of matrix gel into an empty well of the chamber slide as an optional control to observe autofluorescence of leftover matrix.
- Place the chamber slide in a 37 °C incubator for 30–45 min to promote the organoid to adhere to the glass and prevent loss of samples during wash steps.
- Pipetting slowly to prevent detachment from slide, wash organoids with 200 μL of 1x PBS for 5 min at RT while gently shaking.
- Remove the 1x PBS and fix with 200 μL of 4% paraformaldehyde for 30 min at RT while gently shaking. Ensure that the matrix gel appears clear after this step.
NOTE: Methanol or formalin fixation are also possible. Fixation method should be optimized for specific antibodies as certain fixation methods may crosslink the antigens of interest or quench fluorescent-labeling-proteins of interest. - Remove the fixative and wash twice with 200 μL of 1x PBS for 5 min while gently shaking.
NOTE: The length of washing steps should be optimized depending on organoid size. Five minutes is a sufficient starting point, but the washing step can be lengthened as needed. - Quench autofluorescence with 200 μL of 50 mM NH4Cl in 1x PBS for 30 min at RT while gently shaking.
NOTE: This step will quench autofluorescence from debris in the lumen of the organoid and from fluorescent protein expression (such as GFP) that may present from experimental design. It should be included if it is desired to use a fluorophore in the 488 channel but can be skipped if desired to preserve GFP signal. - Remove NH4Cl and wash twice with 200 μL of 1x PBS for 5 min while gently shaking.
- Permeabilize organoids in 200 μL of 0.1% non-ionic detergent-100 in 1x PBS for 30 min at RT while gently shaking.
- Remove the 0.1% non-ionic detergent-100 in 1x PBS and wash twice with 200 μL of 1x PBS for 5 min while gently shaking.
- Block with 5% NHS with 200 μL of 0.1% non-ionic detergent X-100 in PBS and incubate for 60 min at RT while gently shaking.
- Remove the blocking buffer and wash twice with 200 μL of 1x PBS for 5 min while gently shaking.
- Dilute the 1° antibody in 1% BSA with 0.3% Triton X-100 in 1x PBS and add to 200 μL of the chamber slide, then incubate for 1–3 days at 4 °C.
- Remove the 1° antibody and wash twice with 200 μL of 1x PBS for 5 min while gently shaking.
- Dilute the 2° antibody in 1% BSA with 0.3% Triton X-100 in 1x PBS and add to 200 μL of the chamber slide, then incubate for 1–3 days at 4 °C in the dark.
NOTE: The length of incubation times should be optimized for organoid size and antibody. Some will require more time to penetrate through the organoid. Antibody concentration will also need to be optimized. In general, 2x the concentration used for FFPE sections is appropriate for 1° antibodies and the same concentration for FFPE sections is appropriate for 2° antibodies. - Remove the 2° antibody and wash twice with 200 μL of 1x PBS for 5 min while gently shaking.
- Counterstain the actin with phalloidin and the nucleus with DAPI or Hoescht, diluted according to the manufacturer’s recommendations in 1% BSA with 0.3% Triton-X in 1x PBS. Add 200 μL to chamber slide, then incubate at RT for 40 min in the dark.
- Mount in 200 μL of fresh 1x PBS or mounting media and image immediately (fluorescence should be preserved for up to 5 days, if not longer). Representative results are shown in Figure 3C.
NOTE: Sodium azide can be added to samples to prevent contamination if confocal imaging is not readily available. Adding a clearing agent can improve imaging.
7. Whole-mounting of Organoids for Assays and Fluorescent Probes
NOTE: There are commercially available assays and fluorescent probes/dyes (examples are included on the materials list) amendable for use on whole-mounted organoids to observe a variety of useful endpoints19. The following protocol is for the fluorescently-labelled EdU proliferation kit, however any kit can be modified for use with a whole-mounted sample.
- Carefully remove 50 μL of media and replace with 50 μL of 20 μM 2x working EdU solution by pipetting against the side of well, leaving space between media and matrix gel.
- Incubate at 4 °C for 1–3 days (the desired length of time for EdU incorporation should be optimized for cell type of choice).
- Follow steps 6.2–6.8 to transfer the organoids of interest onto a chamber slide or confocal dish.
- Remove PBS and fix with 200 μL of 3.7% paraformaldehyde for 30 min at RT while gently shaking. Ensure that the matrix gel appear clear after this step.
- Remove the fixative and wash twice with 200 μL of 1x PBS for 5 min while gently shaking. Remove PBS and add 200 μL of 0.5% non-ionic detergent-100 in 1x PBS for 30 min at RT while gently shaking.
- Prepare a 1x fluorescent reaction cocktail according to the manufacturer’s specifications.
- Remove 0.5% non-ionic detergent-100 and wash twice with 200 μL of 3% BSA in 1x PBS for 5 min while gently shaking.
- Add reaction cocktail and incubate for 30 min at RT in the dark.
- Remove the fluorescent reaction cocktail and wash once with 200 μL of 3% BSA in 1x PBS for 5 min while gently shaking. Counterstain and mount by following steps 6.21–6.22 (Figure 3D).
Representative Results
Upon successful culture of human primary prostate organoids, morphology and differentiation can be assessed using bright-field image analysis and FFPE and whole-mount staining techniques.
The process of bright-field image capture is illustrated in Figure 1A. Organoids are grown in a 3D matrix and dispersed across different focal planes. To observe organoids in focus across many planes, it is suggested to use an EDF image (Figure 1B, right) instead of a single z-plane (Figure 1B, left). In the lab, organoids grown under different experimental conditions may result in changes in morphology. Using area, circularity (defined by how similar the organoid is to a circle) and max/min radius (measure of length) give users an indication of organoid size and shape and provide a quantifiable readout of morphology. To emphasize the usefulness of these measures, representative images of organoids with similar area but differing morphology are shown in Figure 1C, in which a long organoid will be less circular and have a larger maximum/minimum ratio than a spherical organoid.
Embedding organoids for histology is a useful endpoint to observe the interior of samples and ensure that necrosis is not present within the core of an organoid. A workflow for this process and representative results of the unstained, sectioned samples under a bright-field microscope are provided in Figure 2A,B,C. Figure 3A shows a mishandled organoid (left) compared to properly handed organoids in Figure 3A (right) and Figure 3B. To determine the differentiation-state of the sample, it is recommended to look at basal cell markers (cytokeratin 5 and p63)8 along with luminal cell markers (cytokeratin 8)8. If it is desired to stain organoids in situ, whole-mount staining is a convenient alternative, and these representative results are provided in Figure 3C,D.

Figure 1: Morphology assessment of organoids in 3D culture. (A) Image collection and compression workflow for human primary prostate organoids acquired by a transmitted light inverted microscope with a motorized X/Y scanning stage and companion software. (B) Representative images of a single z-stack (left) vs. an EDF image (right) of a whole-well sample of human primary prostate organoids, showing that more organoids are in focus when multiple z-stacks are combined into a single projected image (scale bar = 1000 µm). (C) Representative images for area, circularity, and max/min ratio of morphologically dissimilar human primary prostate organoids which have the same area (scale bar = 200 µm). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Organoid embedding workflow and representative sectioning results. (A) Human primary prostate organoid embedding workflow. (B) Representative image of an unstained, fresh slide containing human primary prostate organoids under a bright field microscope depicting agar (green arrow), histology gel (black arrow), bubble (blue arrow), organoids (red arrows) (scale bar = 1000 µm). (C) Unstained freshly cut slide (left) vs. unstained dry slide (right), organoids (red arrows) appear translucent when dry (scale bar = 500 µm) and are harder to discern under a bright-field microscope. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Histological staining on sectioned and whole-mounted organoids. (A) H&E staining of a broken human primary prostate organoid from mishandling (aggressive pipetting, wrong processing protocol, left) next to a well-handled organoid (right) (scale bar = 100 µm). (B) Human primary prostate organoid that has been formalin-fixed, paraffin-embedded, sectioned, and stained with basal cytokeratin 5 or luminal cytokeratin 8 (top) or basal p63 and luminal cytokeratin 8 (bottom) and imaged with confocal microscope (scale bar = 100 µm). (C) A whole-mounted human primary prostate organoid stained with basal cytokeratin 5 or luminal cytokeratin 8 and counterstained with phalloidin and DAPI, imaged with confocal microscope (scale bar = 100 µm). (D) A whole-mounted human primary prostate cell organoid stained with fluorescently labeled EdU and counter stained with Hoescht, imaged with confocal microscope (scale bar = 500 µm) Please click here to view a larger version of this figure.
{kind=link}
Discussion
Organotypic culture is an exciting new method for recapitulating tissue with the convenience of an in vitro environment. Currently, labs grow organoids from many types of tissues for various endpoints. The methods described in this paper summarize useful endpoints and highlight new techniques to fully characterize 3D primary prostate cell cultures.
There are a variety of media recipes for cultivating human primary prostate cell organoids5,6,13. While all achieve viable and comparable results, KSFM-based media13 uses minimal additives so it is described here. Additionally, papers have been published using multiple concentrations for matrix gel, from 10% to 75% to culture prostate organoids5,6,13. Because matrix gel is an expensive reagent, and 33% matrix gel has been shown to be sufficient to cultivate long-term (2-3 weeks), viable, unclumped organoids13, this is the recommended concentration. However, matrix gel can vary in protein concentration between lot numbers, so this should be taken into account when plating. It is also recommended to purchase matrix gel in bulk for use across multiple experiments to reduce inconsistency between lots. Other labs have published different formats for plating matrix gel, such as droplets instead of coating a 96-well plate well5. Both methods form viable organoids, but the format described here allows for cells to be plated more sparsely, which has been shown to promote expansion of cells into larger organoids13. However, plating density should be optimized based off patient-specific formation efficiency. Matrix gel is available in many formats including growth factor-reduced, phenol red-free, high concentration, etc. Growth factor-reduced is recommended for defined culture conditions, and phenol red-free is recommended when working with cells that express GFP or in experiments that involve z-stack image capture. It is critical that any matrix gel plating steps be performed with ice-cold reagents, as matrix gel solidifies quickly at room temperature.
Organoids grown under different experimental treatments may result in changes to shape, so bright-field imaging is widely used to study observed morphological phenotypes. Nevertheless, recording and quantifying area or shape is a challenge for two reasons: 1) selection bias during image capture and 2) applying a two-dimensional parameter such as area to a three-dimensional sample. One strategy of image capture is to record a random field and measure a predetermined number of organoids in that field, but this can create bias during selection, and all organoids in the selected field may not be in focus. Whole-well imaging optimized for the 96-well microplate format eliminates this sampling bias by collecting the entire organoid population of interest. However, depending on the microscope objectives on hand, multiple fields may need to be collected and tiled in order to obtain a whole-well image. Some labs have described measuring area from a single z-plane12, but to capture all organoids in focus, it is recommended to collect and stack multiple planes into a single EDF-image13. In some cases, a meniscus in the matrix gel may cause vignetting in the image, and background correction methods may need to be applied using image analysis software. Exact volume is ideally the most appropriate measurement for organoid size; however, this is difficult to precisely obtain, even with multiple z-stack images. Other useful morphological dimensions, such as circularity and maximum/minimum ratio, can easily be calculated from all the organoids in a whole-well EDF image. Together, these methods overcome sampling bias challenges and enable measurement of 2D parameters from 3D objects in a 3D space.
Formalin fixation and paraffin embedding of organoids is a common method to obtain hematoxylin and eosin (H&E), immunohistochemical (IHC), and immunofluorescent (IF) staining for visualization and analysis6,11,20. However, publications that have described this technique lack comprehensive details on the embedding process. Additionally, locating organoids within the paraffin block can be challenging. Some labs pre-stain organoids with trypan blue prior to embedding to aid in location during sectioning11. The method described here incorporates the use of a histological dye for orientation of the organoid/histology gelplug during embedding to promote efficiency of sectioning. H&E slides facilitate examination of necrosis, nuclear texture, and proliferation, and thus it should be a mandatory endpoint for organoid culture to ensure that cells are healthy throughout the sphere. A strength of organoid culture is that samples are comprised of a heterogeneous mixture of cells that better resemble in vivo patient tissue. In the prostate, for example, both basal and luminal cells are present in prostate organoids5, while cells grown in 2D lack luminal differentiation8. To assess organoid differentiation, it is recommended that researchers assess basal markers such as CK5 and p63 and luminal markers such as androgen receptors and CK88. Any other experimental proteins of interest can also be studied using these staining techniques.
Although FFPE sections allow visualization of cells residing in the inner compartment via histologic methods (H&E, IF, IHC), images are limited to cross sections, and organoid shape may be altered by the embedding process. Whole-mount staining enables an organoid to be stained and observed in situ, preserving morphological phenotypes and permitting images of protein localization. Whole-mounting is a tool utilized to stain whole-tissue or whole-animal specimens, such as the zebrafish or mouse embryo, and is easily adapted for organoids. The technique described here was modified from the procedure by Mahéet al.11, which details the initial growth and culture of gastrointestinal organoids directly on a chamber slide for staining, and requires fixation of the entire parent culture. The method outlined here involves the transfer of a single organoid (or organoids) of interest onto a chamber slide at the time of staining. This enables the selection of individual organoids for whole-mount analysis in an ongoing experiment without fixation of the parent culture. When performing whole-mount staining, it is necessary to optimize permeabilization and the incubation time for primary and secondary antibodies to ensure penetration throughout the specimen (anywhere from one or more days is recommended). Once imaged via confocal microscopy, 3D renderings can be produced to enable visualization and localization of a specific protein and calculate the number of positively stained cells present in a sample. Flow cytometry is a useful endpoint to quantify populations of basal, luminal, or stem cells5,14. To do this, cells are recovered from the matrix gel and gently dissociated for staining. Users should carefully select appropriate markers for separation based on the current literature. For human primary prostate epithelial cells, CD26 and CD49f are suitable luminal and basal markers, respectively5,21.
In summary, this protocol details human primary prostate organoid growth, collection, and experimental endpoints. Of note, the mounting technique described can be applied to a variety of other assays that are normally employed for 2D cells, such as fluorescent probe-based experiments looking at proliferation19, apoptosis, and subcellular organelle stains. Additionally, the collection and dissociation method described here could be utilized in preparation for single-cell RNA sequencing18. Collectively, this demonstrates the possibility for a variety of novel, high-fidelity endpoints that researchers can optimize and explore in the future.
Acknowledgements
We thank the UIC Biorespository members, Dr. Klara Valyi-Nagy, and Alex Susma, as well as the urologists, Drs. Michael Abern, Daniel Moreira, and Simone Crivallero, for facilitation of tissue acquisition for the primary cell cultures. We thank the UIC Urology patients for donating their tissue to research. This work was funded, in part, by the Department of Defense Prostate Cancer Research Program Health Disparities Idea Award PC121923 (Nonn) and the UIC Center for Clinical and Translation Science Pre-doctoral Education for Clinical and Translational Scientists (PECTS) Program (McCray and Richards) and by the National Institutes of Health's National Cancer Institute, Grant Numbers U54CA202995, U54CA202997, and U54CA203000, known as the Chicago Health Equity Collaborative (Nonn and Richards). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Defense.
Materials
| Name | Company | Catalog Number | Comments |
| Cells of interest | |||
| Keratinocyte-SFM (KSFM) | ThermoFisher Scientific | 17005042 | |
| Fetal Bovine Serum, charcoal stripped, USDA-approved regions (FBS) | ThermoFisher Scientific | 12676029 | |
| 5α-Dihydrotestosterone (DHT) | Sigma-Aldrich | D-073-1ML | |
| Flat bottom, polystyrene 96-well cell culture treated plate | ThermoFisher Scientific | 161093 | |
| Matrigel (matrix gel) | Corning | Various | Matrix-gel: growth Factor Reduced, Phenol-red free, etc. depending on application |
| Ice Bucket | |||
| Cell Culture Hood | |||
| 1.5 mL micro-centrifuge tubes or 15 mL conical | ThermoFisher Scientific | 05-408-130, 339650 | |
| Centrifuge | |||
| Dispase (neutral protease) | STEMCELL Technologies | 7923 | neutral protease |
| Hanks' balanced salt solution (HBSS) | ThermoFisher Scientific | 14025076 | |
| TrypLE Express (cell dissociation enzymes) | ThermoFisher Scientific | 12605036 | Cell dissociation enzymes (trypsin may also work, depending on cell type, but TrypLE is more gentle and recommended for primary cells) |
| TRIzol | ThermoFisher Scientific | 15596018 | suggested RNA extraction solution |
| RIPA Lysis and Extraction Buffer | ThermoFisher Scientific | 89900 | suggested protein extraction solution |
| DNAzol | ThermoFisher Scientific | 10503027 | suggested DNA extraction solution |
| Organoids | |||
| Flat bottom, polystyrene 96-well cell culture treated plate | ThermoFisher Scientific | 161093 | |
| Brightfield microscope with camera capabilities | ex. EVOS FL Auto Imaging System | ||
| Photo analysis software | ex. Photoshop, CELLESTE, ImageJ, MorphoLibJ, CellProfiler | ||
| Graphing software | ex. Graphpad, Excel, etc | ||
| Organoids | |||
| Ice pack | |||
| Masking tape | |||
| Pipette tips (1000 μL) | |||
| Razor blade | |||
| Dispase | STEMCELL Technologies | 7923 | |
| Agarose | Sigma-Aldrich | A9045 | |
| HistoGel (histology-gel) | ThermoFisher Scientific | HG-4000-012 | |
| Pencil | |||
| "Plunger" from 1 cc insulin syringe | |||
| Tissue Casette | Thomas Scientific | 1202D72 | |
| Container to hold fixative | |||
| 10% neutral buffered formalin (NBF) | Sigma-Aldrich | HT501128 | |
| Histology pen, xylene and EtOH-resistant | Sigma-Aldrich | Z648191-12EA | STATMARK pen |
| Ethanol (histologic grade) | Fisher Scientific | A405P-4 | For fixation and graded dilutions during processing |
| Xylenes | Sigma-Aldrich | 214736-1L | |
| Deionized water | |||
| Paraffin | Leica | various | |
| Tissue processor | |||
| Embedding workstation | |||
| Economy Tissue Float Bath | Daigger | EF4575E XH-1001 | |
| Microtome | |||
| Microtome blades | Ted Pella | 27243 | |
| Positively charged microscope slides | Thomas Scientific | 1158B91 | |
| Laboratory Oven | ThermoFisher Scientific | PR305225G | |
| Hematoxylin and Eosin Stain Kit | Vector Laboratories | H-3502 | Suggested H&E staining kit |
| ABC Peroxidase Standard Staining Kit | ThermoFisher Scientific | 32020 | Suggested immunohistochemistry staining kit |
| Androgen Receptor Primary Antibody (AR) | Cell Signaling Technology | 5153S | Suggested primary antibody for IHC |
| FFPE, sectioned organoid sample baked on a slide | |||
| Ethanol (histologic grade) | Fisher Scientific | A405P-4 | dilutions should be performed using deinoized water |
| Xylenes | Sigma-Aldrich | 214736-1L | |
| Deionized water (DI H2O) | |||
| Antigen retrieval solution | Sigma-Aldrich, Abcam | C9999, ab93684 | |
| 1x Phosphate Buffered Saline - PBS | ThermoFisher Scientific | 10010023 | |
| Triton X-100 | Sigma-Aldrich | X100-1L | |
| Normal Horse Serum | thermoFisher Scientific | 31874 | |
| Bovin Serum Albumin | Sigma-Aldrich | A2058 | |
| Counterstain (DAPI, Hoescht) | Sigma-Aldrich | D9542 | |
| Sodium azide | Sigma-Aldrich | S2002 | suggested for storage but not required |
| Confocal microscope | |||
| Coplin | Fisher Scientific | 19-4 | |
| Staining rack | IHC World | M905-12DGY | |
| Staining dish | IHC World | M900-12B | |
| Decloaking chamber | Biocare Medical | DC2012 | |
| Humidity chamber | Thomas Scientific | 1219D68 | |
| Hydrophobic barrier pen | Vector Laboratories | H-4000 | |
| Cytokeratin 8/18 primary antibody (CK8) | ARP American Research Products | 03-GP11 | suggested primary antibody for IF |
| p63-alpha antibody (p63) | Cell Signaling Technology | 4892S | suggested primary antibody for IF |
| Keratin 5 Polyclonal Antibody (CK5) | Biolegend | 905501 | suggested primary antibody for IF |
| Goat anti-rabbit secondary | ThermoFisher Scientific | A21245 | suggested secondary antibody for p63 or CK5 detection (do not use at same time) |
| Goat anti-guinea pig secondary | ThermoFisher Scientific | A-11075 | suggested secondary antibody for CK8 detection |
| Microscope cover glass | Globe Scientific | 1414-10 | |
| Anti-fade mounting media | ThermoFisher Scientific | S36972 | |
| Organoids | |||
| Pipette tips (1000 μL) | |||
| Pipette tips (200 μL) | |||
| 8-well chamber slide | ThermoFisher Scientific | 154534PK | It is also possible to use a confocal dish, depends on preference of user |
| Cell-Tak | Corning | CB40240 | optional adherent reagent |
| 1x Phosphate Buffered Saline (PBS) | ThermoFisher Scientific | 10010023 | |
| 4% paraformaldehye (PFA) | Biotium | 22023 | other fixatives such as methanol or formalin can be used |
| 50mM NH4Cl | sigma-Aldrich | 254134 | |
| Triton™ X-100 (non-ionic detergent) | sigma-Aldrich | X100-1L | |
| Normal Horse Serum (NHS) | thermoFisher Scientific | 31874 | |
| Bovin Serum Albumin (BSA) | sigma-Aldrich | A2058 | |
| Counterstain (DAPI, Hoescht) | Sigma-Aldrich | D9542 | |
| Counterstain (phalloidin) | thermoFisher Scientific | A22287 | |
| Sodium azide | sigma-Aldrich | S2002 | suggested for storage but not required |
| Confocal microscope | |||
| Cytokeratin 8/18 primary antibody (CK8) | ARP American Research Products | 03-GP11 | suggested primary antibody for IF |
| p63-alpha antibody (p63) | Cell Signaling Technology | 4892S | suggested primary antibody for IF |
| Keratin 5 Polyclonal Antibody (CK5) | Biolegend | 905501 | suggested primary antibody for IF |
| Goat anti-rabbit secondary | ThermoFisher Scientific | A21245 | suggested secondary antibody for p63 or CK5 detection (do not use at same time) |
| Goat anti-guinea pig secondary | ThermoFisher Scientific | A-11075 | suggested secondary antibody for CK8 detection |
| Visikol HISTO-M | Visikol | various | optional clearing agent |
| Organoids | |||
| Pipette tips (1000 μL) | |||
| Pipette tips (200 μL) | |||
| 8-well chamber slide | ThermoFisher Scientific | 154534PK | It is also possible to use a confocal dish, depends on preference of user |
| Cell-Tak | Corning | CB40240 | |
| 1x Phosphate Buffered Saline - PBS | ThermoFisher Scientific | 10010023 | |
| Cell assay of interest | Various | Various | Click-iT EdU Alexa Fluor proliferation assay (fluorescently-labelled EdU proliferation kit), Image-iT Lipid Peroxidation Kit, etc) and fluorescent probes/dyes (ex. HCS mitochondrial Health Kit, CellMask, LIVE/DEAD Viability assays, CellROX reagents, etc) |
| Organoids | |||
| Ice bucket | |||
| Cell culture hood | |||
| 1.5 mL eppendorf tubes or 15 mL conical | |||
| Microcentrifuge | |||
| Dispase | STEMCELL Technologies | 7923 | |
| TrypLE Express (cell dissociation enzymes) | ThermoFisher Scientific | 12605036 | Cell dissociation enzymes (trypsin may also work, depending on cell type, but TrypLE is more gentle and recommended for primary cells) |
| Hanks' balanced salt solution (HBSS) | ThermoFisher Scientific | 14175079 | |
| Flow Tube with Cell Strainer Snap Cap | Fisher Scientific | 08-771-23 | |
| Cytokeratin 5 Antibody - FITC | Millipore Sigma | FCMAB291F | Suggested flow antibody |
| Cytokeratin 8 Antibody - Alexafluor 405 | Abcam | ab210139 | Suggested flow antibody |
| CD49f - Alexafluor 647 | BioLegend | 313609 | Suggested flow antibody |
| CD26 - PE | BioLegend | 320576 | Suggested flow antibody |
References
- Clevers, H. Modeling Development and Disease with Organoids. Cell. 165, 1586-1597 (2016).
- Simian, M., Bissell, M. J. Organoids: A historical perspective of thinking in three dimensions. Journal of Cell Biology. 216 (1), 31-40 (2017).
- Kretzschmar, K., Clevers, H. Organoids: Modeling Development and the Stem Cell Niche in a Dish. Developmental Cell. 38, 590-600 (2016).
- Fatehullah, A., Tan, S. H., Barker, N. Organoids as an in vitro model of human development and disease. Nature Cell Biology. 18 (3), 246-254 (2016).
- Drost, J., et al. Organoid culture systems for prostate epithelial and cancer tissue. Nature Protocols. 11 (2), 347-358 (2016).
- Chua, C. W., et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nature Cell Biology. 16 (10), 951-954 (2014).
- Sobel, R. E., Sadar, M. D. Cell lines used in prostate cancer research: a compendium of old and new lines--part 2. The Journal of Urology. 173 (2), 360-372 (2005).
- Uzgare, A. R., Xu, Y., Isaacs, J. T. In vitro culturing and characteristics of transit amplifying epithelial cells from human prostate tissue. Journal of Cellular Biochemistry. 91 (1), 196-205 (2004).
- Sobel, R. E., Sadar, M. D. Cell lines used in prostate cancer research: a compendium of old and new lines--part 1. The Journal of Urology. 173 (2), 342-359 (2005).
- Puca, L., et al. Patient derived organoids to model rare prostate cancer phenotypes. Nature Communications. 9 (1), 2404 (2018).
- Mahe, M. M., et al. Establishment of Gastrointestinal Epithelial Organoids. Current Protocols in Mouse Biology. 3 (4), 217-240 (2013).
- Borten, M. A., Bajikar, S. S., Sasaki, N., Clevers, H., Janes, K. A. Automated brightfield morphometry of 3D organoid populations by OrganoSeg. Scientific Reports. 8 (1), 5319 (2018).
- Richards, Z., McCray, T., Marsili, J., Zenner, M. L., Manlucu, J. T., Garcia, J., Murray, M., Voisine, C. M., Murphy, A. B., Abdulkadir, S. A., Prins, G. S., Nonn, L. Prostate stroma increases the viability and maintains the branching phenotype of human prostate organoids. iScience. , (2018).
- Hu, W. -. Y., et al. Isolation and functional interrogation of adult human prostate epithelial stem cells at single cell resolution. Stem Cell Research. 23, 1-12 (2017).
- Peehl, D. M. Primary cell cultures as models of prostate cancer development. Endocrine-Related Cancer. 12 (1), 19-47 (2005).
- Peehl, D. M. Growth of prostatic epithelial and stromal cells in vitro. Methods in Molecular Medicine. 81, 41-57 (2003).
- Mihelich, B. L., et al. miR-183-96-182 cluster is overexpressed in prostate tissue and regulates zinc homeostasis in prostate cells. Journal of Biological Chemistry. 286 (52), 44503-44511 (2011).
- Macosko, E. Z., et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 161 (5), 1202-1214 (2015).
- Barrett, C. W., Short, S. P., Choksi, Y. A., Williams, C. S. Whole-mount Enteroid Proliferation Staining. Bio-Protocol. 6 (12), (2016).
- Gao, D., et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 159 (1), 176-187 (2014).
- Karthaus, W. R., et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 159 (1), 163-175 (2014).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved