Use of Single Chain MHC Technology to Investigate Co-agonism in Human CD8+ T Cell Activation
In This Article
Summary
This protocol describes the use of single chain MHC class I complexes to investigate molecular interactions in human CD8+ T cell activation: generation of engineered antigen presenting cells expressing single chain constructs, culture of human CD8+ T cell clone and T cell activation experiments.
Abstract
Non-stimulatory self peptide MHC (pMHC) complexes do not induce T cell activation and effector functions, but can enhance T cell responses to agonist pMHC, through a process termed co-agonism. This protocol describes an experimental system to investigate co-agonism during human CD8+ T cell activation by expressing human MHC class I molecules presenting pre-determined peptides as single polypeptides (single chain MHC) in a xenogeneic cell line. We expressed single chain MHCs under conditions where low levels of agonist single chain p-MHC complexes and high levels of non-stimulatory single chain p-MHC complexes were expressed. Use of this experimental system allowed us to compare CD8+ T cell responses to agonist pMHC in the presence or absence of non-stimulatory pMHC. The protocol describes cell line transfection with single chain MHC constructs, generation of stable cell lines, culture of hepatitis B virus-specific human CD8+ T cells and T cell activation experiments simultaneously quantifying cytokine production and degranulation. The presented methods can be used for research on different aspects of CD8+ T cell activation in human T cell systems with known peptide MHC specificity.
Introduction
MHC class I (MHC-I) molecules present short (8-10 amino acids) peptides derived from proteins synthesized by each cell. MHC-I molecules on healthy cells present self peptides derived from endogenous proteins. Viral infection leads to presentation of non-self virus-derived peptides on MHC-I, but even infected cells present non-self peptides in the presence of large amounts of self peptide-MHC (pMHC). MHC-I is recognized by the T cell receptor (TCR) and the CD8 co-receptor on T cells. TCR binding affinity to peptide pMHC is highly dependent on the sequence of presented peptides, whereas CD8 binding is peptide-independent. TCR binding to non-self agonist pMHC complex leads to T cell activation and effector functions. Non-stimulatory self pMHC complexes do not induce T cell activation and effector functions, but can enhance T cell responses to agonist pMHC, through a process termed co-agonism1,2,3. Co-agonism has been observed in mouse CD4+ T cells4,5,6 as well as in mouse and human CD8+ T cells7,8,9,10,11,12,13. Under physiological conditions, T cells need to recognize limited quantities of agonist pMHC on antigen presenting cells (APCs) co-presenting high levels of non-stimulatory pMHC complexes, suggesting that co-agonism contributes to optimal T cell responses in vivo. Total cell surface MHC class I levels are frequently downregulated during viral infections14 and on tumors15. This immune evasion strategy decreases agonist pMHC presentation, but also reduces the amount of co-agonist pMHC I available for T cell activation enhancement. Moreover, high cell surface expression levels of the MHC class I molecule HLA-C have been shown to correlate with improved HIV control16, suggesting a critical role for total MHC class I expression in T cell activation. Despite the physiological significance of co-agonism, its molecular mechanism is still incompletely understood. This is largely due to technical challenges of experimentally controlling the amount of presented co-agonist pMHC while keeping agonist pMHC constant.
We developed an experimental system to investigate co-agonism during human CD8+ T cell activation by expressing human MHC class I molecules presenting pre-determined peptide as single polypeptide (single chain MHC-I, Figure 1A) in a xenogeneic cell line9,10. Single chain (sc) agonist pMHC was expressed under control of tetracycline-inducible promoter in the hamster-derived T-REx CHO cell line expressing tetracycline repressor; this allows very low “leaky” expression of sc agonist pMHC in the absence of tetracycline and high sc agonist pMHC expression after addition of tetracycline (Figure 1B,C). The sc agonist pMHC-expressing T-REx CHO cells were then supertransfected with non-stimulatory, co-agonist sc pMHC-I under control of a constitutively active promoter (Figure 1B,C). Use of this experimental system allowed us to compare CD8+ T cell responses to agonist pMHC in the presence or absence of high levels of non-stimulatory pMHC. Critically, since peptide and MHC-I heavy chain are covalently linked in the scMHC-I format, this allows introductions of mutations into MHC molecules presenting pre-determined peptides. Use of scMHC-I technology thus allowed us to specifically modulate TCR or CD8-binding properties of agonist or co-agonist pMHC.
Co-agonism in CD8+ T cell activation has been observed using purified MHC-I complexes presenting agonist and co-agonist peptides in the form of heterodimers11,17 or presented on quantum dots12,13. Early work on co-agonism in CD8+ T cell activation in the context of agonist pMHC recognition on APC used TAP2-deficient cell lines as APCs. TAP is required for peptide transport from the cytoplasm to the endoplasmic reticulum, and TAP-deficiency severely reduces the pool of available MHC class I-binding peptides. In the absence of peptides, the nascent complex of MHC class I heavy chain and β2-microglobulin is unstable, resulting in very low MHC-I cell surface expression. Initially, comparison of T cells stimulated with antigen loaded on TAP-sufficient and -deficient mouse RMA and RMA-S cell lines, respectively, as APCs, did not reveal any evidence for co-agonism during mouse T cell activation18. However, use of this experimental system did not allow precise control over the amount of agonist pMHC presented independently of non-stimulatory self pMHC. Moreover, these two cell lines may also differ in expression of other molecules that modulate T cell activation, such as ligands to T cell co-stimulatory or inhibitory receptors, or adhesion molecules.
Subsequently, we developed a protocol for loading TAP2-deficient RMA-S cells with exogenous peptides to allow presentation of a fixed amount of agonist pMHC in the presence or absence of non-stimulatory pMHC. This was achieved through the following: incubation of TAP2-deficient RMA-S cells at lower temperatures (28 °C, instead of the usual 37 °C) to stabilize empty MHC class I heavy chain/β2 microglobulin complexes19; incubation at 28 °C with exogenous agonist peptide; incubation at 28 °C in the presence or absence of exogenous non-stimulatory peptide; and incubation at 37 °C to reduce cell surface expression of empty MHC class I complexes8,9. Use of this experimental set-up revealed that the presence of non-stimulatory pMHC enhanced activation of mouse thymocytes, naïve peripheral CD8+ T cells and CTLs8. Mechanistically, the presence of non-stimulatory pMHC can induce CD8 recruitment to the T cell:APC immune synapse even in the absence of agonist pMHC, and non-stimulatory pMHC can enhance TCR interaction with CD8 coreceptor7,8. However, this experimental system does not allow testing the relative contributions of TCR and CD8 coreceptor binding to agonist and co-agonist pMHC in mediating the activation enhancement, as any modifications altering TCR or CD8 binding to MHC class I would affect all MHC molecules, regardless of the presented peptide. We therefore used MHC class I single chain technology to overcome this problem.
The single chain (sc) MHC class I format has been used before to improve antigenic pMHC presentation for therapeutic strategies, and to answer fundamental questions on mechanisms of TCR and NK cell receptor activation and function20,21. scMHC-I consists of peptide, β2-microglobulin and MHC-I heavy chain joined by two flexible serine/glycine-rich linkers (Figure 1A), several different linker sequences have been developed and tested22. scMHC-I can fold independently of the TAP complex and chaperone proteins required for conventional pMHC complex assembly20. scMHC-I have been shown to fold correctly, as determined using conformation-specific antibody staining22 and by solving sc structure with X-ray crystallography23. The basic scMHC-I design has been modified to improve binding of low affinity peptides24,25.
This protocol describes the use of human scMHC-I to investigate co-agonism during human CTL clone activation. The protocol has been optimized for human CTL clone specific for an epitope of Hepatitis B virus (HBV). HBV is a severe healthcare burden worldwide, as there are 350 million people infected with HBV and chronic HBV infection can lead to development of hepatocellular carcinoma (HCC). HCC cells resulting from HBV infection can usually present HBV-derived epitopes and can be recognized by specific human T cells. HBV and HCC clearance relies on the human T cell responses26, but HCC patients often suffer from T cell exhaustion and reduced T cell responses27. Introduction of HBV-specific TCR into T cells of HBV has been tried as a potential immunotherapy strategy to eliminate chronic HBV infection28. However, it is unknown if this method will be effective in a clinical setting, due to the low levels of surface MHC-I in human hepatocytes29. Therefore, understanding the mechanism of coagonism may provide alternative perspectives for immunotherapy of HBV, possibly by enhancing the presentation of endogenous pMHC to induce co-agonism-mediated T cell responses. The protocol covers all the necessary steps for research on human co-agonism: transfection of T-REx CHO cells with agonist and co-agonist sc pMHC, generation of stable T-REx CHO cell lines, culture of HBV-specific human CTL CD8+ T cell line and CTL activation experiments quantifying cytokine production and degranulation in response to T-REx CHO cells. Although we focus on using the sc MHC class I technology to investigate co-agonism during HBV T cell activation, it must be noted that the presented methods can easily be adapted for research on other aspects of T cell activation in human T cell systems with known pMHC-I specificity.
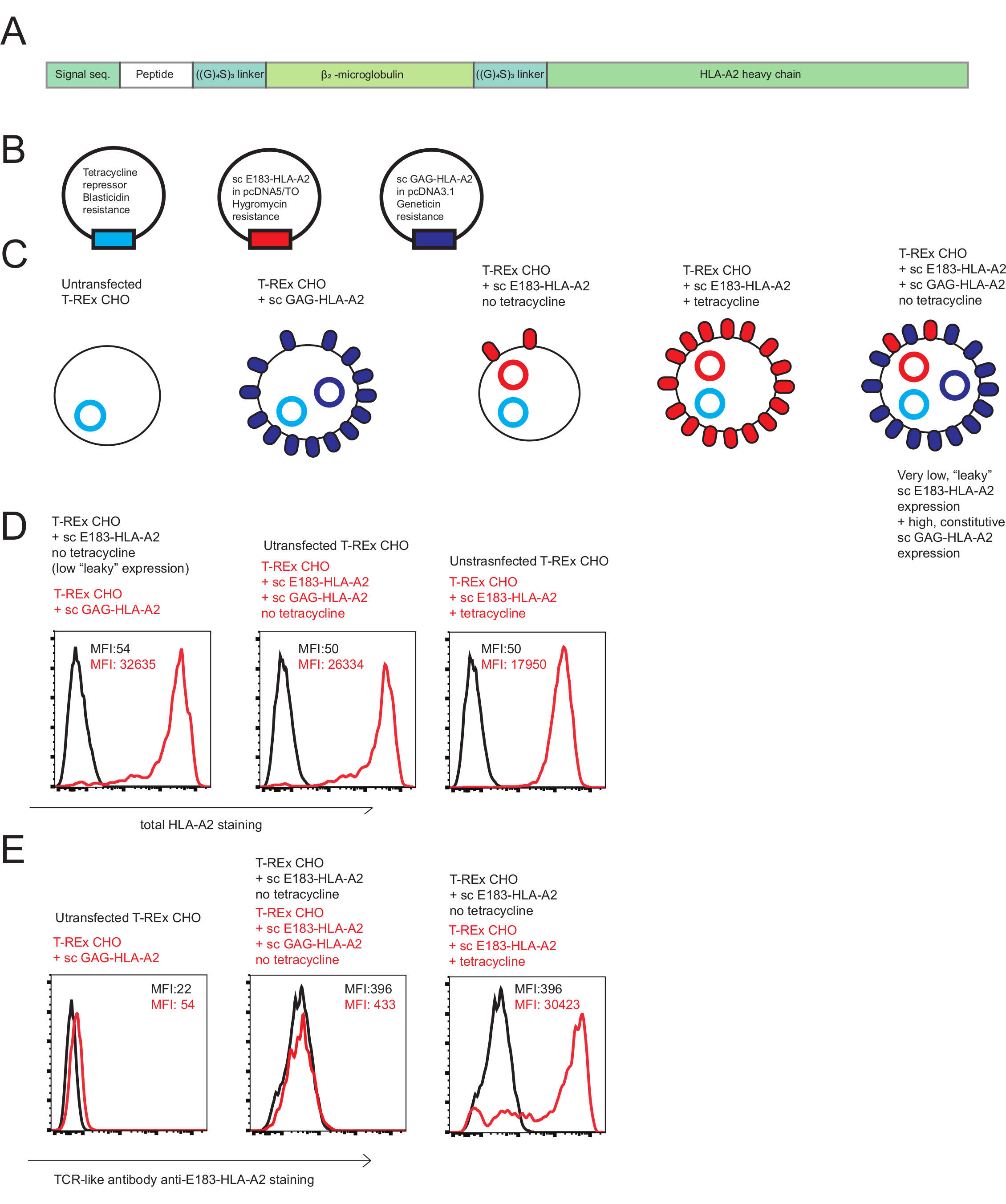
This protocol uses a human CD8+ CTL line specific for HBV-derived peptide E183-91 (FLLTRILTI: "E183")30 presented on HLA-A2. sc HLA molecules consisting of a signal sequence from human β2-microglobulin, a HLA-A2 binding peptide of interest, a glycine/serine linker, a human β2-microglobulin, a glycine/serine linker and a A2 heavy chain can be commercially synthesized (Figure 1A). Plasmids encoding scA2 constructs with agonist E183 peptide, or coagonist HIV GAG (SLYNTVATL)31 peptides are available from the authors upon request. The peptide sequence can be changed using site directed-mutagenesis. A tetracycline-inducible vector pcDNA5/TO was used to express the agonist single chain HLA-A2 with E183 peptide (sc E183-HLA-A2). In the presence of tetracycline repressor, the pcDNA5/TO plasmid allows only very low, “leaky” expression of the protein of interest; high expression can be induced by the addition of tetracycline (Figure 1B,C). Use of a tetracycline-inducible expression system allows control over the amount of cell surface agonist pMHC expression. A constitutive expression plasmid pcDNA3.1 was used to express coagonist scA2 with GAG peptide (sc GAG-HLA-A2). The tetracycline-regulated expression (T-REx) CHO cell line (hamster cell line, no endogenous human MHC) was used to express agonist and co-agonist sc-HLA-A2 constructs. This experimental system allows precise control over pMHC presentation (Figure 1C): no pMHC presentation (untransfected T-REx), a high amount of co-agonist pMHC presentation (constitutive sc GAG-HLA-A2 expression), a low amount of agonist pMHC presentation (sc E183-HLA-A2 in the absence of tetracycline), a high amount of agonist pMHC presentation (sc E183-HLA-A2 in the presence of tetracycline), a low amount of agonist pMHC presentation and high expression of co-agonist pMHC (sc E183-HLA-A2 in the absence of tetracycline, and constitutive sc GAG-HLA-A2 expression). Use of this experimental system allows precise control of cell surface amounts of agonist and coagonist pMHC.
Protocol
NOTE: All steps involving use of live, non-fixed cells should be performed in a biosafety hood, and any biohazardous waste should be discarded according to the local health and safety regulations.
1. CHO Cell Transfection and Generation of Stable CHO Cell Lines
- CHO cell transfection
NOTE: This section describes CHO cell transfection using polyethylenimine (PEI). However, alternative methods can be used, as CHO cells are relatively easy to transfect using commercially available lipid transfection reagents.- Culture CHO cells in complete F12 (cF12, F12 supplemented with 5% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin and 10 µg/mL blasticidin to maintain expression of the tetracycline repressor). Passage the cells every 3-4 days at a 1:10 ratio.
- One day before transfection, prepare the CHO cell suspension. Wash CHO cells in the flask using PBS (10 mL for PBS for a T75 flask, 5 mL PBS for a T25 flask). Add 0.05% Trypsin/0.02% EDTA in PBS (2 mL for a T75 flask, 1 mL for a T25 flask) to the flask and incubate for 5-10 min at room temperature (RT). Using a light microscope, check that the cells have detached, and stop trypsinization by adding cF12 to the flask (8 mL for a T75 flask, 4 mL for a T25 flask).
- Transfer the CHO cell suspension to a 15 mL tube, and centrifuge at 300-400 x g for 5 min at 4 °C or RT. Remove supernatant, re-suspend in cF12 (10 mL for a T75 flask, 5 mL for a T25 flask) and count using a hemocytometer.
- Adjust cell concentration to 60,000-100,000 CHO cells/mL. Add 5 mL of CHO cell suspension (300,000-500,000 CHO cells) per well in a 6 well plate. Incubate overnight at 37 °C, 5% CO2.
- On the day of the transfection, add 400 μL of serum-free F12 media or low serum media to a 1.5 mL tube. Add 3 μg of plasmid to 400 μL of media. Mix by pipetting. Add 6 μg of PEI (6 μL of 1 mg/mL PEI solution) to the media/DNA mix, and vortex immediately for 10 s.
- Incubate the media/DNA/PEI mix at RT for 10-15 min. During this incubation, replace CHO cell media in the 6 well plate with 5 mL of fresh cF12 media.
- Add the media/DNA/PEI solution to CHO cells. Add the solution dropwise and evenly over the well. Incubate overnight at 37 °C, 5% CO2.
- Generation of stable CHO cell lines
- One day after transfection, remove media from the wells and add 5 mL of cF12 containing the appropriate selection antibiotics to each well. Use 0.3 mg/mL hygromycin for pcDNA5TO-based plasmids, and 1 mg/mL geneticin for pcDNA3-based plasmids.
- If the cells reach more than 90% confluency at 24 h post transfection, trypsinize the cells.
- Wash wells with 1 mL of PBS per well, add 1 mL of 0.05% trypsin/0.02% EDTA in PBS and incubate for 5-10 min at RT. Using a light microscope, check that the cells have detached, and stop trypsinization by adding 3-4 mL of cF12 per well.
- Transfer the CHO cell suspension to a 15 mL tube, and centrifuge at 300-400 x g for 5 min at 4 °C or RT. Remove the supernatant and re-suspend in 10 mL of cF12. Add 5 mL of cell suspension per well in 6 well plate. Use two wells to maximize yield of the transfected cells.
- Incubate the 6 well plates at 37 °C, 5% CO2. Monitor cell death and the growth of resistant colonies using a light microscope. Remove media and replace it with fresh media containing the appropriate antibiotics after 4-5 days, or when significant cell death is observed.
- Once the resistant cells reach more than 70% confluence, trypsinize the cells as described in step 1.2.2 and transfer to a T25 flask for expansion. The antibiotic selection takes 1-2 weeks.
- Once the antibiotic-resistant CHO cells in the T25 flask reach 70-80% confluence, perform antibody staining to verify cell surface expression of the constructs of interest. One day before antibody staining, trypsinize the cells as described in step 1.2.2 and adjust the cell concentration to 200,000 cells/mL, and add 1 mL of cell suspension to 12 well plate. For testing of tetracycline-inducible constructs, add 50-60 ng/mL of tetracycline. Incubate overnight at 37 °C, 5% CO2.
- Use cell scraper to detach CHO cells from bottom of the well. This method is preferable to trypsinization, as it reduces the risk of epitope damage due to proteolytic cleavage.
- Transfer the 1 mL of media containing the CHO cells to a FACS tube. Spin down at 300-400 x g, 4 °C for 5 min. Re-suspend in 100 μL of anti-HLA antibody diluted in FWB. Incubate 30 min at 4 °C on ice. Perform this staining with anti-A2 antibody to detect total A2 cell surface expression levels, and TCR-like antibody specific for A2 in complex with E183 peptide to detect agonist pMHC-I cell surface expression levels.
- After verifying MHC expression, FACS-sort the positive cells, and maintain the sorted cells in media with antibiotics to reduce transgene loss. For CHO cells expressing agonist pMHC, single cell sorting is recommended, to ensure comparable agonist pMHC expression with and without supertransfection with co-agonist pMHC. 2-3 weeks are required for CHO cell growth following the single cell sorting.
2. HBV-specific CTL Culture
- Preparation of PBMCs as feeders for HBV-specific human CTL re-stimulation
NOTE: For the A2-E183-specific CTL clone, re-stimulation with freshly isolated PBMCs, rather than thawed PBMCs, gives the best results.
CAUTION: Obtain all the necessary ethical approvals before recruiting volunteers for blood donations. For the purpose of this work, PBMC were collected from healthy volunteers under the protocol approved by NUS IRB. Informed consent was obtained from all donors. Follow all the necessary precautions when working with human blood to reduce risk of transmission of blood borne diseases.- Before starting, set the centrifuge temperature at 19-20 °C and pre-warm the density gradient (e.g., Ficoll) to RT.
- Add 15 mL of room temperature density gradient to 50 mL tube. Centrifuge at 400 x g for 5 min at 19-20 °C. This is to ensure there is no density gradient on the side of the tube.
- Collect 20 mL of blood from a healthy volunteer donor through venipuncture. Add 15 mL of PBS to 20 mL of blood in a 50 mL tube. Mix by pipetting up and down.
- Slowly, add the 35 mL of blood diluted in PBS on top of the 15 mL of density gradient from step 2.1.2. Make sure that the blood and the density gradient do not mix. Centrifuge the tubes at 19 °C, 1,200 x g for 20 min with the brakes off.
- Remove approximately 70% of the plasma layer. Carefully aspirate the layer containing the PBMC (the cloudy layer on top of the clear density gradient layer) using a Pasteur pipette and place it in a new 50 mL tube.
- Add PBS to PBMCs to obtain a 50 mL total volume. Centrifuge tube at 4 °C, 300-400 x g for 10 min. Carefully remove the supernatant using the vacuum aspirator and sterile glass Pasteur pipette, or by decanting.
- Re-suspend the cells in 50 mL of PBS. Centrifuge tube at 4 °C, 300 x g for 10 min. Carefully remove the supernatant using the vacuum aspirator and sterile glass Pasteur pipette, or by decanting.
- Re-suspend cells in 2 mL of complete human T cell media (serum free media supplemented with 2% human serum). Count the cells using hemocytometer. This protocol gives on average 1-2 x 106 PBMCs per 1 mL of blood used.
- CTL re-stimulation using PBMCs as feeder cells
- Irradiate PBMCs at 30 Gy to inhibit proliferation in response to subsequent stimulation.
CAUTION: Ensure adequate training and compliance with the laboratory safety requirements when using the irradiator. - Re-suspend the irradiated PBMCs in complete human T cell media at 2 x 106 cells/mL. Add cytokines and lectin from Phaseolus vulgaris (PHA) for the CTL re-stimulation at 2x concentration into the PBMC suspension. The final cytokine and PHA concentrations are: 20 U/mL recombinant human IL-2, 10 ng/mL recombinant IL-7, 10 ng/mL recombinant human IL-15 and 1.5 mg/mL PHA.
- Thaw the frozen CTL vial in a 37 °C water bath. Transfer the thawed cells into a 15 mL tube with 10 mL of complete human T cell media. Spin down at 300-400 x g at 4 °C for 5 min. Carefully remove the supernatant using the vacuum aspirator and sterile glass Pasteur pipette, or by decanting.
- Re-suspend CTLs in 2 mL of complete human T cell media. Count the cells using hemocytometer. Add complete human T cell media to obtain cell concentration of 106 cells/mL.
- In a 24 well plate, add 0.5 mL of irradiated PBMCs (106 cells) with 2x cytokines and PHA (step 2.2.2), and 0.5 mL of CTLs (0.5 x 106 cells) per well. The number of wells used depends on the total number of CTLs or PBMCs available. Incubate at 37 °C, 5% CO2.
- After 4-5 days, when CTL blasts are visible and the media becomes yellow, transfer the cultures into 6 well plates, and supplement the initial 1 mL volume with 4 mL of complete human T cell media with 20 U/mL IL-2, 10 ng/mL IL-7 and 10 ng/mL IL-15 (no PHA).
- After 2-3 days, transfer the cultures into a T75 flask, add 40 mL of complete human T cell media with cytokines (step 2.2.2, no PHA) and culture in an upright position.
- Maintain CTLs at a concentration of 1-2 x 106 cells/mL by adding fresh media supplemented with cytokines (step 2.2.2, no PHA) at a volume equal to that of the existing culture volume at every other day. CTLs can be used for experiments 7-14 days after re-stimulation.
- Irradiate PBMCs at 30 Gy to inhibit proliferation in response to subsequent stimulation.
3. T Cell Activation Experiments
NOTE: A typical experiment requires several different T-RExCHO lines: untransfected T-REx CHO cells (negative control), T-REx CHO cells expressing non-stimulatory pMHC only (scA2-GAG; negative control), T-REx CHO cells expressing low amounts of agonist pMHC-I (scA2-ENV without tetracycline, experimental sample), T-REx CHO cells expressing low amounts of agonist pMHC and high amounts of non-stimulatory co-agonist pMHC (scA2-ENV and scA2-GAG, without tetracycline, experimental sample), and T-REx CHO cells expressing high amounts of agonist pMHC-I scA2-ENV with tetracycline, positive control), as shown on Figure 1C,D.
- Plating CHO cells as antigen presenting cells
- Culture CHO cells in T75 flask, as described in step 1.1.1. Use cells at 70-90% confluence for best results. One day before the T cell activation experiment, trypsinize the cells as described in section 1.1.2. After trypsinization, transfer the cell suspension into a 15 mL tube, centrifuge at 400 x g for 5 min at 4 °C or RT, remove supernatant by pipetting or decanting.
- Re-suspend the cell pellet in 5 mL of cF12. Count CHO cells using a hemocytometer.
- Adjust the cell concentration to 2 x 105/mL using cF12. Add 50-60 ng/mL of tetracycline to the agonist only samples to induce high amounts of agonist pMHC-I expression as positive control (Figure 1C). CHO cells will settle down in 15 mL tubes, so make sure to mix them by pipetting or vortexing before plating.
- For T cell activation assay, add 100 μL of the CHO cell suspension per well to U-bottom 96 well plate, and use triplicates per CHO cell line. For CHO cell anti-MHC staining, add 1 mL of cell suspension to 12 well plate, and use triplicates per cell line. Incubate overnight at 37 °C, 5% CO2.
- T cell activation assay
NOTE: This T cell activation assay allows simultaneous quantification of T cell degranulation (CD107a staining, a measure of T cell cytotoxicity) and cytokine production.- The day before the experiment, centrifuge CTLs cells at 400 x g at 4 °C for 5 min, count using hemocytometer and re-suspend the cells at 106 cells/mL in complete human T cell media without cytokines or PHA. Incubate at 37 °C, 5% CO2. The purpose of this rest step is to reduce CTL sensitivity, as we have observed maximum activation in response to low amounts of antigen without this step.
- On the day of the experiment, centrifuge T cells at 400 x g at 4 °C for 5 min, remove the supernatant by decanting or pipetting, and re-suspend the cells in 2 mL complete serum-free media (without cytokines or PHA). Count live T cells using a hemocytometer, and adjust T cell concentration to 106 live cells/mL using complete human T cell media (without cytokines or PHA).
- Add anti-human CD107a antibody at a 1:100 dilution (final concentration of 2.5 μg/mL), add brefeldin A at a 1:1000 dilution (final concentration of 1 μg/mL).
- Remove media from the CHO cells in the 96 well plate by aspirating with glass Pasteur pipette or by flicking the plate. Per well, add 200 μL of the T cell suspension (0.2 x 106 cells) containing anti-CD107a and brefeldin A. Co-culture T cells with CHO cells for 3-4 h.
- At the end of the co-culture, perform T cell surface antigen staining. Spin down the 96 well plate at 300-400 x g, 5 min at 4 °C, and remove the supernatant by aspirating or flicking. Add 2.5 μL of CD3 and 2.5 μL of CD8 antibodies in 50 μL of 0.5% BSA in PBS (flow wash buffer, FWB) per well for cell surface antigen staining. Incubate samples on ice for 30 min in the dark.
- Wash samples by adding 150 μL of FWB per well. Spin down the 96 well plate at 300-400 x g, 5 min at 4 °C, and remove the supernatant by aspirating or flicking.
- Perform fixation/permeabilization steps using a fixation/permeabilization kit. Add 100 µL of fixation/permeabilization solution (from the fixation/permeabilization kit) per well, mix by pipetting. Incubate on ice for 20 min.
- Centrifuge at 300-400 x g and 4 °C for 5 min, and remove the supernatant by aspirating or flicking. Add 200 μL of 1x perm/wash buffer (dilute the 10x perm/wash stock buffer from the kit prior to use) per well. Repeat this step.
- Re-suspend the cell pellet in 50 μL of 1x perm/wash buffer containing anti-IFN-γ antibody at 0.3 μg/mL. Incubate on ice for 30 min in dark.
- Add 150 μL of 1x perm/wash buffer. Centrifuge at 300-400 x g for at 4 °C for 5 min, and remove the supernatant by aspirating or flicking. Add 200 μL of 1x perm/wash buffer, centrifuge 300-400 x g for at 4 °C for 5 min, and remove the supernatant by aspirating or flicking.
- Re-suspend each sample in 200 μL of FWB. Proceed with flow cytometric analysis.
- CHO cell MHC class I staining
NOTE: It is critical to verify scMHC-I cell surface levels on cells used on each experiment, as cell lines may lose transgene expression. When preparing 96 well plate for T cell stimulation, set up 12 well plate for CHO cell staining, as outlined in Section 3.1.4.- Use a cell scraper to detach CHO cells from the bottom of the well. This method is preferable to trypsinization, as it reduces the risk of epitope damage due to proteolytic cleavage.
- Transfer the 1 mL of media containing the CHO cells to the FACS tube. Spin down at 300-400 x g at 4 °C for 5 min. Re-suspend in 100 μL of anti-HLA antibody diluted in FWB. Incubate for 30 min at 4 °C on ice.
- Perform this staining with anti-A2 antibody to detect total A2 cell surface expression levels, and TCR-like antibody specific for A2 in complex with E183 peptide32 to detect agonist pMHC-I cell surface expression levels.
- Add 1 mL of FWB to each CHO sample, spin down at 300-400 x g, 4 °C, 5 min, and then re-suspend the sample in 0.3 mL of FWB. Proceed with flow cytometry analysis.
Representative Results
The sc MHC class I technology used here allows precise control of cell surface expression of MHC class I with known, covalently linked peptides, to induce T cell activation. We have generated an engineered xenogeneic APC system (Figure 1A,B,C) that allows presentation of low levels of agonist pMHC in the presence or absence of co-agonist pMHC (Figure 1D,E). Critically, we have used a TCR-like antibody specific for HLA-A2 presenting E183 peptide to show that presentation of the agonist sc E183-HLA-A2 is not altered by co-expression of co-agonist sc GAG-HLA-A2 (Figure 1E). However, it must be noted that the E183-HLA-A2 specific TCR-like antibody shows some binding to HLA-A2 presenting GAG peptide (Figure 1E). Similar engineered antigen presenting cell systems can be constructed using different peptides or MHC molecules to investigate molecular requirements for TCR and/or coreceptor interactions in human T cells with different specificities.
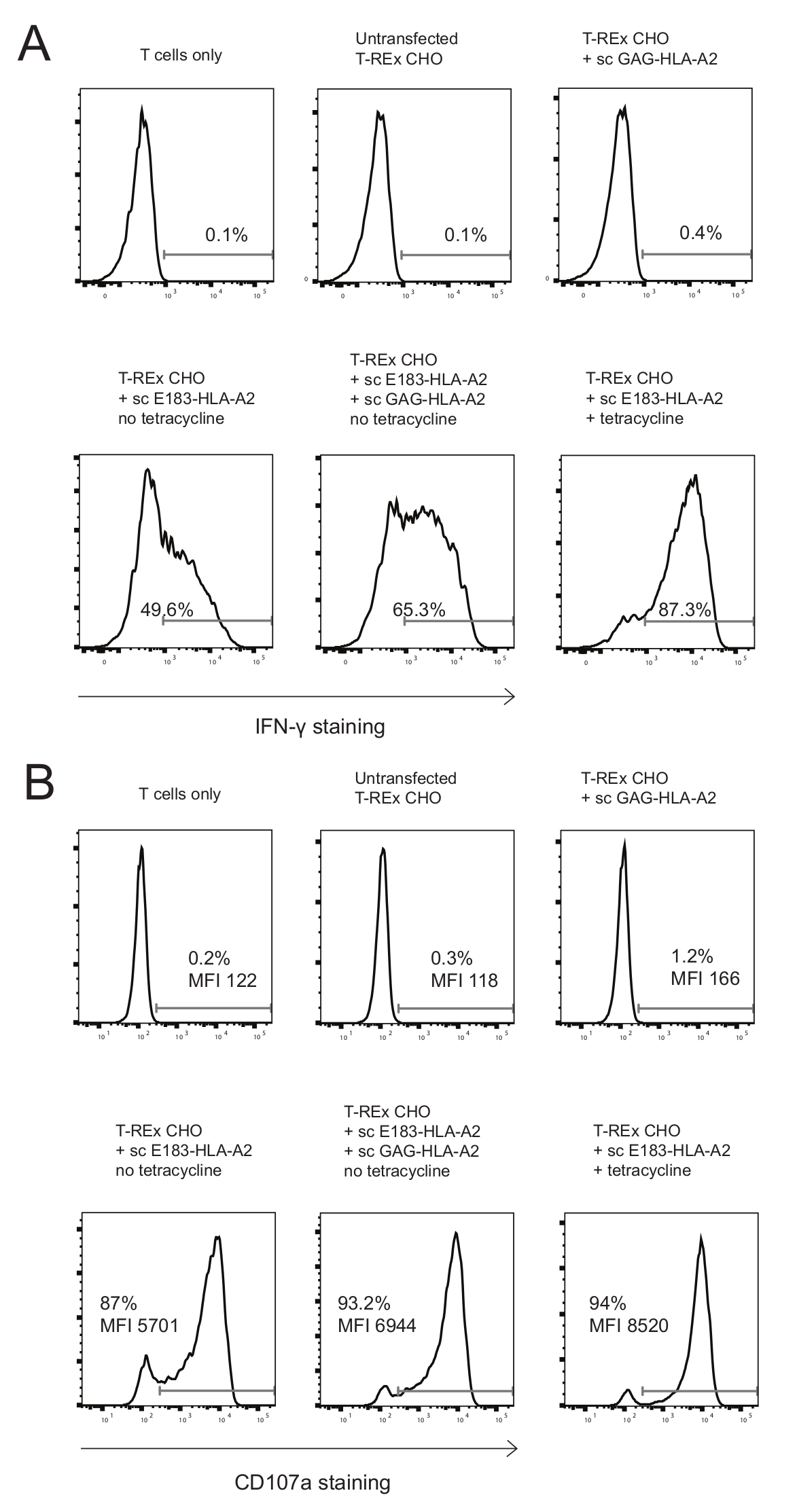
We used these engineered APCs to stimulate an E183-HLA-A2-specific human CD8+ T cell clone. Three negative controls were used: T cells only, untransfected T-REx CHO cells, and T-REx CHO cells expressing high amounts of co-agonist sc GAG-HLA-A2 to test for potential T cell activation by any molecules expressed on T-REx CHO cells (untransfected T-REx CHO cells as compared to T cell only), and for T cell activation by sc-GAG-HLA-A2 (T-REx CHO cells expressing high levels of sc-GAG-HLA-A2 as compared to untransfected T-REx CHO cells). Untransfected T-REx CHO cells or T-REx CHO cells expressing sc-GAG-HLA-A2 did not induce activation of E183-HLA-A2-specific CD8+ CTL clone, as determined by quantifying IFN-γ production and expression of degranulation marker CD107a as a measure of T cell cytotoxicity (Figure 2A,B). Expression of high levels of agonist sc E183-HLA-A2, used as positive control, induced very efficient IFN-γ production and degranulation (Figure 2A,B), demonstrating that the sc HLA-A2 construct can be recognized by a specific TCR to activate T cell effector functions. Expression of low levels of sc E183-HLA-A2 induced lower levels of IFN-γ production and degranulation, and this was enhanced by presence of co-agonist sc GAG-HLA-A2 (Figure 2A,B). It must be noted that very high percentages of CD107a+ T cells were observed even in response to low levels of antigenic pMHC (Figure 2B), consistent with previous reports of relatively low requirements for the amount of agonist pMHC for induction of T cell cytotoxicity33. The CD107a MFI, indicating the amount of degranulation on a single cell basis, provides a better dynamic range (Figure 2B). The T cell activation assay used allows simultaneous quantification of two major effector functions: cytokine production and cytotoxicity, and provides very sensitive readout with good dynamic range.
We used the single chain technology to test whether TCR binding to co-agonist pMHC is required for co-agonist pMHC-dependent activation enhancement. We mutated valine at position 152 at the edge of the HLA-A2 peptide binding groove (Figure 3B) to glutamic acid to create V152E mutant, as this mutation has been reported to abolish TCR binding to HLA-A234. We then tested if the V152E mutation can abolish TCR binding for the E183-HLA-A2 CTL clone used, by introducing this mutation into agonist sc E183-HLA-A2 and expressing the mutant agonist sc construct in T-REx CHO cells. Wild type, but not V152E, sc E183-HLA-A2 induced activation of the E183-HLA-A2-specific CTL clone used (Figure 3A). It must be noted that any TCR-binding mutation on MHC class I needs to be separately tested for each individual TCR/T cell clone used, as the effect of mutation will depend on the precise molecular interactions between TCR and pMHC complex, and these will differ between different TCRs. For example, we tested eight mutations, either previously reported TCR-binding mutations or mutations predicted to disrupt TCR binding without abrogating peptide binding into HLA-A2. Of these, V152E was the only mutation that abolished activation of the E183-specific T cell clone used10. We then introduced the V152E mutation into the co-agonist sc GAG-HLA-A2, and co-expressed WT or V152E sc GAG-HLA-A2 with low levels of agonist sc E183-HLA-A2. Both WT and V152E sc GAG-HLA-A2 enhanced IFN-γ production and degranulation (Figure 3C,D), suggesting that TCR binding to co-agonist pMHC is not required for co-agonist pMHC-dependent CD8+ T cell activation enhancement. Single chain MHC class I technology, as presented here, can be used to modify TCR and/or coreceptor binding to MHC class I with known peptide to investigate the molecular requirements for T cell antigen recognition and activation.

Figure 1: Single chain HLA-A2 constructs used to investigate co-agonist in human CD8+ T cell activation. (A) Schematic diagram of a scHLA-A2 construct, consisting of covalently signal sequence from fused human β2 microglobulin, peptide of choice, flexible glycine-serine linker (GGGGSGGGGS GGGGS), human β2 microglobulin, flexible glycine-serine linker and HLA-A2 heavy chain. (B) Schematic diagram of the plasmids used to generate engineered antigen presenting cells to investigate co-agonism in human CD8+ T cell activation: tetracycline repressor, agonist sc E183-HLA-A2 under control of tetracycline-inducible promoter, non-stimulatory co-agonist sc GAG-HLA-A2 under control of constitutively active promoter. (C) Schematic diagram of the engineered antigen presenting cells used to investigate co-agonism: T-REx CHO cells (no human MHC expression), T-REx CHO cells expressing constitutively high levels of sc GAG-HLA-A2 (non-stimulatory co-agonist pMHC), T-REx CHO cells expressing low “leaky” levels of sc E183-HLA-A2 in the absence of tetracycline (low level of agonist peptide), T-REx CHO cells expressing high levels of sc E183-HLA-A2 in the presence of tetracycline (high level of agonist peptide), T-REx CHO cells expressing low levels of sc E183-HLA-A2 and high levels of sc GAG-HLA-A2 (low level of agonist peptide and high level of co-agonist peptide). (D) Cell surface staining of the engineered antigen presenting cell lines using anti-HLA-A2 antibody. The numbers shown denote MFI values for MHC stain in the live cell gate. Data representative of 5 independent experiments (E) Cell surface staining of the engineered antigen presenting cell lines using TCR-like antibody specific against HLA-A2 presenting E183 peptide. The numbers shown denote MFI values for MHC stain in the live cell gate. Data representative of 5 independent experiments. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Presence of co-agonist pMHC enhanced cytokine production and degranulation of E183-HLA-A2-specific human CD8+ CTL clone. (A) Intracellular IFN-γ staining of HLA-A2-specific human CD8+ CTL clone after 3h stimulation with the indicated engineered antigen presenting cells. The numbers shown denote the percentage positive for IFN- γ staining. Data representative of 3 independent experiments, each performed with technical triplicates. (B) Cell surface CD107a staining of HLA-A2-specific human CD8+ CTL clone after 3h stimulation with the indicated engineered antigen presenting cells. The numbers shown denote the percentage positive for CD107a staining or the CD107a MFI for the CD8+ T cell population. Data representative of 3 independent experiments, each performed with technical triplicates. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Co-agonist – mediated T cell activation enhancement does not require TCR binding to the co-agonist pMHC complex. (A) V152E mutation in sc E183-HLA-A2 abolishes activation of the E183-HLA-A2-specific human CD8+ CTL clone. T-REx CHO cells expressing high levels (tetracycline induction) of the WT or V152E mutant sc E183-HLA-A2 were used to stimulate E183-HLA-A2-specific human CD8+ CTL clone for 3h. T cell activation was quantified using intracellular IFN-γ staining. The numbers shown denote the percentage positive for IFN- γ staining. Data representative of 3 independent experiments, each performed with technical triplicates. (B) Location of V152E mutation within the peptide binding groove of HLA-A2. (C) V152E mutation on sc GAG-HLA-A2 co-agonist pMHC complex does not abolish co-agonist-dependent activation enhancement. Intracellular IFN-γ staining of HLA-A2-specific human CD8+ CTL clone after 4h stimulation with the indicated engineered antigen presenting cells. The numbers shown denote the percentage positive for IFN- γ staining. Data representative of 3 independent experiments, each performed with technical triplicates. (D) V152E mutation on sc GAG-HLA-A2 co-agonist pMHC complex does not abolish co-agonist-dependent activation enhancement. Cell surface CD107a staining of HLA-A2-specific human CD8+ CTL clone after 3h stimulation with the indicated engineered antigen presenting cells. The numbers shown denote the percentage positive for CD107a staining or the CD107a MFI for the CD8+ T cell population. Data representative of 3 independent experiments, each performed with technical triplicates. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The presented protocol provides a robust tool for investigation of molecular interactions during human CD8+ T cell activation. A critical step for investigation of co-agonism during human T cell activation is control over agonist pMHC cell surface expression to ensure equal agonist pMHC presentation on the APCs with or without co-agonist pMHC expression. In our experimental system, this is achieved by using a single cell clone expressing low amounts of agonist pMHC, followed by supertransfection with a co-agonist pMHC construct and agonist pMHC quantification using TCR-like antibody10,32. As agonist sc pMHC expression can change over time, it is critical to quantify agonist pMHC surface expression on APCs used in each experiment using TCR-like antibodies. If no specific TCR-like antibody is available, agonist scMHC-I can be expressed as fluorescent protein fusion, and its cell surface expression can then be quantified using a sensitive microscopy method, such as total internal reflection fluorescence microscopy35,36. Moreover, cell surface expression of co-agonist pMHC decreases over time, due to methylation-dependent and -independent silencing of the CMV promoter37, and should be monitored using antibody staining and flow cytometry analysis, with repeated FACS-sorting when needed. We have cultured CHO cells for up to 5 weeks with relatively limited loss of sc pMHC expression. We highly recommend freezing the CHO cells soon after selection/sorting to ensure a reliable stock of cells with known sc MHC expression.
The several steps of the presented protocols can be modified, depending on the availability of equipment, or depending on the specific research aims. For example, PBMC proliferation potential can be inhibited (Step 3.2.1) using alternative methods that do not require a gamma (or X-) irradiator, such as treatment with mitomycin C38. A non-enzymatic cell dissociation buffers containing metal chelators39 can be used instead of cell scraping in step 3.3.1. Moreover, the antigen presenting system can be modified to make it more suitable for human CTL clones expressing different TCRs. CHO cells express hamster MHC class I molecules, and although these are irrelevant for the CTL line studied here (Figure 2A and Figure 3A, we observed no T cell responses to untransfected CHO cells), there is a possibility that other human TCRs may show some xenoreactivity. In that case, hamster MHC class I expression can be reduced by knocking out hamster β2-microglubulin or TAP using CRISPR/Cas9; or a human APC line can be used after CRISPR/Cas9-mediated β2-microglubulin or TAP knock out.
The experimental system presented here allows precise control of TCR and CD8 interactions with MHC molecules presenting pre-defined peptides. For example, we have previously shown that mutations abolishing CD8 binding40 to coagonist pMHC eliminated coagonist-mediated activation enhancement in murine and human CD8+ T cells9,10, whereas abrogating TCR interaction with co-agonist pMHC had no effect on coagonist-mediated activation enhancement10. However, the use of single chain MHC constructs has several limitations. For example, it is time and labor-consuming to test multiple co-agonist peptide sequences using this experimental system, as this involves generation of multiple plasmids encoding sc MHC with different peptides, and subsequent transfections and cell sorting. Previously, we used the murine TAP-deficient cell line RMA-S to test multiple co-agonist peptides in mouse T cell activation8,9, but this approach was not successful with the TAP-deficient human T2 cell line10, most likely due to presentation of TAP-independent peptides41. Another limitation of our experimental system is that is does not provide a fast method for altering the amount of co-agonist pMHC molecules for the purpose of determining the critical amount of co-agonist pMHC required to trigger T cell activation. Moreover, the tetracycline-inducible system used does not allow titration of agonist pMHC quantity at the single cell level by changing the tetracycline concentration, as varying tetracycline concentration alters the proportion of HLA-A2 positive cells, rather than HLA-A2 levels on per cell basis. An alternative approach that we are currently investigating is to use supported lipid bilayers42, previously used to investigate co-agonism during murine CD4+ T cell activation4 or beads presenting fixed amount of agonist pMHC in the presence of varying amounts of co-agonist pMHC. When used in conjunction with UV-cleavable peptide technology4,43, these alternative experimental systems should allow fast testing of multiple co-agonist peptide sequences as well as different amounts of co-agonist pMHC.
The sc MHC technology can be also applicable to investigation of co-agonism in human or murine CD4 T cells, as single chain MHC class II molecules presenting pre-determined peptides have been successfully expressed on mammalian cell surfaces44. Moreover, the use of sc MHC technology can be extended to investigation of non-classical MHC molecules such as HLA-E. Sc HLA-E has been described before, and its expression has been shown to inhibit human T cell activation45. Another non-classical MHC molecule of interest is CD1, which presents lipids instead of peptides46. Sc constructs consisting of CD1 heavy chain and β2-microglobulin have been shown to be expressed on the cell surface and to bind relevant lipid ligands47. Single chain MHC technology has great potential as a research tool for investigating molecular interactions during T and NK cell activation.
Acknowledgements
This research was supported by the Singapore Ministry of Health’s National Medical Research Council under its CBRG/0064/2014 to N.R.J.G., by CREATE under its R571-002-012-592 to P.A.M., National Research Foundation Investigatorship R571-000-272-281 to P.A.M., and by a Singapore Translational Research (STaR) Investigator Award (NMRC/STaR/013/2012) to A.B. We are grateful to Dr. Paul Hutchinson and Mr. Teo Guo Hui from NUS Immunology Programme Flow Core facility for the expert cell sorting. We are grateful to Elijah Chen for critical reading of the manuscript.
Materials
| Name | Company | Catalog Number | Comments |
| Aim-V (serum free media) | Gibco | 12055091 | |
| blasticidin | Gibco | R21001 | |
| Cytofix/Cytoperm (fixation/permealibisation kit) | BD | 554714 | |
| F12 | Gibco | 10565-018 | |
| Fetal bovine serum (FBS) | HyClone | SH3007103 | |
| geneticin | Gibco | 10131035 | |
| GolgiPlug (Brefeldin A) | BD | 555029 | |
| Human serum | Sigma-Aldrich | H4522 | |
| hygromycin | Gibco | 10687010 | |
| Lectin from Phaseolus vulgaris (PHA) | Sigma-Aldrich | L4144 | |
| Opti-MEM (low serum media) | Bibco | 31985070 | |
| 10 X PBS | Vivantis | PB0344 – 1L | |
| Penicillin/Streptomycin | HyClone | SV30010 | |
| polyethylenimine (PEI) | Sigma-Aldrich | 408727 | |
| recombinant human IL2 | R&D Systems | 202-IL | |
| recombinant human IL7 | R&D Systems | 207-IL | |
| recombinant human IL15 | R&D Systems | 247-ILB | |
| tetracycline | Sigma-Aldrich | T7660 | |
| T-REx CHO cell line | Thermo Fisher Scientific | R71807 | |
| 10 x Trypsin/EDTA | Gibco | 15400054 | |
| Antibodies | |||
| CD3 | BD | 347347 | Clone SK7, RRID:AB_400287 |
| CD8α | BD | 563795 | Clone RPA-T8, RRID:AB_2722501 |
| CD107a | BD | 566261 | Clone H4A3, RRID:AB_2739639 |
| HLA-A2 | eBioscience | 17-9876-41 | Clone BB7.2, RRID:AB_11151522 |
| IFN-γ | eBioscience | 12-7319-41 | Clone 4S.B3, RRID:AB_1311250 |
References
- Gascoigne, N. R. Do T cells need endogenous peptides for activation?. Nature Reviews. Immunology. 8 (11), 895-900 (2008).
- Gascoigne, N. R., Zal, T., Yachi, P. P., Hoerter, J. A. Co-receptors and recognition of self at the immunological synapse. Current Topics in Microbiology and Immunology. 340, 171-189 (2010).
- Krogsgaard, M., Juang, J., Davis, M. M. A role for "self" in T-cell activation. Seminars in Immunology. 19 (4), 236-244 (2007).
- Krogsgaard, M., et al. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 434 (7030), 238-243 (2005).
- Ebert, P. J., Jiang, S., Xie, J., Li, Q. J., Davis, M. M. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nature Immunology. 10 (11), 1162-1169 (2009).
- Lo, W. L., et al. An endogenous peptide positively selects and augments the activation and survival of peripheral CD4+ T cells. Nature Immunology. 10 (11), 1155-1161 (2009).
- Yachi, P. P., Ampudia, J., Gascoigne, N. R., Zal, T. Nonstimulatory peptides contribute to antigen-induced CD8-T cell receptor interaction at the immunological synapse. Nature Immunology. 6 (8), 785-792 (2005).
- Yachi, P. P., Lotz, C., Ampudia, J., Gascoigne, N. R. T cell activation enhancement by endogenous pMHC acts for both weak and strong agonists but varies with differentiation state. The Journal of Experimental Medicine. 204 (11), 2747-2757 (2007).
- Hoerter, J. A., et al. Coreceptor affinity for MHC defines peptide specificity requirements for TCR interaction with coagonist peptide-MHC. The Journal of Experimental Medicine. 210 (9), 1807-1821 (2013).
- Zhao, X., et al. Nonstimulatory peptide-MHC enhances human T-cell antigen-specific responses by amplifying proximal TCR signaling. Nature Communications. 9 (1), 2716 (2018).
- Juang, J., et al. Peptide-MHC heterodimers show that thymic positive selection requires a more restricted set of self-peptides than negative selection. The Journal of Experimental Medicine. 207 (6), 1223-1234 (2010).
- Anikeeva, N., et al. Quantum dot/peptide-MHC biosensors reveal strong CD8-dependent cooperation between self and viral antigens that augment the T cell response. Proceedings of the National Academy of Sciences of the United States of America. 103 (45), 16846-16851 (2006).
- Anikeeva, N., Gakamsky, D., Scholler, J., Sykulev, Y. Evidence that the density of self peptide-MHC ligands regulates T-cell receptor signaling. PloS One. 7 (8), e41466 (2012).
- Schuren, A. B., Costa, A. I., Wiertz, E. J. Recent advances in viral evasion of the MHC Class I processing pathway. Current Opinion in Immunology. 40, 43-50 (2016).
- Bubenik, J. MHC class I down regulation, tumour escape from immune surveillance and design of therapeutic strategies. Folia Biologica. 51 (1), 1-2 (2005).
- Apps, R., et al. Influence of HLA-C expression level on HIV control. Science. 340 (6128), 87-91 (2013).
- Cebecauer, M., et al. CD8+ cytotoxic T lymphocyte activation by soluble major histocompatibility complex-peptide dimers. The Journal of Biological Chemistry. 280 (25), 23820-23828 (2005).
- Sporri, R., Reis e Sousa, C. Self peptide/MHC class I complexes have a negligible effect on the response of some CD8+ T cells to foreign antigen. European Journal of Immunology. 32 (11), 3161-3170 (2002).
- Ljunggren, H. G., et al. Empty MHC class I molecules come out in the cold. Nature. 346 (6283), 476-480 (1990).
- Kotsiou, E., Brzostek, J., Gould, K. G. Properties and applications of single-chain major histocompatibility complex class I molecules. Antioxidants & Redox Signaling. 15 (3), 645-655 (2011).
- Hansen, T. H., Connolly, J. M., Gould, K. G., Fremont, D. H. Basic and translational applications of engineered MHC class I proteins. Trends in Immunology. 31 (10), 363-369 (2010).
- Yu, Y. Y., Netuschil, N., Lybarger, L., Connolly, J. M., Hansen, T. H. Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. Journal of Immunology. 168 (7), 3145-3149 (2002).
- Mitaksov, V., et al. Structural engineering of pMHC reagents for T cell vaccines and diagnostics. Chemistry & Biology. 14 (8), 909-922 (2007).
- Truscott, S. M., et al. Disulfide bond engineering to trap peptides in the MHC class I binding groove. Journal of Immunology. 178 (10), 6280-6289 (2007).
- Truscott, S. M., et al. Human major histocompatibility complex (MHC) class I molecules with disulfide traps secure disease-related antigenic peptides and exclude competitor peptides. The Journal of Biological Chemistry. 283 (12), 7480-7490 (2008).
- Bertoletti, A., Ferrari, C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Postgraduate Medical Journal. 89 (1051), 294-304 (2013).
- Boni, C., et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. Journal of Virology. 81 (8), 4215-4225 (2007).
- Gehring, A. J., et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. Journal of Hepatology. 55 (1), 103-110 (2011).
- Gehring, A. J., et al. The level of viral antigen presented by hepatocytes influences CD8 T-cell function. Journal of Virology. 81 (6), 2940-2949 (2007).
- Bertoni, R., et al. Human histocompatibility leukocyte antigen-binding supermotifs predict broadly cross-reactive cytotoxic T lymphocyte responses in patients with acute hepatitis. The Journal of Clinical Investigation. 100 (3), 503-513 (1997).
- Goulder, P. J., et al. Patterns of immunodominance in HIV-1-specific cytotoxic T lymphocyte responses in two human histocompatibility leukocyte antigens (HLA)-identical siblings with HLA-A*0201 are influenced by epitope mutation. The Journal of Experimental Medicine. 185 (8), 1423-1433 (1997).
- Sastry, K. S., et al. Targeting hepatitis B virus-infected cells with a T-cell receptor-like antibody. Journal of Virology. 85 (5), 1935-1942 (2011).
- vanden Berg, H. A., et al. Cellular-level versus receptor-level response threshold hierarchies in T-cell activation. Frontiers in Immunology. 4, 250 (2013).
- Moots, R. J., Matsui, M., Pazmany, L., McMichael, A. J., Frelinger, J. A. A cluster of mutations in HLA-A2 alpha 2 helix abolishes peptide recognition by T cells. Immunogenetics. 34 (3), 141-148 (1991).
- Crites, T. J., Chen, L., Varma, R. A TIRF microscopy technique for real-time, simultaneous imaging of the TCR and its associated signaling proteins. Journal of Visualized Experiments. (61), (2012).
- Axmann, M., Schutz, G. J., Huppa, J. B. Single Molecule Fluorescence Microscopy on Planar Supported Bilayers. Journal of Visualized Experiments. (105), e53158 (2015).
- Mariati, E. Y., Koh, J. H., Yeo, S. C., Ho, Y., Yang, Toward stable gene expression in CHO cells. Bioengineered. 5 (5), 340-345 (2014).
- Matsushita, S., Tanaka, Y., Matsuoka, T., Nakashima, T. Clonal expansion of freshly isolated CD4T cells by randomized peptides and identification of peptide ligands using combinatorial peptide libraries. European Journal of Immunology. 31 (8), 2395-2402 (2001).
- Heng, B. C., Cowan, C. M., Basu, S. Comparison of enzymatic and non-enzymatic means of dissociating adherent monolayers of mesenchymal stem cells. Biological Procedures Online. 11, 161-169 (2009).
- Smith, K., et al. Sensory adaptation in naive peripheral CD4 T cells. The Journal of Experimental Medicine. 194 (9), 1253-1261 (2001).
- Wei, M. L., Cresswell, P. HLA-A2 molecules in an antigen-processing mutant cell contain signal sequence-derived peptides. Nature. 356 (6368), 443-446 (1992).
- Vardhana, S., Dustin, M. Supported Planar Bilayers for the Formation of Study of Immunological Synapses and Kinapse. Journal of Visualized Experiments. (19), (2008).
- Bakker, A. H., et al. Conditional MHC class I ligands and peptide exchange technology for the human MHC gene products HLA-A1, -A3, -A11, and -B7. Proceedings of the National Academy of Sciences of the United States of America. 105 (10), 3825-3830 (2008).
- Rhode, P. R., et al. Single-chain MHC class II molecules induce T cell activation and apoptosis. Journal of Immunology. 157 (11), 4885-4891 (1996).
- Crew, M. D., Cannon, M. J., Phanavanh, B., Garcia-Borges, C. N. An HLA-E single chain trimer inhibits human NK cell reactivity towards porcine cells. Molecular Immunology. 42 (10), 1205-1214 (2005).
- Van Kaer, L., Wu, L., Joyce, S. Mechanisms and Consequences of Antigen Presentation by CD1. Trends in Immunology. 37 (11), 738-754 (2016).
- Im, J. S., et al. Direct measurement of antigen binding properties of CD1 proteins using fluorescent lipid probes. The Journal of Biological Chemistry. 279 (1), 299-310 (2004).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved