Cell-free Protein Expression Using the Rapidly Growing Bacterium Vibrio natriegens
In This Article
Summary
Cell-free expression systems are powerful and cost-efficient tools for the high-throughput synthesis and screening of important proteins. Here, we describe the preparation of cell-free protein expression system using Vibrio natriegens for the rapid protein production using plasmid DNA, linear DNA, and mRNA template.
Abstract
The marine bacterium Vibrio natriegens has garnered considerable attention as an emerging microbial host for biotechnology due to its fast growth rate. A general protocol is described for the preparation of V. natriegens crude cell extracts using common laboratory equipment. This high yielding protocol has been specifically optimized for user accessibility and reduced cost. Cell-free protein synthesis (CFPS) can be carried out in small scale 10 μL batch reactions in either a 96- or 384-well format and reproducibly yields concentrations of > 260 μg/mL super folder GFP (sfGFP) within 3 h. Overall, crude cell extract preparation and CFPS can be achieved in 1−2 full days by a single user. This protocol can be easily integrated into existing protein synthesis pipelines to facilitate advances in bio-production and synthetic biology applications.
Introduction
Cell-free protein synthesis is a versatile and cost-effective method for the expression of valuable proteins or peptides1,2,3,4. Historically, cell-free protein synthesis has been performed using Escherichia coli expression systems; however, there has been a recent surge in using alternative, non-model organisms with novel properties as chassis for cell-free expression5,6,7,8,9,10,11,12. Organisms with unique metabolic profiles are prime candidates as alternatives to E. coli cell-free systems. For example, the marine bacterium Vibrio natriegens is the fastest growing of all known organisms with an observed doubling time of less than 10 min13. This has garnered V. natriegens considerable attention as an emerging microbial host for research and biotechnology14,15,16,17,18. Given that the rapid growth rate of V. natriegens has been linked to high rates of protein synthesis and metabolic efficiency19,20,21, harnessing its cellular machinery for cell-free synthesis may significantly expand the toolkit for rapid protein production and high-throughput screening.
Recently, a cell-free V. natriegens expression system has been demonstrated which is capable of producing super folder GFP (sfGFP) at concentrations of > 260 μg/mL in 3 h with a T7 promoter9. The overall aim of developing this method was to provide users with a highly accessible, cost-efficient, reproducible, and high-yielding cell-free protein expression system that can be prepared using common lab equipment in a short amount of time. This protocol utilizes 1 L cultures in shake flasks, cell lysis by pulse sonication, and small-scale batch reactions in a 96- or 384-well format to maximize parallelization and screening throughput. A long, sustained protein expression is made possible by supplementation of 3-phosphoglyceric acid (3-PGA) as an energy source8,22,23. Upon successfully completing this protocol, a user will have the capability to express a desired protein or set of proteins in a cell-free format using V. natriegens crude cell extract.
Starting from a glycerol stock, V. natriegens crude cell extracts are prepared from cells harvested at an optical density at 600 nm (OD600) of 1.0. A 1 L culture will yield approximately 2−3 mL of extract, which is sufficient for more than 800 cell-free reactions at 25% crude cell extract. Proteins can be expressed using plasmid DNA, linear DNA, or mRNA template; however, linear DNA template degradation by endogenous nucleases remains a major drawback when using wild type V. natriegens cell-free system9. Starting from V. natriegens cultures, protein useable for downstream applications can be achieved by a single user in 1−2 full days.
Protocol
1. Preparation of V. natriegens Crude Cell Extracts – Bacterial Culture
- Prepare V. natriegens bacterial growth media LB-V2 as per Table 1. Sterilize the growth media by autoclaving. Allow media to reach to room temperature (RT). Store excess media at RT.
CAUTION: Wear proper personal protection equipment (PPE) and consult lab specific instructions when operating an autoclave. - Use a glycerol stock of wild type V. natriegens to inoculate 3 mL of LB-V2 media. Grow overnight at 30 °C while shaking at 225 rpm.
- Wash 1 mL of the overnight culture by centrifugation at 10,600 x g on a benchtop centrifuge for 1 min. Aspirate the supernatant without disturbing the resulting pellet and resuspend in 1 mL of fresh LB-V2 media.
- In an autoclaved 4 L baffled Erlenmeyer flask with sterile cover, add 1 L of fresh LB-V2 growth media. Inoculate using 1 mL of washed overnight culture (1:1,000 dilution ratio). Grow culture at 30 °C while shaking at 225 rpm.
NOTE: V. natriegens cultures can be scaled up or down while maintaining the 1:1,000 dilution ratio. For example, add 250 μL of washed overnight culture to 250 mL of fresh LB-V2 growth media in a 2 L baffled Erlenmeyer flask with sterile cover. - Monitor the culture’s OD600 using a spectrophotometer. When culture reaches OD600 = 1.0 ± 0.2, harvest culture via centrifugation at 3,500 x g for 20 min at 4 °C. Place pellet on ice.
NOTE: V. natriegens grows rapidly, so close monitoring of the culture is necessary. Growth to OD600 = 1.0 should take approximately 1.5−2 h at the 1:1,000 dilution ratio. - Aspirate the supernatant and immediately store the resultant bacterial pellet at -80 °C or directly proceed to cell lysis described in section 2.
NOTE: This step is a good stopping point; however, it is recommended that the pellet be processed immediately or within 1−2 days for best results.
2. Preparation of V. natriegens Crude Cell Extracts – Cell Lysis
- Prepare S30 lysis buffer as per Table 1 in sterile deionized (DI) water and adjust pH to 7.7 using glacial acetic acid.
CAUTION: Glacial acetic acid should be handled with proper PPE when adjusting the pH. - Cool S30 lysis buffer to approximately 4 °C in a refrigerator or on ice before beginning the cell lysis procedure.
NOTE: For a quick cool down, lysis buffer can be placed at -20 °C. Do not allow the buffer to freeze. - Place cell pellets on ice for 10−20 min or until completely thawed. Resuspend all pellets resulting from the same 1 L culture using 10 mL of cold S30 lysis buffer, then transfer suspension to a 50 mL tube. If pellets are not frozen in the freezer at -80 °C in step 1.6, proceed directly to resuspension.
NOTE: Increase the volume of S30 lysis buffer used to initially resuspend pellets as needed. - Centrifuge suspension at 3,500 x g for 10 min at 4 °C. Aspirate the supernatant without disturbing the pellet. Wash the pellet a second time using 10 mL of cold S30 lysis buffer. Place pellet on ice.
- In a cold room, add 500 μL of cold S30 lysis buffer to the pellet in the 50 mL tube. Using a wide-bore pipette tip, resuspend the pellet and carefully transfer the entire pellet suspension to a 2 mL tube.
NOTE: If a wide bore pipette is not available, use a pair of scissors to cut the end of a 1 mL pipette tip to increase the bore for pellet transfer.- Transfer as much pellet as possible without significantly increasing the volume; however, the pellet should be resuspended in enough liquid to be sonicated, as indicated by a homogenous suspension in the 2 mL tube. Do not overfill the 2 mL tube. The suspension should not exceed 1.5 mL; split into multiple tubes if necessary.
- Keep the cell pellet on ice and work in a cold room. Fill a 600 mL beaker with ice and place a 2 mL tube holder on top of the ice.
NOTE: It is helpful to place the tube holder near the side of the beaker, so the pellet suspension will be visible in the ice to monitor sonication progress (see Figure 1). - Vortex suspended pellet in 2 mL tube briefly to homogenize the cells, flick tubes to remove any cells on the bottom of the cap, and place into tube holder with cap open. Lower sonicator tip into the suspension so that it is just under the liquid surface.
- Prepare the sonication set-up as depicted in Figure 2 using a sonicator and probe with a ⅛-inch tip diameter. Input the following settings into the sonicator control: 20 kHz frequency and 50% amplitude, pulse ON time: 10 s, pulse OFF time: 60 s.
- Run the pulse sonication protocol for three cycles. If the total volume of the pellet suspension is > 500 μL, run the pulse sonication protocol six times.
NOTE: Generally, 3−6 pulses are sufficient to lyse V. natriegens pellets; additional pulses may be required depending on sonicator. During sonication, use the adjustment knob platform to move the probe up and down to lyse any pellet that may have settled to the bottom of the tube. The tube can be removed from the holder and briefly vortexed to re-homogenize the suspension in between pulses. See discussion below for desired consistency of sonicated cells.
CAUTION: Wear appropriate hearing protection when sonicator is active. - Following sonication, centrifuge crude cell extract at 16,000 x g for 30−45 min at 4 °C or until lysate is free of any cellular debris as depicted in Figure 3.
- In a cold room, aliquot 50 μL of the resulting supernatants to new 2 mL tubes without disturbing the pellet.
NOTE: Any debris that is transferred accidently into the new 2 mL tubes will drastically reduce extract’s capacity for high yielding protein synthesis.- Optionally, set aside 10 μL of cell extract for post-lysis protein quantification in a separate 2 mL tube (see step 2.13 below).
- Flash freeze crude cell extracts by placing tubes into a tube holder with a dipping string attached as depicted in Figure 4. Submerge tubes into a Dewar containing liquid nitrogen and immediately place in a freezer at -80 °C until use.
CAUTION: Use appropriate PPE for handling liquid nitrogen including lab coat, safety glasses, face shield, cryogen apron, and gloves. - Optionally, quantify the total protein of the cell lysate using 10 μL set aside from step 2.11.1 by diluting sample 1:100 in 1x phosphate-buffered saline (PBS) and using any standard total protein quantification assay such as Bradford assay, bicinchoninic acid assay (BCA), etc.
3. Preparation of Cell-free Reaction Components
- General reaction components
- Prepare working stocks of Mg-glutamate and K-glutamate in 50 mL tubes with sterile DI water at concentrations of 100 mM and 2000 mM, respectively.
- Prepare a working stock of 50% (w/v) polyethylene glycol (PEG)-8000 by adding 100 mL of sterile DI water to a 250 mL beaker. Place a small magnetic stirrer into the beaker. Weigh out 50 g of PEG-8000 and add to the 100 mL of water in the beaker.
- Place the beaker on a heated stir plate set to 100 °C and stir at 250 rpm until PEG-8000 is in solution. Allow PEG-8000 liquid mixture to cool before transferring to 50 mL tubes.
- Energy solution master mix
- Prepare a 5 M solution of KOH by adding 140 g of KOH pellets to 500 mL of sterile DI water.
CAUTION: Prepare this solution in a chemical hood and wear PPE when handling strong bases. The solution will become hot so the bottle cap must be loose to prevent pressure build-up. Allow the KOH solution to reach RT before using. - Prepare a 1750 mM HEPES-KOH buffer by adding 20.85 g of HEPES to a 100 mL bottle. Slowly add sterile DI water until the volume reaches 40 mL. Vortex bottle to dissolve the HEPES. Use the 5 M KOH solution to adjust the pH to 8.0 and then bring the solution volume to 50 mL.
CAUTION: KOH should be handled with proper PPE when adjusting the pH. - Prepare the remaining 10x energy master mix stock components at the concentrations indicated in Table 2 in sterile DI water and place each stock on ice. Thaw the 100 mM ATP, GTP, CTP, and UTP stocks at RT and place on ice.
NOTE: A thermomixer set to 37 °C and 350 rpm can be used to dissolve reagents into solution if necessary. Do not overheat or leave reagents on the thermomixer for an extended period of time. - In a 15 mL tube, add each energy solution master mix component in accordance to the order and volume specified in Table 2. Vortex the solution after each component is added. This will make 5 mL of 10x energy solution master mix.
- Divide the 10x energy solution master mix into 200 μL aliquots in 2 mL tubes. Flash freeze each aliquot as performed in step 2.12. Immediately place them into the -80 °C freezer until use.
- Prepare a 5 M solution of KOH by adding 140 g of KOH pellets to 500 mL of sterile DI water.
- Amino acid master mix
- To prepare fresh 4x amino acid master mix, begin by thawing each amino acid stock at RT and then placing each on ice. Use a vortex and/or thermomixer set at 37 °C and 350 rpm to ensure all amino acid stocks are fully dissolved.
NOTE: Cysteine may not fully dissolve; it can be added to the amino acid master mix as a suspension. Do not overheat or leave reagents on the thermomixer for an extended period of time. - In a 15 mL tube, add the appropriate volume amino acids to sterile DI water so that final concentration of each is 8 mM in the following order: ALA, ARG, ASN, ASP, GLN, GLU, GLY, HIS, IIE, LYS, MET, PHE, PRO, SER, THR, VAL, TRP, TYR, LEU, and CYS. After adding each amino acid, vortex the master mix solution. The volumes listed in Table 2 will make up 2.4 mL of amino acid master mix.
- Divide the 4x amino acid master mix into 200 μL aliquots in 2 mL tubes. Flash freeze each aliquot as performed in step 2.12. Immediately place into a freezer at -80 °C until use.
- To prepare fresh 4x amino acid master mix, begin by thawing each amino acid stock at RT and then placing each on ice. Use a vortex and/or thermomixer set at 37 °C and 350 rpm to ensure all amino acid stocks are fully dissolved.
- Production of reaction-ready plasmid DNA template
NOTE: Cell-free protein expression in this system has been optimized using the super folder green fluorescent protein (GFP) expression vector T7-pJL1-sfGFP (Table of Materials). It is recommended to use this plasmid as a control for cell-free reaction efficiency and the pJL1 backbone for the cloning and expression of other protein sequences. Other plasmid DNA templates can be used; however, it is important to note that transcription is controlled by a T7 promoter sequence and the presence of T7 RNA polymerase. A simple protocol for the large-scale production of any plasmid DNA template from transformed E. coli is described below.- Purify desired vector using a plasmid purification kit as per manufacturer’s instruction (Table of Materials).
NOTE: Concentrating the plasmid DNA template as much as possible is recommended to meet the tight volume constraints of the cell-free reaction. In general, aim for a 750−1500 ng/μL working stock.
- Purify desired vector using a plasmid purification kit as per manufacturer’s instruction (Table of Materials).
- Production of reaction-ready mRNA template
NOTE: This section is optional. Protein expression has been tested using mRNA template generated from the in vitro transcription of the plasmid T7-pJL1-sfGFP by the listed T7 RNA polymerase (Table of Materials).- Prepare mRNA template from plasmid DNA encoding the protein of interest using the in vitro transcription reaction components in Table 3. Incubate reactions for 1 h at 37 °C in a thermocycler.
NOTE: It is recommended to perform 8−10x of these reactions in parallel to generate enough material. - Pool all transcription reactions. Purify using an mRNA purification and concentrator kit as per manufacturer’s instruction (Table of Materials). Elute mRNA template into sterile DI water. Store mRNA template at -80 °C until use.
- Prepare mRNA template from plasmid DNA encoding the protein of interest using the in vitro transcription reaction components in Table 3. Incubate reactions for 1 h at 37 °C in a thermocycler.
4. Performing Cell-free Protein Eexpression Reactions Using V. natriegens Crude Extract
- Cell-free protein expression using plasmid or linear DNA template
- Remove 10x energy solution master mix and 4x amino acid master mix aliquots from the -80 °C freezer, thaw at RT, and place on ice. Remove the T7 RNA polymerase and RNase inhibitor stocks from the -20 °C freezer and place on ice. Thaw DNA template at RT and place on ice.
- Prepare a cell-free reaction master mix as per Table 4 by adding each component in the following order to a 2 mL tube on ice: amino acid master mix, energy solution master mix, Mg-glutamate, K-glutamate, DNA template, PEG-8000, T7 RNA polymerase, and RNase inhibitor. Gently flick the tube after each addition to the master mix.
NOTE: If linear template is to be used for cell-free protein expression, add 5−10x more material as compared to plasmid template to obtain appreciable yields of protein. - Remove V. natriegens crude cell lysate prepared in step 2.12 from the -80 °C freezers and place on ice for 10−20 min until thawed. Add the appropriate volume crude cell extract to the cell-free reaction master mix as per Table 4 and gently mix by flicking or pipetting up and down.
- End-point cell-free protein expression using thermocycler
- Pipette 10 μL of the cell-free reaction master mix to the bottom of a 96- or 384-well PCR plate. In between each transfer to the PCR plate, mix the master mix by flicking the tube gently.
NOTE: Cell-free reaction master mix should be well mixed at all times to maximize reaction reproducibility and cell-free protein expression in all samples.
- Pipette 10 μL of the cell-free reaction master mix to the bottom of a 96- or 384-well PCR plate. In between each transfer to the PCR plate, mix the master mix by flicking the tube gently.
- Briefly centrifuge the plate at 1,000 x g for 10 s to pool any master mix that may have become stuck on the sides of the wells. Seal the wells with a plate adhesive to prevent evaporation and then place the PCR plate into a thermocycler set at 26 °C with a heated lid set at 105 °C.
NOTE: The even heat distribution and heated lid of a thermocycler greatly improves protein expression yields. - Incubate the cell-free reactions for a minimum of 3 h. After incubation, expressed proteins can be purified, quantified, and used for downstream processes.
NOTE: Expressed proteins can be directly quantified in the cell-free reaction using a method of the user’s choice. For example, fluorescent proteins can be quantified using an external standard curve or radioactivity can be measured if using a radiolabeled amino acid in the cell-free reaction. UV-visible spectroscopy or total protein assays are generally not recommended for directly measuring protein production in cell-free reactions without an initial purification. - Alternatively, monitor cell-free protein expression kinetics using a plate reader.
- Pipette 10 μL of the cell-free reaction master mix to the bottom of a black 384-well assay plate with clear glass bottoms. Keep the assay plate on ice or work in a cold room while adding the master mix to ensure that the full kinetic profile is obtained. Seal the wells with a clear plate adhesive to prevent evaporation and then place the assay plate into a plate reader set at 26 °C.
- Incubate the cell-free reactions for 3−6 h while monitoring the appropriate fluorescent excitation/emission wavelengths corresponding to the expressed protein. For example, monitor at Ex/Em = 485 nm/528 nm for sfGFP.
- Cell-free protein expression using mRNA template
- Thaw mRNA template prepared in step 3.5.2 at RT and place on ice.
- Perform step 4.1.1 through step 4.1.6 as previously specified for linear or plasmid DNA template using Table 4 to prepare an alternative cell-free reaction master mix for mRNA template.
5. Calibration of V. natriegens Cell-free Reactions with sfGFP
NOTE: This section is optional. The optimal cell-free reaction ion concentration can vary slightly for each crude extract preparation based on the conditions used for cell lysis. Consider performing the optional cell-free reaction ion calibration protocol described below using sfGFP if reaction yields are significantly lower than expected (concentrations < 1.0 μg/mL).
- Prepare the Mg2+ and K+ ion calibration solutions as specified in Table 5 in sterile DI water from the 100 mM Mg-glutamate and 2,000 mM K-glutamate stocks prepared in step 3.1.1. Place calibration solutions on ice until needed.
- Following the calibration map in Table 6, pipette 1 μL of the Mg-glutamate and 2 μL of K-glutamate ion calibration solutions into the appropriate wells in a 384-well PCR plate.
- The order of operations of this step is as follows: Pipette the Mg-glutamate ion calibration solution into the bottom of each well followed by the K-glutamate ion calibration solution onto the side of well without touching the liquid already present in the wells.
- Seal the wells by placing an adhesive on top of the plate to prevent evaporation while preparing the calibration cell-free reaction master mix. Gently tap the 384-well plate to mix the Mg- and K-glutamate calibration solutions.
NOTE: A repeater pipette is highly recommended for completing this step reproducibly and quickly.
- Prepare the calibration cell-free reaction master mix as specified in Table 5 using T7-pJL1-sfGFP plasmid DNA template in a 2 mL tube. Add each component in the following order to the master mix: amino acid master mix, energy solution master mix, T7-pJL1-sfGFP DNA template, PEG-8000, T7 RNA polymerase, and RNase inhibitor. Gently flick the tube to mix after each component addition.
NOTE: Do not add Mg2+ or K+ to the calibration cell-free master mix. - Remove the adhesive from the plate. Carefully pipette 7 μL of the calibration cell-free reaction master mix on to the sides of the wells without touching the mixed ion calibration solution already present in the wells. Reseal the wells with the adhesive and gently tap the 384-well PCR plate to mix the cell-free master mix with the ion calibration solution.
NOTE: A repeater pipette is highly recommended for completing this step reproducibly and quickly. - Briefly centrifuge the plate 1,000 x g for 10 s to pool any unmixed liquid that may have become stuck on the sides of the wells. Place the 384-well plate into a thermocycler set at 26 °C with a heated lid set at 105 °C.
- Incubate the cell-free reactions for 3 h. After incubation, transfer the contents of each well to a black 384-well assay plate with glass bottoms with a multi-channel pipette and read the fluorescence of sfGFP at Ex/Em = 485 nm/528 nm using a plate reader to determine the ion concentration combination that yields the highest amount of protein.
Representative Results
The described protocol for protein production using V. natriegens cell-free expression system can be executed in 1−2 days by a single user, starting from inoculation of culture to protein available for downstream applications. Preparation of crude cell extracts and master mix stocks comprise a significant portion of this time; however, once prepared, most bulk reagents can be stored long term (Table 7) and used as needed, shortening the time needed to complete the protocol.
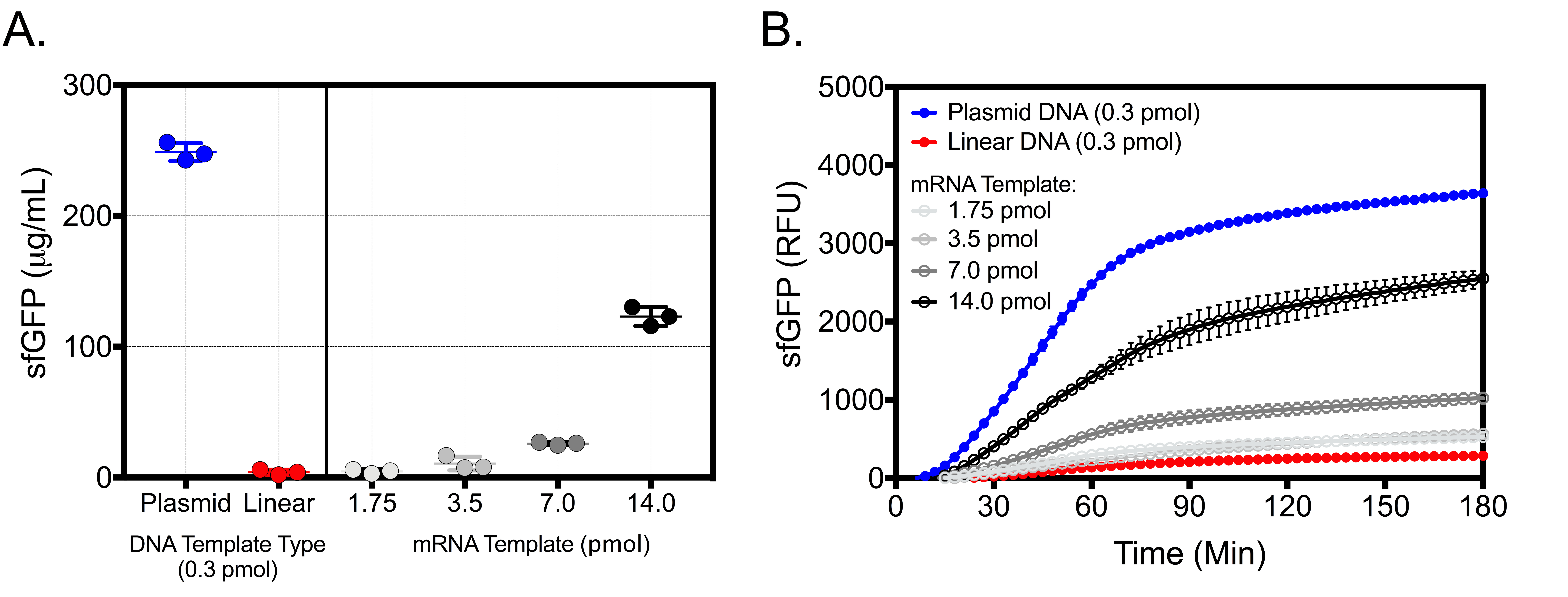
The expression system described is best used with plasmid DNA template or mRNA generated by in vitro transcription reactions. While linear DNA can also be used as template for protein production, it yields significantly lower amount of protein. For example, at optimal reactions conditions at 26 °C, a single 10 μL cell-free reaction can produce > 260 μg/mL of sfGFP in 3 hours using 0.3 pmol of plasmid DNA template or a comparable > 125 μg/mL of sfGFP using 14 pmol of mRNA transcript (Figure 5A,B). However, 0.3 pmol of linear DNA will produce significantly less protein (< 20 μg/mL). For each template type, the majority of the protein will be produced within 1−1.5 h; however, it is recommended that reaction is run for a minimum of 3 hours. Cell-free reaction concentrations of sfGFP were determined via linear regression using a purified sfGFP standard curve measuring fluorescence at Ex/Em = 485 nm/528 nm.
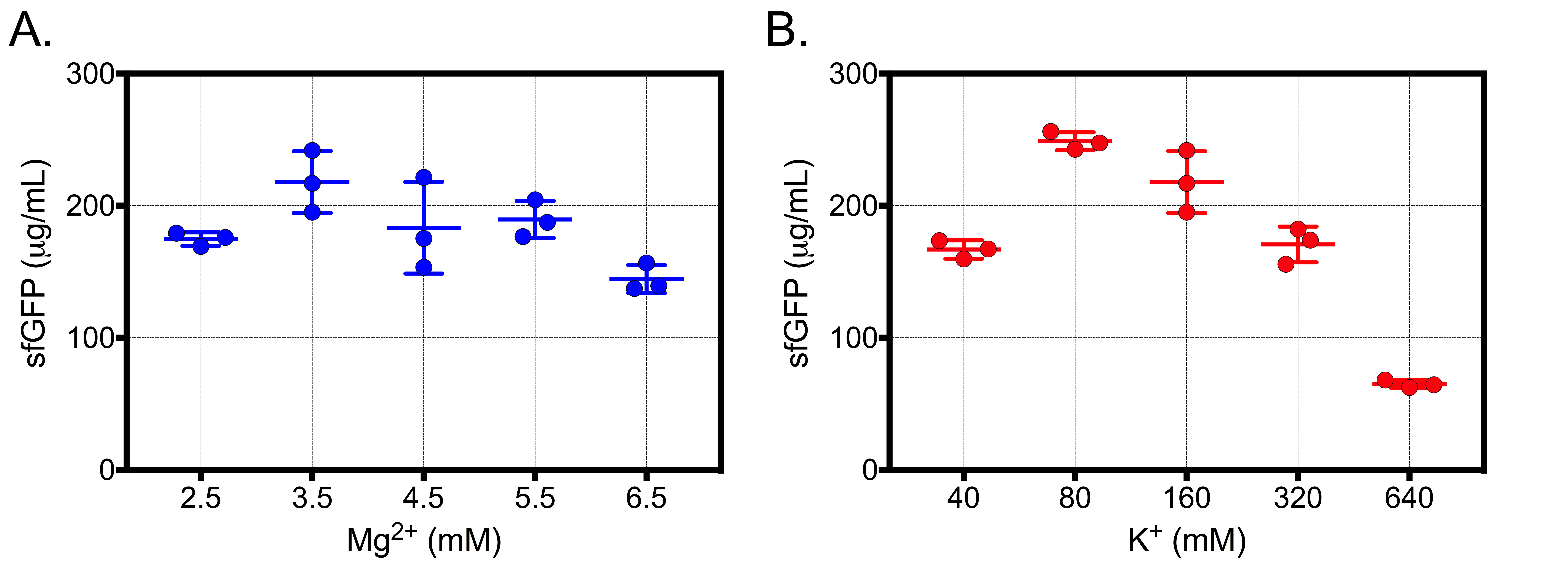
The yield of protein production is significantly affected by the concentration of potassium and magnesium ions (K+ and Mg2+, respectively). Under optimized conditions, it was found that the optimal Mg2+ and K+ ion concentrations will be 3.5 mM and 80 mM, respectively (Figure 6A,B). Deviations from the optimal ion concentrations may result in a decreased capacity for the cell-free expression system to produce protein with appreciable yields. Additional calibration may be necessary if reaction yields are significantly lower than expected. An optional protocol for ion calibration is described in section 5. This allows for some flexibility in compensating for crude cell extract variability incurred from individual cell lysis equipment and conditions.

Figure 1: A close-up view of the beaker and tube holder on the sonication platform. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Sonication equipment setup for the preparation of crude cell extract. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: A view of the pellet after sonication and centrifugation. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Flash freezing dip for extract storage. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative results for V. natriegens cell-free protein synthesis using different template types. (A) endpoint assay of sfGFP production using equimolar concentrations of plasmid and linear DNA template (0.3 pmol) as well as increasing concentrations of mRNA template. Cell-free sfGFP concentration was determined using a standard curve of purified sfGFP measured at Ex/Em = 485 nm/528 nm after 180 minutes of incubation at 26 °C at optimal reaction conditions in 10 μL volumes. (B) Kinetic assay of sfGFP production using equimolar concentrations of plasmid and linear DNA template as well as increasing concentrations of mRNA template. sfGFP measurements were taken every 3 minutes at Ex/EM = 485 nm/528 nm over 180 minutes of total incubation time at optimal reaction conditions in 10 μL volumes. For both endpoint and kinetic assays, samples were blank corrected using cell-free reactions supplemented with all components except template. The mean and standard deviations are shown (n = 3). Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Representative results for ion concentration calibration. (A) V. natriegens cell-free reactions were supplemented with increasing concentrations of Mg2+ and incubated at 26 °C for 180 minutes in 10 μL reactions. The total concentration of K+ was 160 mM for all Mg2+ concentrations. Cell-free sfGFP concentration was determined using a standard curve of purified sfGFP measured at Ex/Em = 485 nm/528 nm. (B) V. natriegens cell-free reactions were supplemented with increasing concentrations of K+ and incubated at 26 °C for 180 min in 10 μL reactions. The total concentration of Mg2+ was 3.5 mM for all K+ concentrations. For both calibrations, samples were blank corrected using cell-free reactions supplemented with all components except template. The mean and standard deviations are shown (n = 3). This figure has been modified from Wiegand et al.9. Reprinted with permission from Wiegand, D.J., Lee, H.H., Ostrov, N., Church, G.M. Establishing a Cell-Free Vibrio natriegens Expression System. ACS Synthetic Biology. 7 (10), 2475−2479 (2018). Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
{kind=link}
| Table 1 A | Preparation of LB-V2 Bacterial Growth Media | ||
| Component | Quantity (g) | Final Concentration (mM) | Final Volume (L) |
| LB Broth (Miller) | 25 | 1 | |
| NaCl | 11.69 | 200 | |
| MgCl2 | 2.20 | 23.1 | |
| KCl | 0.31 | 4.2 | |
| Table 1 B | Preparation of S30A Lysis Buffer | ||
| Component | Quantity (g) | Final Concentration (mM) | Final Volume (L) |
| Tris Solution (pH 8.0) - 1 M (mL)* | 25 | 50 | 0.5 |
| Mg-glutamate | 2.72 | 14 | |
| K-glutamate | 6.10 | 60 | |
| Dithiothreitol (DTT) - 1 M (mL)* | 1 | 2 | |
Table 1: Reagents for the preparation of 1 L of LB-V2 bacterial growth media and 0.5 L of S30A cell lysis buffer. Please click here to download this file in Excel.
| Preparation of 10x Energy Solution Master Mix | ||||
| Component | Stock Concentration (mM) | Final Concentration (mM) | Quantity (µL) | Final Volume (µL) |
| HEPES-KOH pH 8 | 1750 | 500 | 1428.57 | 5000 |
| ATP | 100 | 15 | 750.00 | |
| GTP | 100 | 15 | 750.00 | |
| CTP | 100 | 9 | 450.00 | |
| UTP | 100 | 9 | 450.00 | |
| tRNA from E. coli MRE 600 (mg/mL)* | 100 | 2 | 100.00 | |
| Coenzyme A Hydrate | 200 | 2.6 | 65.00 | |
| NAD | 200 | 3.3 | 82.50 | |
| cAMP | 650 | 7.5 | 57.69 | |
| Folinic Acid | 100 | 0.7 | 35.00 | |
| Spermidine | 1600 | 10 | 31.25 | |
| 3-PGA | 2000 | 300 | 750.00 | |
| Sterile Deionized Water | 49.99 | |||
| Preparation of 4x Amino Acid Master Mix | ||||
| Component | Stock Concentration (mM) | Final Concentration (mM) | Quantity (µL) | Final Volume (µL) |
| ALA | 168 | 8 | 114.3 | 2400 |
| ARG | 168 | 8 | 114.3 | |
| ASN | 168 | 8 | 114.3 | |
| ASP | 168 | 8 | 114.3 | |
| GLN | 168 | 8 | 114.3 | |
| GLU | 168 | 8 | 114.3 | |
| GLY | 168 | 8 | 114.3 | |
| HIS | 168 | 8 | 114.3 | |
| IIE | 168 | 8 | 114.3 | |
| LYS | 168 | 8 | 114.3 | |
| MET | 168 | 8 | 114.3 | |
| PHE | 168 | 8 | 114.3 | |
| PRO | 168 | 8 | 114.3 | |
| SER | 168 | 8 | 114.3 | |
| THR | 168 | 8 | 114.3 | |
| VAL | 168 | 8 | 114.3 | |
| TRP | 168 | 8 | 114.3 | |
| TYR | 168 | 8 | 114.3 | |
| LEU | 140 | 8 | 137.1 | |
| CYS | 168 | 8 | 114.3 | |
| Sterile Deionized Water | 91.4 | |||
Table 2: Reagents for the preparation of 5 mL of 10x energy solution master mix and 2.4 mL of 4x amino acid master mix. Please click here to download this file in Excel.
| T7 RNA Polymerase In Vitro Transcription Reactions | ||||
| Component | Stock (mM) | Final Concentration (mM) | Quantity (µL) 1x Reaction | Quantity (µL) 50x Reactions |
| 10x RNAPol Reaction Buffer | 1.00 | 50 | ||
| ATP | 100 | 0.5 | 0.10 | 5 |
| GTP | 100 | 0.5 | 0.10 | 5 |
| CTP | 100 | 0.5 | 0.10 | 5 |
| UTP | 100 | 0.5 | 0.10 | 5 |
| DNA Template (ng/µL)* | 1000 | 500 | 0.50 | 25 |
| T7 RNA Polymerase | 200 | 2.6 | 2.00 | 100 |
| Rnase Inhibitor, Murine | 200 | 3.3 | 0.50 | 25 |
| Sterile Deionized Water | 15.60 | 780 | ||
| Reaction Volume (µL): | 20 | |||
Table 3: In vitro transcription components for mRNA generation. Please click here to download this file in Excel.
| Cell-free Reaction Master Mix | ||||
| Component | Stock Concentration (mM) | Final Concentration (mM) | Quantity (µL) 1x Reaction | Quantity (µL) 50x Reactions |
| Extract (%)* | 25 | 2.50 | 125.00 | |
| Mg-glutamate | 100 | 3.5 | 0.35 | 17.50 |
| K-glutamate | 2000 | 80 | 0.40 | 20.00 |
| 4x Amino Acid Master Mix | 8.0 | 2 | 2.50 | 125.00 |
| 10x Energy Solution Master Mix | 1.00 | 50.00 | ||
| Plasmid DNA (ng/µL)* | 1000 | 500 | 0.50 | 25.00 |
| 50% PEG-8000 (%)* | 50 | 2 | 0.40 | 20.00 |
| T7 RNA Polymerase | 1.00 | 50.00 | ||
| RNase Inhibitor, Murine | 0.10 | 5.00 | ||
| Sterile Deionized Water | 1.25 | 62.50 | ||
| Reaction Volume (µL): | 10 | |||
| Alternative Cell-free Reaction Master Mix for mRNA Template | ||||
| Component | Stock Concentration (mM) | Final Concentration (mM) | Quantity (µL) 1x Reaction | Quantity (µL) 50x Reactions |
| Extract (%)* | 25 | 2.50 | 125.00 | |
| Mg-glutamate | 100 | 3.5 | 0.35 | 17.50 |
| K-glutamate | 2000 | 80 | 0.40 | 20.00 |
| 4x Amino Acid Master Mix | 8.0 | 2 | 2.50 | 125.00 |
| 10x Energy Solution Master Mix | 1.00 | 50.00 | ||
| mRNA Template (ng/µL)* | 2000 | 4000 | 2.00 | 100.00 |
| 50% PEG-8000 (%)* | 50 | 2 | 0.40 | 20.00 |
| RNase Inhibitor, Murine | 0.10 | 5.00 | ||
| Sterile Deionized Water | 0.75 | 37.50 | ||
| Reaction Volume (µL): | 10 | |||
Table 4: Components of optimized V. natriegens cell-free reaction master mix for DNA template and mRNA template. Please click here to download this file in Excel.
| Mg2+ Calibration | Quantity (µL) 1x Reaction | Quantity (µL) 1x Reaction | Quantity (µL) 100x Reactions | Quantity (µL) 100x Reactions | |
| Final (mM) | Stock | 100 mM Mg-Glu | diH20 | 100 mM Mg-Glu | Deionized H2O |
| 2.5 | 0 | 0.00 | 1.00 | 0 | 100 |
| 3.5 | 1 | 0.10 | 0.90 | 10 | 90 |
| 4.5 | 2 | 0.20 | 0.80 | 20 | 80 |
| 5.5 | 3 | 0.30 | 0.70 | 30 | 70 |
| Reaction Volume (µL): | 10 | ||||
| K+ Calibration | Quantity (µL) 1x Reaction | Quantity (µL) 1x Reaction | Quantity (µL) 100x Reactions | Quantity (µL) 100x Reactions | |
| Final (mM) | Stock | 2000 mM K-Glu | diH20 | 2000 mM K-Glu | Deionized H2O |
| 40 | 30 | 0.15 | 1.85 | 12 | 148 |
| 80 | 70 | 0.35 | 1.65 | 28 | 132 |
| 160 | 150 | 0.75 | 1.25 | 60 | 100 |
| 320 | 310 | 1.55 | 0.45 | 124 | 36 |
| Reaction Volume (µL): | 10 | ||||
| Ion Calibration Cell-free Reaction Master Mix | |||||
| Component | Stock Concentration (mM) | Final Concentration (mM) | Quantity (µL) 1x Reaction | Quantity (µL) 50x Reactions | |
| Extract (%)* | 25 | 2.50 | 125.00 | ||
| Mg-glutamate | Variable | Variable | 1.00 | 50.00 | |
| K-glutamate | Variable | Variable | 2.00 | 100.00 | |
| 4x Amino Acid Master Mix | 8.0 | 2 | 2.50 | 125.00 | |
| 10x Energy Solution Master Mix | 1.00 | 50.00 | |||
| Plasmid DNA (ng/µL)* | 1000 | 250 | 0.25 | 12.50 | |
| 50% PEG-8000 (%)* | 50 | 2 | 0.40 | 20.00 | |
| T7 RNA Polymerase | 1.00 | 50.00 | |||
| RNase Inhibitor, Murine | 0.10 | 5.00 | |||
| Sterile Deionized Water | 0.00 | 0.00 | |||
| Reaction Volume (µL): | 10 | ||||
Table 5: Ion concentration calibration master mixes. Please click here to download this file in Excel.
| CF Ion Calibration Map | 40 mM K+ | 80 mM K+ | 160 mM K+ | 320 mM K+ | |||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| 2.5 mM Mg2+ | A | ||||||||||||
| 3.5 mM Mg2+ | B | ||||||||||||
| 4.5 mM Mg2+ | C | ||||||||||||
| 5.5 mM Mg2+ | D | ||||||||||||
Table 6: Ion calibration map. Please click here to download this file in Excel.
| Storage Conditions and Shelf Lives of CF Components | ||
| Component | Storage Location | Shelf Life |
| Sterile LB-V2 Growth Media | 4 °C | 3-6 months |
| V. natriegens Crude Cell Extract | -80 °C | 1-3 weeks |
| 100 mM Mg-Glutamate | Room Temp | 6 months |
| 2000 mM K-Glutamate | Room Temp | 6 months |
| 50% PEG-8000 | Room Temp | 6 months |
| 10x Energy Solution Master Mix | -80 °C | 3-6 months |
| 4x Amino Acid Master Mix | -80 °C | 3-6 months |
| Plasmid/Linear DNA Template | -20 °C | 6-12 months |
| mRNA Template | -80 °C | 3-4 weeks |
Table 7: Cell-free storage conditions and shelf lives. Please click here to download this file in Excel.
Discussion
This protocol has been optimized for wild-type V. natriegens and bacterial growth media comprised of LB supplemented with V2 salts (Table of Materials). Other strains of V. natriegens can be similarly cultured to generate crude cell extracts for cell-free reactions; however, their use requires additional optimization of this protocol. Additionally, this cell-free protein expression system has been optimized using 3-phosphoglyceric acid (3-PGA) as the primary energy regeneration source. Other energy regeneration sources may be used; however, optimization of reagents and calibration will likely be required to obtain high yielding protein expression10,11.
Specific attention to several critical steps in this protocol will ensure maximal extract productivity for high-yielding cell-free protein production. First, crude cell extract must be prepared from V. natriegens cell cultures harvested in a mid-exponential phase of growth; protein yield is maximal when cultures reach an OD600 = 1.0 ± 0.2. While cell-free protein production is possible from cells harvested at a range of optical densities, we have previously found that cells harvested in an exponential state of growth yield significantly more protein9. The observed effects of optical density on crude cell extract performance are consistent with those reported for other cell-free expression systems derived from cells grown in batch culture conditions1. Because V. natriegens grows at a rapid rate, it is critical to closely monitor cultures’ optical densities. In general, it is expected that V. natriegens cultures should reproducibly reach an OD600 of 1.0 within 1−1.5 h using this protocol; however, individual growth conditions such as the use of baffled versus non-baffled flasks, which affects aeration, or air versus water incubation, which affects the rate and stability of incubation temperature, may alter growth time. Furthermore, it is generally recommended to culture at least 250 mL of V. natriegens in a 1 L baffled flask to ensure a large cell pellet at harvest for easy manipulation and transfer. This greatly improves the success of crude cell extract preparation as well as increases the total volume of extract produced from a pellet. When using smaller scale preparation, culture conditions and reagents can be adjusted appropriately. For large-scale fermentation, further optimization of culture conditions may be required. Finally, to ensure high protein yield, it is critical that cell pellets are processed immediately after harvesting, or within 1−2 days of storing at -80 °C.
The proper lysis of the cell pellet by pulse sonication is critical to the success of cell-free protein expression and is often the most difficult aspect of this protocol for new users. Typically, a well lysed pellet will yield a significant volume of liquid extract that is free of debris. The extract should be slightly viscous but can be easily pipetted when aliquoting into storage tubes before flash freezing in liquid nitrogen. Figure 3 depicts a representation of a well lysed pellet (Figure 3A) in comparison to a poorly lysed pellet (Figure 3B) after the post lysis centrifugation step. A major indication of complete cell lysis is a crude cell extract total protein concentration > 20 mg/mL as determined by a total protein assay (step 2.13). Over-sonication or excessive heating of the crude cell extract will damage the cellular machinery, which cannot be determined without performing a cell-free reaction. Thus, it is highly beneficial to test extract efficiency with a control reaction before dedicating significant time and effort to downstream protein expression applications. While additional optimization may be necessary for different sonication equipment, the pulse sonication steps described have been highly reproducible in our hands.
The use of linear DNA template derived from PCR amplification, restriction enzyme digest, or commercial gene synthesis can significantly increase the capacity for high-throughput and rapid protein production in cell-free expression systems24. While protein production from PCR amplified linear template has been demonstrated, the yield of these reactions are approximately 13.5-fold lower compared to reactions using plasmid DNA template at equimolar ratios9. This is primarily due to the instability of the linear DNA template which is likely degraded by endogenous nucleases present in V. natriegens crude cell extract. While lambda phage protein GamS has been previously used to protect Linear DNA template24,25, it was found to be incompatible with V. natriegens extracts9. Additionally, while increasing the concentration of linear DNA template may allow for a higher protein yield, its fast degradation in crude cell extract will still be a major problem.
A solution to overcoming linear DNA template degradation may be to supplement cell-free reactions with mRNA template generated from in vitro transcription of linear DNA. On the other hand, the use of an RNase inhibitor offers significant protection against mRNA transcript degradation and appreciable protein yields can be obtained in the 10 μL cell-free reaction format (Figure 5A,B). Cloning of linear DNA into a circular template through TA ligation, TOPO cloning, Golden Gate assembly, or other recombination methods may be used to circumvent template degradation. Nevertheless, further approaches for inhibition of nuclease activity will be necessary for efficient protein expression using linear DNA template.
To date, several different approaches have been proposed for preparation of crude extract for cell-free protein expression9,10,11,26. In developing this protocol, we sought to maximize user accessibility, reduce overall cost, and minimize time-consuming steps. For example, a high protein yield is achieved using a simple two-step sonication-centrifugation process, and does not require cell homogenizers, lengthy dialysis steps or run-off reaction. It is simple to execute in a short period of time and does not require high level of laboratory expertise. Thus, it can help facilitate cell-free expression as a standard for translational academic research and industrial process design.
This protocol expands the toolkit available for investigation and utility of V. natriegens, a non-model organism with unique biological properties. Higher protein yield can be achieved by employing semi- or fully-continuous cell-free reactions, to allow for energy regeneration, resupplying of amino acids, and the removal of waste products3,5,27. Furthermore, the engineering of wild-type V. natriegens to produce DNAse- or RNAse-deficient strains, removal of deleterious and competing metabolic pathways, and expression of additional tRNAs could greatly enhance the production of proteins in this system28,29. As we unravel the biology underlying its rapid growth, further development of V. natriegens cell-free systems may accelerate bioproduction capabilities and enable robust expression of therapeutic peptides, small molecules, and synthetic materials.
Acknowledgements
This work was funded by the National Institute of General Medical Sciences 1U01GM110714-01 and Department of Energy DE-FG02-02ER63445. The authors would like to thank Dr. Richard Kohman, Dr. Jenny Tam, and Dr. Edgar Goluch for helpful advice on constructing the protocol section of this manuscript.
Materials
| Name | Company | Catalog Number | Comments |
| 15 mL Tubes | Corning | 352196 | |
| 2 mL Tubes | Eppendorf | 22363352 | |
| 384-well Black Assay Plates | Corning | 3544 | |
| 384-well PCR Plates | Eppendorf | 951020702 | |
| 50 mL Tubes | Corning | 352070 | |
| 96-well PCR Plates | Eppendorf | 30129300 | |
| Adenosine 3',5'-cyclic monophosphate sodium salt monohydrate | Sigma | A6885 | |
| Adenosine 5'-triphosphate - 100 mM | NEB | N0450L | |
| Applied Biosystems Veriti 384-well Thermocycler | ThermoFisher | 4388444 | |
| Assay Plate Adhesives | BioRad | MSB1001 | |
| β-Nicotinamide adenine dinucleotide hydrate (NAD) | Sigma/Roche | 10127965001 | |
| Coenzyme A hydrate | Sigma | C4283 | |
| Cytidine 5'-triphosphate - 100 mM | NEB | N0450L | |
| D-(-)-3-Phosphoglyceric acid disodium salt (3-PGA) | Sigma | P8877 | |
| Dewar Flask - 4L | ThermoScientific | 10-194-100C | |
| DL-Dithiothreitol solution - 1 M | Sigma | 42816 | |
| Folinic acid calcium salt hydrate | Sigma | 47612 | |
| Glacial Acetic Acid | Sigma | A6283 | |
| Guanosine 5'-triphosphate - 100 mM | NEB | N0450L | |
| HEPES | Sigma | H3375 | |
| L-Glutamic acid hemimagnesium salt tetrahydrate | Sigma | 49605 | |
| L-Glutamic acid potassium salt monohydrate | Sigma | G1149 | |
| LB Broth (Miller) | Sigma | L3522 | |
| Magnesium chloride (MgCl2) | Sigma | M8266 | |
| Plasmid pJL1-sfGFP | Addgene | 69496 | |
| Plasmid Plus Maxi kit | Qiagen | 12963 | |
| Poly(ethylene glycol) (PEG)-8000 | Sigma | 89510 | |
| Potassium chloride (KCl) | Sigma | P9333 | |
| Potassium hydroxide Pellets | Sigma/Roche | 1050121000 | |
| Q125 Sonicator and CL-18 probe with a ⅛-inch tip | Qsonica | 4422 | |
| RNA Clean and Concentrator Kit | Zymo | R1013 | |
| RNase Inhibitor, Murine | NEB | M0314 | |
| RTS Amino Acid Sampler | Biotechrabbit | BR1401801 | |
| Sodium chloride (NaCl) | Sigma | S7653 | |
| Spermidine | Sigma | S0266 | |
| T7 RNA Polymerase | NEB | M0251 | |
| Tris Solution (pH 8.0) - 1 M | Invitrogen | AM9856 | |
| tRNA from E. coli MRE 600 | Sigma/Roche | 10109541001 | |
| Uridine 5'-triphosphate - 100 mM | NEB | N0450L | |
| Vibrio natriegens (Wild-type) Lyophilized Stock | ATCC | 14048 |
References
- Kwon, Y. C., Jewett, M. C. High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Scientific Reports. 5, 8663 (2015).
- Schoborg, J. A., et al. A cell-free platform for rapid synthesis and testing of active oligosaccharyltransferases. Biotechnology and Bioengineering. 115 (3), 739-750 (2018).
- Caschera, F., Noireaux, V. Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie. 99, 162-168 (2014).
- Henrich, E., Hein, C., Dötsch, V., Bernhard, F. Membrane protein production in Escherichia coli cell-free lysates. FEBS Letters. 589 (15), 1713-1722 (2015).
- Li, J., Wang, H., Kwon, Y. C. Establishing a high yielding streptomyces‐based cell‐free protein synthesis system. Biotechnology and Bioengineering. 114 (6), 1343-1353 (2017).
- Moore, S. J., Lai, H. E., Needham, H., Polizzi, K. M., Freemont, P. S. Streptomyces venezuelae TX-TL--a next generation cell-free synthetic biology tool. Biotechnology Journal. 12 (4), 1600678 (2017).
- Wang, H., Li, J., Jewett, M. C. Development of a Pseudomonas putida cell-free protein synthesis platform for rapid screening of gene regulatory elements. Synthetic Biology. 3 (1), ysy003 (2018).
- Kelwick, R., Webb, A. J., MacDonald, J. T., Freemont, P. S. Development of a Bacillus subtilis cell-free transcription-translation system for prototyping regulatory elements. Metabolic Engineering. 38, 370-381 (2016).
- Wiegand, D. J., Lee, H. H., Ostrov, N., Church, G. M. Establishing a Cell-Free Vibrio natriegens Expression System. ACS Synthetic Biology. 7 (10), 2475-2479 (2018).
- Des Soye, B. J., Davidson, S. R., Weinstock, M. T., Gibson, D. G., Jewett, M. C. Establishing a High-Yielding Cell-Free Protein Synthesis Platform Derived from Vibrio natriegens. ACS Synthetic Biology. 7 (9), 2245-2255 (2018).
- Failmezger, J., Scholz, S., Blombach, B., Siemann-Herzberg, M. Cell-Free Protein Synthesis From Fast-Growing Vibrio natriegens. Frontiers in Microbiology. 9, 1146 (2018).
- Moore, S. J., MacDonald, J. T., Wienecke, S. Rapid acquisition and model-based analysis of cell-free transcription-translation reactions from nonmodel bacteria. Proceedings of the National Academy of Sciences of the United States of America. 115 (19), E4340-E4349 (2018).
- Eagon, R. G. Pseudomonas natriegens, a marine bacterium with a generation time of less than 10 minutes. Journal of Bacteriology. 83, 736-737 (1962).
- Weinstock, M. T., Hesek, E. D., Wilson, C. M., Gibson, D. G. Vibrio natriegens as a fast-growing host for molecular biology. Nature Methods. 13 (10), 849-851 (2016).
- Lee, H. H., et al. Vibrio natriegens, a new genomic powerhouse. bioRxiv. , 058487 (2016).
- Lee, H. H., Ostrov, N., Gold, M. A., Church, G. M. Recombineering in Vibrio natriegens. bioRxiv. , 130088 (2017).
- Schleicher, L., et al. Vibrio natriegens as Host for Expression of Multisubunit Membrane Protein Complexes. Frontiers in Microbiology. 9, 2537 (2018).
- Fernández-Llamosas, H., Castro, L., Blázquez, M. L., Díaz, E., Carmona, M. Speeding up bioproduction of selenium nanoparticles by using Vibrio natriegens as microbial factory. Scientific Reports. 7 (1), 16046 (2017).
- Aiyar, S. E., Gaal, T., Gourse, R. L. rRNA promoter activity in the fast-growing bacterium Vibrio natriegens. Journal of Bacteriology. 184 (5), 1349-1358 (2002).
- Hoffart, E., et al. High substrate uptake rates empower Vibrio natriegens as production host for industrial biotechnology. Applied and Environmental Microbiology. 83, e01614-e01617 (2017).
- Long, C. P., Gonzalez, J. E., Cipolla, R. M., Antoniewicz, M. R. Metabolism of the fast-growing bacterium Vibrio natriegens elucidated by 13C metabolic flux analysis. Metabolic Engineering. 44, 191-197 (2017).
- Shin, J., Noireaux, V. Efficient cell-free expression with the endogenous E. Coli RNA polymerase and sigma factor 70. Journal of Biological Engineering. 4, 8 (2010).
- Sitaraman, K., et al. A novel cell-free protein synthesis system. Journal of Biotechnology. 110 (3), 257-263 (2004).
- Sun, Z. Z., Yeung, E., Hayes, C. A., Noireaux, V., Murray, R. M. Linear DNA for rapid prototyping of synthetic biological circuits in an Escherichia coli based TX-TL cell-free system. ACS Synthetic Biology. 3 (6), 387-397 (2014).
- Seki, E., Matsuda, N., Yokoyama, S., Kigawa, T. Cell-free protein synthesis system from Escherichia coli cells cultured at decreased temperatures improves productivity by decreasing DNA template degradation. Analytical Biochemistry. 377 (2), 156-161 (2008).
- Failmezger, J., Rauter, M., Nitschel, R., Kraml, M., Siemann-Herzberg, M. Cell-free protein synthesis from non-growing, stressed Escherichia coli. Scientific Reports. 7 (1), 16524 (2017).
- Shirokov, V. A., Simonenko, P. N., Biryukov, S. V., Spirin, A. S. Continuous-Flow and Continuous-Exchange Cell-Free Translation Systems and Reactors. Cell-Free Translation Systems. , 91-107 (2002).
- Michel-Reydellet, N., Woodrow, K., Swartz, J. Increasing PCR fragment stability and protein yields in a cell-free system with genetically modified Escherichia coli extracts. Journal of Molecular Microbiology and Biotechnology. 9 (1), 26-34 (2005).
- Swartz, J. R. Expanding biological applications using cell-free metabolic engineering: An overview. Metabolic Engineering. 50, 156-172 (2018).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved