Mass Spectrometry-Based Proteomics Analyses Using the OpenProt Database to Unveil Novel Proteins Translated from Non-Canonical Open Reading Frames
In This Article
Summary
OpenProt is a freely accessible database that enforces a polycistronic model of eukaryotic genomes. Here, we present a protocol for the use of OpenProt databases when interrogating mass spectrometry datasets. Using OpenProt database for analysis of proteomic experiments allows for discovery of novel and previously undetectable proteins.
Abstract
Genome annotation is central to today's proteomic research as it draws the outlines of the proteomic landscape. Traditional models of open reading frame (ORF) annotation impose two arbitrary criteria: a minimum length of 100 codons and a single ORF per transcript. However, a growing number of studies report expression of proteins from allegedly non-coding regions, challenging the accuracy of current genome annotations. These novel proteins were found encoded either within non-coding RNAs, 5' or 3' untranslated regions (UTRs) of mRNAs, or overlapping a known coding sequence (CDS) in an alternative ORF. OpenProt is the first database that enforces a polycistronic model for eukaryotic genomes, allowing annotation of multiple ORFs per transcript. OpenProt is freely accessible and offers custom downloads of protein sequences across 10 species. Using OpenProt database for proteomic experiments enables novel proteins discovery and highlights the polycistronic nature of eukaryotic genes. The size of OpenProt database (all predicted proteins) is substantial and need be taken in account for the analysis. However, with appropriate false discovery rate (FDR) settings or the use of a restricted OpenProt database, users will gain a more realistic view of the proteomic landscape. Overall, OpenProt is a freely available tool that will foster proteomic discoveries.
Introduction
Over the past decades, mass spectrometry (MS-)based proteomics has become the golden technique to decipher proteomes of eukaryotic cells1,2,3,4,5. This method relies on current genome annotations to generate a reference protein sequence database that outlines the scope of possibilities6,7,8. However, genome annotations hold arbitrary criteria for ORF annotation, such as a minimum length of 100 codons and a single ORF per transcript9,10. An increasing number of studies challenge the current annotation model and report discoveries of unannotated functional ORFs in eukaryotic genomes8,11,12,13,14. These novel proteins are found encoded in allegedly non-coding RNAs, in the 5' or 3' untranslated regions (UTR) of mRNAs, or overlapping the canonical coding sequence (cCDS) in an alternative frame. Although most of these discoveries have been serendipitous, they demonstrate the caveats of current genome annotations and the polycistronic nature of eukaryotic genes8.
Here, we highlight the use of OpenProt databases for MS-based proteomics. OpenProt is the first database to hold a polycistronic annotation model for eukaryotic transcriptomes. It is freely available at www.openprot.org15. A proportion of these predicted ORFs would be random and non-functional, which is why OpenProt cumulates experimental and functional evidence to increase confidence. Experimental evidence include protein expression (by MS) and translation evidence (by ribosome profiling)15. Functional evidence include protein orthology (with an In-Paranoid like approach) and functional domain prediction15.
OpenProt offers the possibility to download several databases, from containing only well-supported proteins to custom-made databases. Here, we will present a pipeline for the use of OpenProt databases and will offer insights into which database to choose considering the experimental aim. The proteomics analysis pipeline presented here is supported by the Galaxy framework as it is open-access and easy-to-use, but the databases can work with any workflow16,17,18. We will also present how to use the OpenProt website for gathering further information on novel proteins detected by MS. Using OpenProt databases will provide a more exhaustive view of the proteomic landscape and will foster proteomics and biomarkers discoveries in a more systematic way than current methods.
This protocol highlights the use of OpenProt databases15 when interrogating MS datasets; it will not review the design of the experiment itself, which has been thoroughly reviewed elsewhere20,21,22. In an effort to remain fully open-source, the protocol is freely available (Supplementary Material S1-S4). For easier reading, all terms used in OpenProt and hereby throughout this protocol are defined in Table 1.
Protocol
1. OpenProt database download
NOTE: Custom databases based on RNA-seq data for example can also be obtained and the procedure is detailed in the second section of this protocol. If a custom database is needed, please skip to the next section.
- Go to the OpenProt website: www.openprot.org and open the Downloads page using the link from the top page menu.
- Click on the species of interest based on the analyzed experimental data.
- Click on the protein type desired.
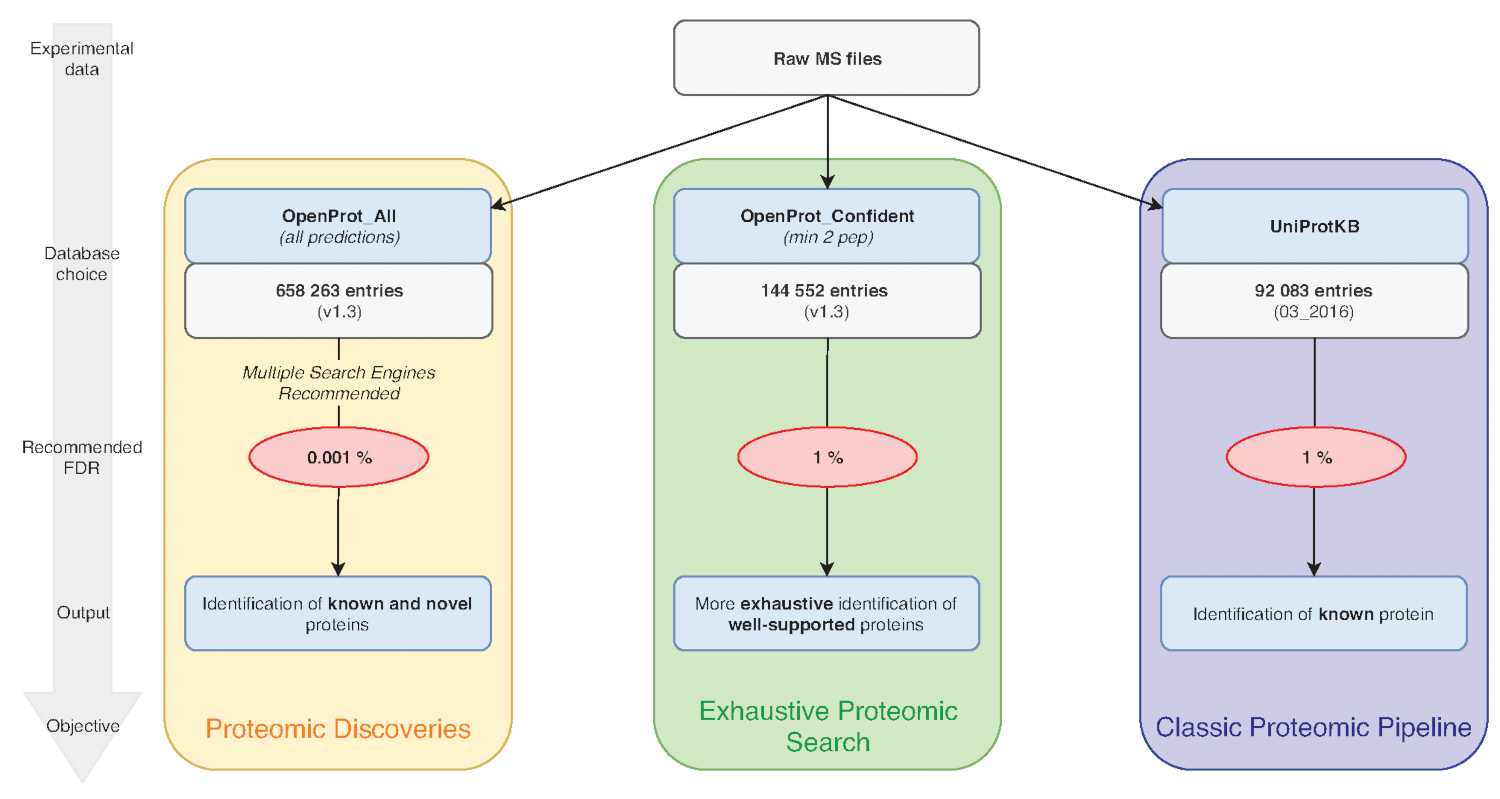
NOTE: OpenProt offers three classifications: RefProt, Isoforms and AltProt. As shown in Figure 1, this parameter will vary based on the research objective.- Click on RefProt alone to generate files containing only known proteins.
- Click on AltProt and Isoforms to generate files containing only novel proteins - either novel isoforms of known proteins (Isoforms) or coded by an alternative ORF (AltProts). Please note that OpenProt enforces a minimum ORF length of 30 codons15.
- Click on AltProts, Isoforms and RefProts to generate files containing all protein types present in the OpenProt database - known and novel proteins.
- If available, click on the annotation from which protein sequences are drawn.
NOTE: OpenProt offers a more exhaustive proteomic landscape by combining multiple annotations. Transcriptome annotations have a minimal overlap; thus, the selected annotation can substantially affect the visualized proteomic profile15,23. - Click on the level of supporting evidence necessary for protein consideration. As shown in Figure 1, this parameter will vary based on the research objective.
- Click on minimum of two unique peptides detected to generate files containing only the most confident proteins.
NOTE: A criterion of two unique peptides is currently considered a gold standard in proteomics for protein expression. If the experimental aim is to detect known and well-supported proteins, the use of this parameter is recommended. - Click on minimum of one unique peptides detected to generate files containing proteins that have already been seen at least once among the mass spectrometry experiments re-analyzed by OpenProt.
NOTE: This allows for consideration of the shorter length of AltProts and the probability that some of them may contain only one unique tryptic peptide8,11. - Click on all predicted to generate files containing all of OpenProt predictions.
NOTE: This setting is recommended only if the experimental aim is to discover novel proteins (Figure 1). The subsequent substantial increase in the search space calls for an adapted analysis pipeline as discussed below7,15.
- Click on minimum of two unique peptides detected to generate files containing only the most confident proteins.
- Click on the desired file format to download. For proteomic analyses, choose the Fasta (protein) file. The readme file contains all necessary information on the file format.
2. Custom OpenProt database download
NOTE: This section details how to obtain a custom database. If no custom database is needed, skip to the next section.
- Go to the OpenProt website (www.openprot.org) and open the Search page using the link from the top page menu.
- Click on the species of interest based on the experimental data analyzed.
- Enter a list of genes or transcripts of interest.
- When using a list of genes, enter it in the Gene query box.
- When using a list of transcripts, enter it in the Transcript query box.
- Tick any box that applies to the desired database.
- Do not click on any box to obtain a table containing all types of protein supported by OpenProt: RefProt, Isoforms and AltProts.
- Click on Show only proteins with experimental evidence to obtain a table containing all types of proteins (RefProts, Isoforms and AltProts) that have been detected at least once by MS and/or for which translation evidence has been collected from ribosome profiling data.
- Similarly, click on Show only proteins detected by MS or on Show only proteins detected by ribosome profiling to obtain a table containing all types of proteins that have been detected at least once by MS or by ribosome profiling respectively.
- Click on Show only AltProts or on Show only isoforms to obtain a table containing only AltProts or only Isoforms respectively.
- Click on both Show only AltProts and Show only Isoforms to obtain a table containing both types of proteins.
NOTE: All combinations of filters are possible.
- Once all desired parameters are set, click on Search. The table output will appear below the search query fields.
- Click on the Download Fasta button at the right top corner of the output table. This will generate a Fasta file containing all proteins resulting from the queried list of genes or transcripts.
- Please note that for computational reasons, OpenProt holds a maximum of 2,000 elements to be queried (genes or transcripts) at a time. In the event of a list above that limit, several fasta can be generated and then concatenated (as detailed below); or simply download the whole OpenProt database and filter the obtained file as desired.
- Bin the whole list of genes or transcripts into sub-lists of 2,000 entries or less. For each sub-list, download a Fasta file as described above (step 3.3 to 3.6).
- Log in to the European Galaxy instance (or any other instance where proteomics tools are available), https://usegalaxy.eu/.
- Create a new history and import all of the downloaded OpenProt databases (one per sub-list of genes or transcripts) by clicking on the upload logo at the left top of the screen.
- Use the Fasta Merge Files and Filter Unique Sequences tool developed by the GalaxyP developers (https://github.com/galaxyproteomics/). Select the Merge all Fasta option and input all of the imported OpenProt databases.
NOTE: Each tool can be searched by using the query box on the left side of the screen - Select the accession only option to assess sequence unicity and copy the OpenProt identifier parse rule (>(.*)\|), then click on Execute.
- Note that all files have been concatenated into a unique Fasta file with no redundancy that now appears in the history panel on the right side of the screen. This constitutes the working database.
3. Database handling
NOTE: From now on, the Galaxy platform will be used, but the same principles can be applied to other proteomic software.
- Log in to the European Galaxy instance (or any other instance where proteomics tools are available), https://usegalaxy.eu/.
- Create a new history and import the downloaded OpenProt database by clicking on the upload logo at the left top of the screen.
- Go to the workflow page and import the Database Handling workflow (Supplementary Material S1) by clicking on the upload logo at the left top of the middle panel.
- Click on Run the workflow and select the imported OpenProt database as input.
NOTE: This workflow will append the CRAPome repository to the OpenProt fasta and generate decoy sequences (reverse sequences)24. If a shuffle decoy list is desired, it can be done by changing this parameter on the DecoyDatabase tool. - Rename the obtained Fasta file to something meaningful. The database is ready to be used for proteomics analyses.
4. Mass spectrometry file preparation
NOTE: Most of the proteomics tools available on Galaxy instances use the mzML format, and peptide search engines prefer data in centroid mode.
- Open the freely available MSConvert tool from the ProteoWizard suite and upload the data file to be analyzed25.
- Choose the directory for the output and the desired file format to mzML.
- Set a peak picking filter using the wavelet based algorithm (CWT) on MS1 and MS2 levels, and start the conversion26.
5. Peptide and protein identification/quantification
NOTE: This part of the pipeline uses tools from the OpenMS suite, a versatile and easy-to-use framework18.
- Log in to the European Galaxy instance (or any other instance where proteomics tools are available), https://usegalaxy.eu/.
- Create a new history and transfer the previously created database (step 3.5) to this new history with a drag-and-drop.
- Import the transformed mzML data file (step 4.3) by clicking on the Upload logo at the left top of the screen.
- Go to the workflow page and import the desired workflow by clicking on the upload logo at the left top of the middle panel.
NOTE: MS experiments are differently designed based on the desired final output. Workflows are provided here for two frequent designs: protein identification and protein quantification based on stable isotope labeling (SIL). However, the Galaxy instance contains many other tools that will support other types of proteomic analyses27,28.- For a protein identification design, import the workflow provided in Supplementary Material S2. When using this workflow, please do not use the zlip compression when converting your files (step 4.2)
- For a protein quantification based on stable isotope labeling design, import the workflow provided in Supplementary Material S3.
- Select run the workflow and review the different parameters.
- Select the imported mzML data file as input, and the previously created database (step 3.5) as the database Fasta file.
- Since the workflow uses the X!Tandem search engine, import the X!Tandem default configuration file (provided in Supplementary Material S4)29 by clicking on the upload logo at the left top of the screen.
- The workflow uses multiple search engines (MS-GF+ and X!Tandem). Append other search engines or choose a single one simply by adding or removing the tools from the workflow30,31.
NOTE: Using multiple search engines is recommended as it increases sensibility and sensitivity of the analysis32. - In order to account for the substantial increase in size when using the whole OpenProt database, use a stringent FDR15. By default, the provided workflow is set for a 0.001% FDR, adequate for the use of the whole OpenProt database. For other databases, this can be edited to any desired value.
NOTE: Be sure to adapt the parameters of the different tools depending on the mass spectrometer used and the experimental protocol (precursor ion and fragment error, fixed and variable modifications, used enzyme, etc.).
- Optionally, download output for each step of the workflow for storage or quality control analysis by clicking on the chosen step from the history panel, then clicking on the Save logo that will appear underneath.
6. Quality control
NOTE: Because MS-based proteomics is the result of a complex process where each step needs to be optimized to produce reproducible results, quality control is a necessary procedure in the workflow33.
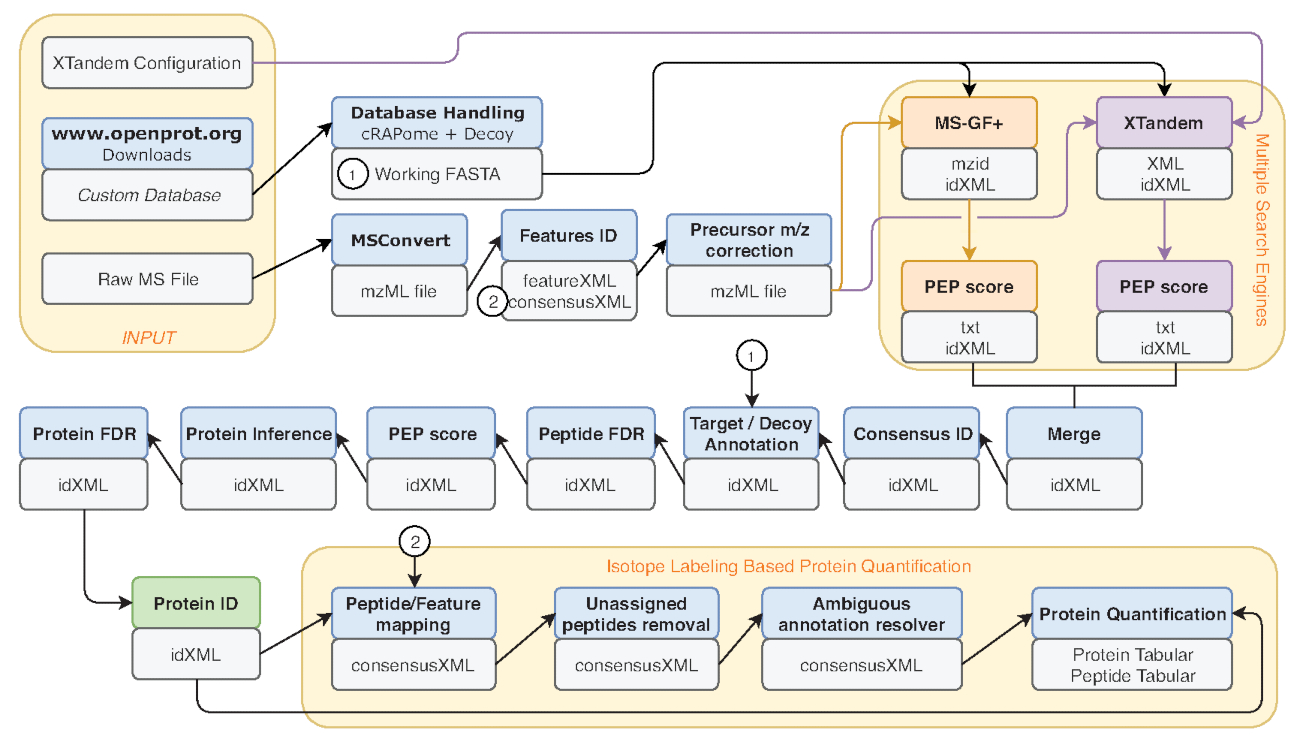
- Several metrics are common benchmark of performance, such as the number of peptide-spectrum matches (PSM), the number of identified peptides and proteins. Run the File Info tool on the IDFilter output (indicated in green in Figure 2) to provide such metrics.
- Although not applicable to every identification, especially with large datasets, reports of novel proteins should always be carefully evaluated. Inspection of the protein score, the sequence coverage, and the spectra supporting the finding is of vital importance. Use the TOPPview tool from the OpenMS framework to do this; it is freely available and well documented18,34,35.
7. OpenProt database mining
NOTE: Once a confident identification of a novel protein predicted by OpenProt (accession numbers starting with IP_ for AltProts and II_ for novel Isoforms) has been made, more biological information can be gathered from the OpenProt website15.
- Go to the OpenProt website: www.openprot.org and open the Search page using the link at the top page menu.
- Click on the species of interest (same as the one in which the protein was identified) and enter the protein accession number in the Protein query box.
- Click on search and a table containing basic information on the queried protein will appear. The table features: the protein length (in amino acid), its molecular weight (kDa) and isoelectric point, supporting experimental evidence by MS or ribosome profiling (Translation Evidence, TE), and functional predictions such as predicted domains and protein orthology (across the 10 species supported by OpenProt, v1.3). The table also contains information about the related gene and transcript and the localization of the protein within the transcript.
- Click on the Details link to gather further information. The newly opened page contains a genome browser which is centered on the queried protein, and information such as the genomic and transcriptomic coordinates and the presence of a Kozak or high-efficiency translation initiation site (TIS) motif36,37.
- Click on the Protein or DNA links from the info tab, to obtain protein or DNA sequences respectively.
- Browse detailed information about MS evidence, ribosome profiling detection, conservation and identified protein domains by clicking onto the top tabs15.
Representative Results
The workflow described above was applied to a MS dataset available on the PRIDE repository38,39. The original study developed a method (iMixPro), using stable isotope labeling of amino acids in cell culture (SILAC), to eliminate false positives from affinity-purification MS (AP-MS) experiments38. In brief, an AP-MS experiment consists of using beads-bound antibodies to fetch a protein of interest (bait) and its interactors (preys). The collected proteins are then digested and prepared for MS. The sample preparation method and the instrument settings are described in the original study and on the PRIDE repository (PXD004246). A challenge in such experiments is the abundance of false positives, notably from proteins binding to the beads but not the bait. Here, we used SILAC to generate different isotope ratios between true preys and false positives: 3 control samples (no bait) cultured in light medium, 1 sample expressing the bait cultured in light medium, and 1 sample expressing the bait cultured in heavy medium are processed with the beads and further mass spectrometry analysis. With such design, non-specific proteins binding to the beads will have an heavy-to-light ratio of 1:4; when true preys will have a ratio of 1:138.
We re-analyzed their AP-MS data using the OpenProt database; the baits included three endogenous proteins (PTPN14, JIP3 and IQGAP1), and two over-expressed proteins (RAF1 and RNF41). Since the experiments used SILAC, the Galaxy workflow for protein quantification was used (Supplementary Material S3, Figure 2). The workflow was run using the whole OpenProt database (OpenProt_all) or a restricted OpenProt database (OpenProt_2pep, including only proteins previously detected with a minimum of two unique peptides).
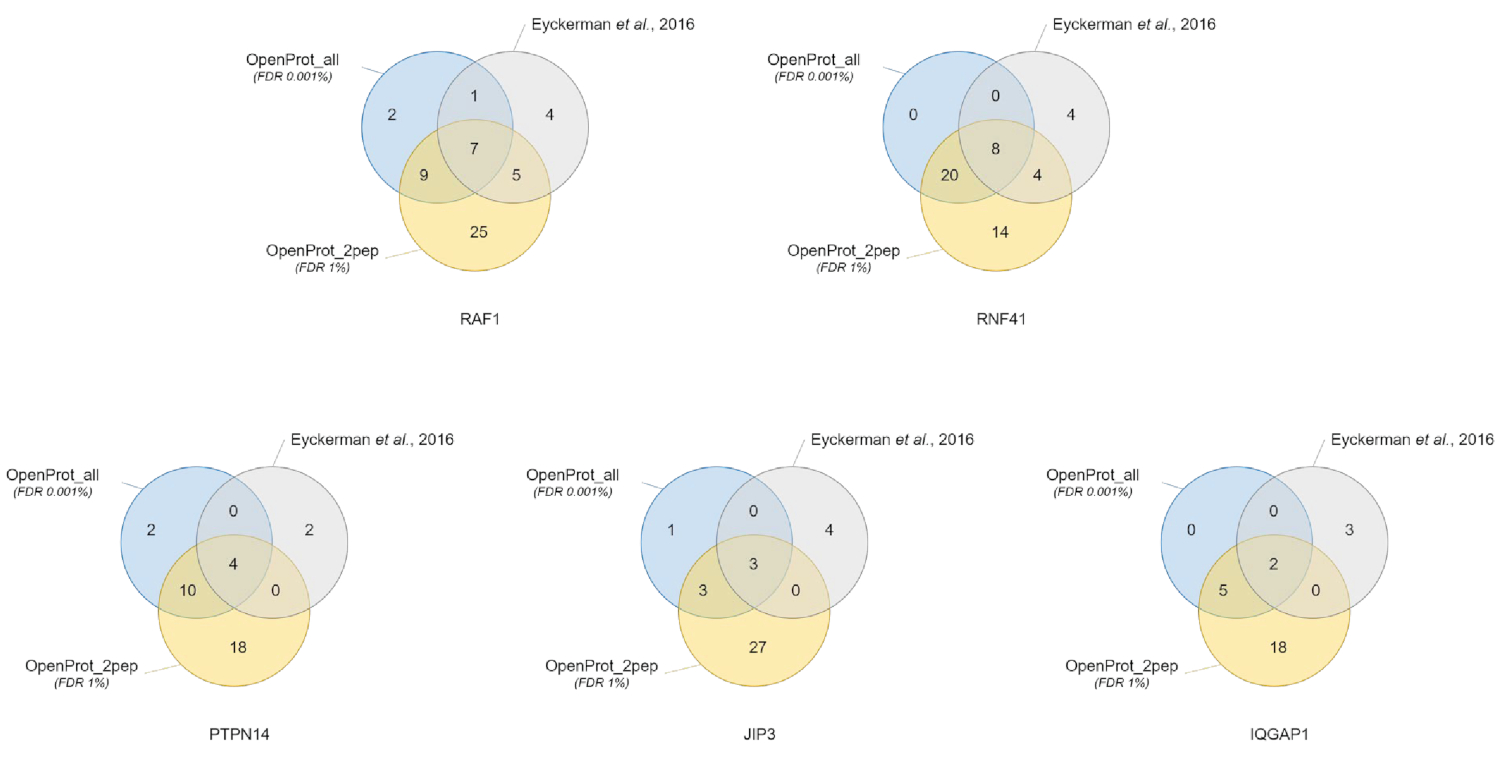
Protein identification and quantification were good and reproducible across the different used databases. As shown in Figure 3, most proteins identified in the original paper were also identified using either the OpenProt_2pep or OpenProt_all database (a detailed list is available in Supplementary Material S5). This result shows that the pipeline described here and the OpenProt databases are able to produce protein identification and quantification comparable to that of current procedures based on the UniProtKB databases40. However, the use of OpenProt databases has the unique advantage of allowing detection of novel and previously undetectable proteins, as demonstrated in this case study.
11 well-supported proteins (1 Isoform and 10 AltProts), yet currently not annotated in databases, were identified across all datasets, with confident peptides, using the OpenProt_2pep database (all protein accessions, along with the number of supporting peptides, are available in Supplementary Material S5). This database allows the use of a traditional 1% FDR as the search space increase remains moderate. These 11 proteins were not identified in the original study as they were absent from the database.
29 novel proteins (16 isoforms and 13 AltProts) were discovered across all datasets, with confident peptides, using the OpenProt_all database (all protein accessions, along with the number of supporting peptides, are available in Supplementary Material S6). As shown in Figure 3, the recommended stringent FDR did not affect the most confident protein identifications, although it did decrease the total number of identified proteins. Comparatively to the OpenProt_2pep database, a higher number of novel proteins can be confidently identified. All of these novel proteins are absent from the OpenProt_2pep database. This highlights the crucial role of the chosen database for MS-based proteomics.
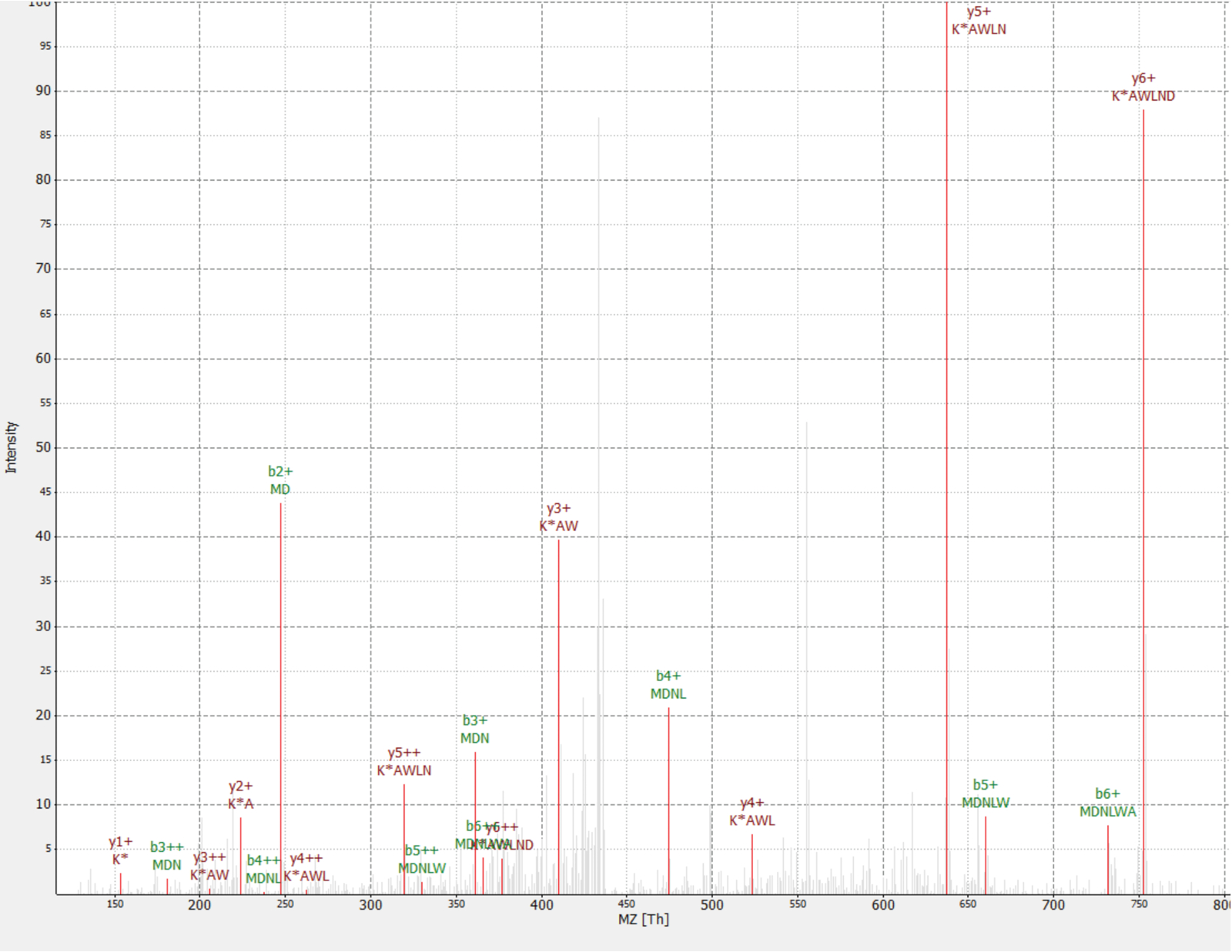
One novel protein was discovered as an interactor of the RAF1 protein (IP_637643). Using the OpenProt website, one can see this protein had not been detected by MS nor ribosome profiling until now (OpenProt v1.3). The protein is 46 amino acids long and can only give two unique peptides upon tryptic digestion. The peptide detected in the RAF1 AP-MS dataset (fraction 18) had a good quality spectrum, as shown in Figure 4, and displayed a heavy-to-light ratio of 1,09. The protein is encoded in the NANOGNBP1 gene, which is a pseudogene of NANOGNB. The transcript (ENST00000448444), currently annotated as non-coding, was detected across several tissues according to the GTEx portal40. The protein contains a predicted functional domain associated with DNA binding (Gene Ontology GO:0003677)41.

Figure 1: Database choice for proteomics analyses chart. Analyses of MS data, notably the database choice, depend on the research objectives. Three common objectives are outlined in blue (classic proteomic pipeline), green (exhaustive proteomic search) and orange (proteomic discovery). Each objective depends on an appropriate database and pipeline. A single identification tool may be used for an exhaustive and classical proteomics pipelines. For the proteomic discovery pipeline, we strongly recommend using multiple identification engines. Recommended FDRs are indicated in red, and protein database sizes are indicated in grey boxes. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Graphical representation of the Galaxy workflow used. Step-by-step representation of the proteomic analysis workflow used for re-analysis of Eyckerman et al. data38. Input files, peptide search, and protein quantification are indicated by orange boxes. Blue boxes correspond to the tools used and grey boxes correspond to the output files generated. The different search engines (MS-GF+ and X!Tandem) are indicated by different colors (respectively red and purple) as well as the arrows indicating their necessary inputs and outputs. The green box highlights the tool generating a list of protein identifications. When multiple outputs are generated, the one used for downstream steps is indicated as the closest to the arrow. This workflow is freely available in Supplementary Material S2. The X!Tandem default parameters configuration file is available in Supplementary Material S4. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Comparison of interactor identification per bait using different databases. Venn diagrams of protein identifications using the most confident OpenProt database (in orange, supporting evidence of minimum 2 unique peptides, OpenProt_2pep) with a 1% FDR, or the whole OpenProt database (in blue, OpenProt_all) with a 0.001% FDR, or as reported in the original paper (in grey)38. Each diagram corresponds to identified interactors for the mentioned bait: RAF1, RNF41, PTPN14, JIP3 and IQGAP1. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: MS/MS spectrum of identified MDNLWAK(13C6) peptide from novel protein IP_637643. Intensity is relative (0 to 100%). Selected peaks are indicated in red, y ions annotations are in dark red and b ions annotations in green. Extracted from the TOPPview software34. Precursor Error = 2.70 ppm, PEP score = 0.12. Please click here to view a larger version of this figure.
{kind=link}
| Term | Definition | Reference |
| Alternative ORF (AltORF) | non-canonical ORF currently not annotated in genome annotations, but annotated in OpenProt. | 15 |
| Reference ORF (RefORF) | canonical ORF annotated in genome annotations and OpenProt. | 15 |
| Alternative protein (AltProt) | novel protein coded by an AltORF, with no significant similarity with a RefProt. Accession prefix: IP_. | 15 |
| Reference protein (RefProt) | protein currently annotated in protein sequence databases such as UniProtKB, Ensembl or NCBI RefSeq, and also in OpenProt. | 15 |
| Novel Isoform | novel protein coded by an AltORF, with a significant similarity with a RefProt. Accession prefix: II_. | 15 |
| OpenProt_2pep database | contains the sequence of all RefProts and novel proteins predicted by OpenProt, already detected with a minimum of 2 unique peptides. | 15 |
| OpenProt_1pep database | contains the sequence of all RefProts and novel proteins predicted by OpenProt, already detected with a minimum of 1 unique peptide. | 15 |
| OpenProt_all database | contains the sequence of all RefProts and novel proteins predicted by OpenProt. | 15 |
Table 1: Definition of terms used in OpenProt and throughout the protocol
Supplementary Material S1: Galaxy workflow for database handling. This will append the CRAPome and decoy sequences (reverse) to the input database. Output is a Fasta file. Please click here to download.
Supplementary Material S2: Galaxy workflow for protein identification. This will identify proteins from a mass spectrometry data file using two search engines (MS-GF+ and X!Tandem). Each parameter can be tuned as desired before running the workflow. Please click here to download.
Supplementary Material S3: Galaxy workflow for protein quantification using stable isotope labeling (SIL). This will identify and quantify proteins from a mass spectrometry data file using two search engines (MS-GF+ and X!Tandem). Each parameter can be tuned as desired before running the workflow. Please click here to download.
Supplementary Material S4: X!Tandem default parameters configuration file. This XML file is necessary for running the X!TandemAdapter tool on the Galaxy platform. Please click here to download.
Supplementary Material S5: Quantified proteins from iMixPro datasets. Data files from Eyckerman et al. 201638 were processed using OpenProt databases and quantified proteins are listed for each condition. Baits are PTPN14, JIP3, IQGAP1, RAF1 and RNF41. Gene names indicated in green correspond to proteins also identified in the original paper38. Gene names indicated in orange correspond to known interactors according to BioGrid that were not reported in the original paper. Gene names indicated in light blue correspond to novel proteins identified as interactors (the corresponding protein accession number is indicated in brackets). Gene names indicated in light grey and italics correspond to likely contaminants (keratin proteins). Please click here to download.
Supplementary Material S6: Identified novel proteins from iMixPro datasets. Data files from Eyckerman et al. 201638 were processed using OpenProt databases and novel identified proteins are listed for each condition. Baits are PTPN14, JIP3, IQGAP1, RAF1 and RNF41. Protein accession numbers are listed, starting with II_ for novel isoforms of a known protein, and with IP_ for novel proteins from an alternative ORF (AltProt).The number of supporting peptides are indicated in brackets. Please click here to download.
Discussion
When analyzing data from mass spectrometers, the quality of protein identification partly relies on the accuracy of the used database6,20. Current approaches traditionally use UniProtKB databases, yet these support the genome annotation model of a single ORF per transcript and a minimum length of 100 codons (with the exception of previously demonstrated examples)40. Multiple studies relate the shortcomings of such databases with the discovery of functional ORFs from allegedly non-coding regions8,11,12,13. Now, OpenProt allows for more exhaustive protein identification as it draws protein sequences from multiple transcriptome annotations. OpenProt retrieves NCBI RefSeq (GRCh38.p7) and Ensembl (GRCh38.83) transcriptomes and UniProtKB annotations (UniProtKB-SwissProt, 2017-09-27)40,42,43. As current annotations present little overlap, OpenProt thus displays a more exhaustive view of the potential proteomic landscape than when limited to one annotation15.
Furthermore, as OpenProt enforces a polycistronic model, it allows for multiple protein annotations per transcript. For statistical and computational reasons, OpenProt still holds a minimum length threshold of 30 codons15. Yet, it predicts thousands of novel protein sequences, thereby widening the scope of possibilities for protein identification. With this approach, OpenProt supports proteomic discoveries in a more systematic manner.
The quality of protein identification can also be affected by the parameters that are used. MS-based proteomics analyses typically hold a 1% protein FDR. However, the whole OpenProt database contains about 6 times more entries (Figure 1). To account for this substantial increase in the search space, we recommend using a more stringent FDR of 0.001%. This parameter was optimized using benchmark studies and manual evaluation of randomly selected spectra15. False positive are still a possibility, though, and we encourage thorough inspection and validation of supporting evidence for a novel protein. A recommended standard could be the identification of a protein from two different MS runs, as background data and false positives vary between datasets15.
The pipeline provided here and used for the case study can be modified as pleased to fit the experimental design and parameters. We would recommend using multiple search engines as it increases sensibility and sensitivity of peptide identification32. Furthermore, we encourage using the database corresponding best to the experimental aim (Figure 1). As using the whole OpenProt database comes with a stringent FDR, true identifications may be lost. Thus, the whole database should be intended for discovery of novel proteins, whilst classical proteomics profiling should be using the smaller OpenProt databases (such as OpenProt_2pep used in the case study above).
OpenProt currently predicts sequences starting with an ATG codon, whereas several studies highlighted translation initiation at other codons44,45. When a novel protein is identified by one or several unique peptides, it is possible the true initiation codon is not the presumed ATG. Users can look for translation evidence on the OpenProt website. Currently, OpenProt only reports translation events if they concern the entire predicted protein sequence (100% overlap)15. Thus, absence of translation evidence would not mean the protein is not translated, but that the start codon may not be the alleged ATG.
Despite its current limitations, OpenProt offers a more exhaustive view of eukaryotic genomes' coding potential. OpenProt databases foster proteomic discoveries and the understanding of proteomic functions and interactions. Future developments of the OpenProt database will include annotation of other species, translation evidence from non-ATG start codon and development of a pipeline to include novel proteins in whole genome and exome sequencing studies.
Acknowledgements
We thank Vivian Delcourt for his help, discussions and advice on this work. X.R. is a member of the Fonds de Recherche du Québec Santé (FRQS)-supported Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke. This research was supported by a Canada Research Chair in Functional Proteomics and Discovery of Novel Proteins to X.R. and CIHR grant MOP-137056. We thank the team at Calcul Québec and Compute Canada for their support with the use of the supercomputer mp2 from Université de Sherbrooke. Operation of the mp2 supercomputer is funded by the Canada Foundation of Innovation (CFI), le ministère de l'Économie, de la science et de l'innovation du Québec (MESI) and les Fonds de Recherche du Québec - Nature et technologies (FRQ-NT). The Galaxy server that was used for some proteomics calculations is in part funded by Collaborative Research Centre 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and German Federal Ministry of Education and Research (BMBF grants 031 A538A/A538C RBC, 031L0101B/031L0101C de.NBI-epi, 031L0106 de.STAIR (de.NBI)).
Materials
| Name | Company | Catalog Number | Comments |
| OpenProt website | open source | n/a | www.openprot.org |
| Galaxy Server | open source | n/a | https://usegalaxy.eu/ |
| TOPPview software | open source | n/a | www.openms.de |
References
- Kim, M. S., et al. A draft map of the human proteome. Nature. 509 (7502), 575-581 (2014).
- Wilhelm, M., et al. Mass-spectrometry-based draft of the human proteome. Nature. 509 (7502), 582-587 (2014).
- Hein, M. Y., et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 163 (3), 712-723 (2015).
- Huttlin, E. L., et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 162 (2), 425-440 (2015).
- Huttlin, E. L., et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 545 (7655), 505-509 (2017).
- Kumar, D., Yadav, A. K., Dash, D. Choosing an Optimal Database for Protein Identification from Tandem Mass Spectrometry Data. Proteome Bioinformatics. , 17-29 (2017).
- Jeong, K., Kim, S., Bandeira, N. False discovery rates in spectral identification. BMC Bioinformatics. 13 (Suppl 16), (2012).
- Brunet, M. A., Levesque, S. A., Hunting, D. J., Cohen, A. A., Roucou, X. Recognition of the polycistronic nature of human genes is critical to understanding the genotype-phenotype relationship. Genome Research. , (2018).
- Brent, M. R. Genome annotation past, present, and future: how to define an ORF at each locus. Genome Research. 15 (12), 1777-1786 (2005).
- Harrow, J., et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Research. 22 (9), 1760-1774 (2012).
- Samandi, S., et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. eLife. 6, e27860 (2017).
- Saghatelian, A., Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nature Chemical Biology. 11 (12), 909-916 (2015).
- Delcourt, V., Staskevicius, A., Salzet, M., Fournier, I., Roucou, X. Small Proteins Encoded by Unannotated ORFs are Rising Stars of the Proteome, Confirming Shortcomings in Genome Annotations and Current Vision of an mRNA. Proteomics. , (2017).
- Plaza, S., Menschaert, G., Payre, F. In Search of Lost Small Peptides. Annual Review of Cell and Developmental Biology. 33 (1), (2017).
- Brunet, M. A., et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Research. , (2018).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 44 (W1), W3-W10 (2016).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research. 46, W537-W544 (2018).
- Sturm, M., et al. OpenMS – An open-source software framework for mass spectrometry. BMC Bioinformatics. 9 (1), 163 (2008).
- Carithers, L. J., et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation and Biobanking. 13 (5), 311-319 (2015).
- Aebersold, R., Mann, M. Mass spectrometry-based proteomics. Nature. 422 (6928), 6928 (2003).
- Domon, B., Aebersold, R. Mass Spectrometry and Protein Analysis. Science. 312 (5771), 212-217 (2006).
- Hu, J., Coombes, K. R., Morris, J. S., Baggerly, K. A. The importance of experimental design in proteomic mass spectrometry experiments: Some cautionary tales. Briefings in Functional Genomics. 3 (4), 322-331 (2005).
- Wu, P. Y., Phan, J. H., Wang, M. D. Assessing the impact of human genome annotation choice on RNA-seq expression estimates. BMC Bioinformatics. 14 (11), S8 (2013).
- Mellacheruvu, D., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10 (8), 730-736 (2013).
- Adusumilli, R., Mallick, P. Data Conversion with ProteoWizard msConvert. Proteomics: Methods and Protocols. , 339-368 (2017).
- French, W. R., et al. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard’s msConvert. Journal of Proteome Research. 14 (2), 1299-1307 (2015).
- Kuenzi, B. M., et al. APOSTL: An Interactive Galaxy Pipeline for Reproducible Analysis of Affinity Proteomics Data. Journal of Proteome Research. 15 (12), 4747-4754 (2016).
- Hoekman, B., Breitling, R., Suits, F., Bischoff, R., Horvatovich, P. msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies. Molecular & Cellular Proteomics: MCP. 11 (6), (2012).
- Bjornson, R. D., et al. X!!Tandem, an improved method for running X!tandem in parallel on collections of commodity computers. Journal of Proteome Research. 7 (1), 293-299 (2008).
- Kim, S., Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nature Communications. 5, 5277 (2014).
- Vaudel, M., Barsnes, H., Berven, F. S., Sickmann, A., Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 11 (5), 996-999 (2011).
- Shteynberg, D., Nesvizhskii, A. I., Moritz, R. L., Deutsch, E. W. Combining results of multiple search engines in proteomics. Molecular & Cellular Proteomics: MCP. 12 (9), 2383-2393 (2013).
- Bittremieux, W., et al. Quality control in mass spectrometry-based proteomics. Mass Spectrometry Reviews. 37 (5), 697-711 (2018).
- Bertsch, A., Gröpl, C., Reinert, K., Kohlbacher, O. OpenMS and TOPP: Open Source Software for LC-MS Data Analysis. Data Mining in Proteomics: From Standards to Applications. , 353-367 (2011).
- Pfeuffer, J., et al. OpenMS – A platform for reproducible analysis of mass spectrometry data. Journal of Biotechnology. 261, 142-148 (2017).
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 299 (1-2), 1-34 (2002).
- Noderer, W. L., et al. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Molecular Systems Biology. 10, 748 (2014).
- Eyckerman, S., et al. Intelligent Mixing of Proteomes for Elimination of False Positives in Affinity Purification-Mass Spectrometry. Journal of Proteome Research. 15 (10), 3929-3937 (2016).
- Vizcaíno, J. A., et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Research. 44 (D1), D447-D456 (2016).
- Bateman, A., et al. UniProt: the universal protein knowledgebase. Nucleic Acids Research. 45 (D1), D158-D169 (2017).
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research. 45 (D1), D331-D338 (2017).
- O’Leary, N. A., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research. 44, D733-D745 (2016).
- Zerbino, D. R., et al. Ensembl 2018. Nucleic Acids Research. 46 (D1), D754-D761 (2018).
- Andreev, D. E., et al. Translation of 5’ leaders is pervasive in genes resistant to eIF2 repression. eLife. 4, e03971 (2015).
- Jackson, R., et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature. 564, 434-438 (2018).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved