
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Medicine
Características de la imagen de la enfermedad pulmonar intersticial asociada a la esclerosis sistémica
Aquí, presentamos recomendaciones prácticas para realizar tomografía computarizada torácica de alta resolución para diagnosticar y evaluar la enfermedad pulmonar intersticial relacionada con la esclerosis sistémica.
El diagnóstico precoz de la enfermedad pulmonar intersticial relacionada con la esclerosis sistémica (SSc-ILD) es importante para permitir que el tratamiento se administre con un retraso mínimo. Sin embargo, el diagnóstico de SSc-ILD es difícil porque los síntomas clave no son específicos. La tomografía computarizada de alta resolución (HRCT) del tórax se reconoce como un método de diagnóstico de imágenes sensible para diagnosticar y evaluar la SSc-ILD. La exposición de los pacientes a la radiación ionizante puede considerarse como una limitación, aunque se pueden tomar medidas metodológicas para moderar esto. Presentamos recomendaciones prácticas para realizar escaneos HRCT e interpretar los resultados. Las características clave de SSc-ILD en HRCT incluyen un patrón de neumonía intersticial no específica (NSIP) con opacidades periféricas de vidrio molido y bronquiectasia de tracción extensa. A pesar de las similitudes entre SSc-ILD y la fibrosis pulmonar idiopática (IPF), el HRCT se puede utilizar para diferenciar entre estas condiciones: en SSc-ILD en comparación con el IPF, hay una mayor proporción de opacidad del vidrio molido y la fibrosis es menos gruesa. Un esófago dilatado, lleno de aire con diámetro >10 mm, que sugiere dismotilidad esofágica se observa comúnmente en SSc-ILD. El tamaño de la arteria pulmonar mayor que la aorta ascendente adyacente sugiere hipertensión pulmonar coexistente. Los nódulos deben ser monitoreados debido al mayor riesgo de cáncer de pulmón. Una gran cantidad de enfermedades en el HRCT (20 %) o una puntuación alta de fibrosis sugiere un mayor riesgo de mortalidad. El HRCT es fundamental para diagnosticar la SSc-ILD, y las evaluaciones en serie pueden ser útiles para monitorear la progresión de la enfermedad o la respuesta al tratamiento.
La esclerosis sistémica (SSc) es una enfermedad compleja, heterogénea y autoinmune. Puede manifestarse como vasculopatía, fenómeno de Raynaud y fibrosis de la piel y órganos internos1. SSc se clasifica en subtipos de la siguiente manera: cutáneo limitado, cutáneo difuso, esclerodermia sinustinada (sin afectación de la piel) y síndrome de superposición SSc1.
SSc no se hereda en la moda mendeliana, pero los factores genéticos parecen influir en la susceptibilidad a la enfermedad. Las tasas de incidencia difieren entre los grupos étnicos y se incrementan entre los individuos con antecedentes familiares de la enfermedad2,3. También parecen existir factores de riesgo ambientales, con una alta exposición a sílice o disolventes orgánicos que parecen aumentar la aparición de SSc4. La prevalencia mundial de SSc es de alrededor de 1 de cada 10.0001. Más hembras que machos se ven afectadas por SSc, con proporciones femeninas:masculinas reportadas que oscilan entre 3:1 y 8:1, y el grupo de edad con mayor incidencia de la enfermedad es de 45-54 años5.
El pulmón es el segundo órgano visceral más comúnmente afectado en pacientes con SSc6. Hay dos manifestaciones pulmonares principales de SSc: enfermedad pulmonar intersticial (ID), e hipertensión pulmonar7. La ENFERMEDAD por ILE suele ser fibrosa; ocurre en aproximadamente el 80% de los pacientes con SSc y es más frecuente en la esclerodermia cutánea difusa que en la forma limitada de la enfermedad1,,8. La hipertensión pulmonar puede manifestarse como hipertensión arterial pulmonar aislada (HAP, que tiene una prevalencia del 13-35% en SSc) o hipertensión pulmonar resultante de la afectación ventricular izquierda/disfunción diastólica o ILD/hipoxemia7. Los perfiles de anticuerpos difieren entre los pacientes con SSc-ILD y los que tienen SSc-HAP. Por ejemplo, la presencia de anticuerpos anti-Scl-70 se asocia con SSc-ILD8,mientras que los anticuerpos anticentromere son más frecuentes en pacientes con SSc con HAP que en aquellos sin HAP9.
Los síntomas de la SSc-ILD incluyen disnea, tos, dolor en el pecho y limitación del ejercicio. ILD es un importante contribuyente a la morbilidad en SSc10,11,12. Como consecuencia, se ha informado que los costos anuales de atención médica por todas las causas son más altos en pacientes con SSc-ILD que en aquellos con SSc y sin ILD: $31,285–55,446 frente a $18,513–23,268, respectivamente13.
SSc-ILD es la principal causa de mortalidad en pacientes con SSc, representando entre el 30 y el 35% de las muertes en este grupo10,,14. Se ha informado que la mediana de supervivencia entre los pacientes con SSc-ILD es de 5 a 8 años10,,15; en comparación, aproximadamente el 76% de la población general con SSc sobrevive durante más de 10 años desde la aparición de la enfermedad16. Los predictores significativos de mortalidad en SSc-ILD incluyen la edad, la capacidad vital forzada (CVF), la capacidad de difusión basal del pulmón para el monóxido de carbono (DLCO), el grado de enfermedad en la tomografía computarizada de alta resolución (HRCT), la presencia de hipertensión pulmonar y los niveles de Antígeno Kerbs von den Lungren 6 (KL-6) antígeno17,,18.
El diagnóstico precoz es importante para permitir que el tratamiento se administre con un retraso mínimo y, en pacientes con un fenotipo progresivo, la progresión de la enfermedad puede ralentizarse. Sin embargo, el diagnóstico de SSc-ILD es difícil porque los síntomas no específicos de tos, disnea y fatiga pueden confundirse con otros aspectos de la SSc, como la enfermedad cardíaca y la afectación musculoesquelética. Las evaluaciones para el diagnóstico de las ENFERMEDADes intelectuales incluyen: presentación clínica, historia, estado de tabaquismo, función pulmonar, imágenes y, en algunos casos, biopsia pulmonar. La afirmación del diagnóstico SSc-ILD requiere varias investigaciones, que a menudo se utilizan en la combinación19. Las evaluaciones más utilizadas incluyen pruebas de función pulmonar y HRCT20,21,22,23. También se pueden emplear otros métodos de diagnóstico por imágenes, como la radiografía de tórax y las imágenes que ahorran radiación (por ejemplo, imágenes por resonancia magnética [RM], ultrasonido pulmonar)22. Las pruebas de la función pulmonar se utilizan para evaluar la gravedad de la ILD y monitorear su curso. Sin embargo, el uso de pruebas de función pulmonar por sí solo es de uso limitado para el diagnóstico de SSc-ILD24,25. El HRCT del tórax es visto como el medio no invasivo más sensible para facilitar el diagnóstico diferencial de SSc-ILD19. Los resultados básicos del HRCT, así como los cambios a lo largo del tiempo, se pueden utilizar para predecir el curso futuro de la enfermedad pulmonar y la respuesta potencial a la terapia26.
La exposición a la radiación con HRCT se considera a veces como un factor limitante para el cribado regular27,28; limitar el número de rodajas es un método potencial para reducir el riesgo de radiación, y la dosis también puede reducirse disminuyendo la tensión o la corriente29,30,31. Alternativamente, se pueden considerar diferentes métodos de evaluación. Por ejemplo, la RMN parece tener cierto potencial para la evaluación y el seguimiento de los pacientes con IDA22. En un estudio utilizando imágenes de RESONANCIA magnética ponderadas por T2 con sincronización respiratoria, HRCT se realizó en paralelo como la evaluación de "estándar de oro"; Se notificaron sensibilidades al 100% y 60% de especificidad con RMN para determinar la presencia de ILD32. En otro estudio33se notificó un acuerdo similar entre la RMN y el HRCT en la detección y categorización de la DI. A pesar de los resultados prometedores, la RMN es actualmente una metodología de investigación y aún no está lista para el uso clínico generalizado.
Aquí, proporcionamos una visión práctica de la interpretación de los resultados de la imagen, con un enfoque en el HRCT, para diagnosticar la participación pulmonar en SSc, determinar el pronóstico, y también explorar los desarrollos futuros que pueden mejorar los métodos de diagnóstico por imágenes y la interpretación de los resultados. Las imágenes HRCT de casos representativos se incluyen en el documento.
1. Escaneo HRCT
- Realizar el escaneo volumétrico de adquisición HRCT del tórax36. Los agentes de contraste no son necesarios36,37.
- Obtenga las siguientes adquisiciones con los parámetros que se muestran en el Cuadro 136,37.
- Adquiera una exploración inspiratoria supina (volumétrica) desde los ápices pulmonares hasta la base pulmonar.
- Adquiera una exploración espiratoria supina (secuencial con espacios de 10-20 mm) desde 2 cm por debajo de los ápices pulmonares hasta la base pulmonar.
- Adquirir un inspiratorio propenso opcional (secuencial con espacios de 10-20 mm) desde la carina hasta la base pulmonar.
- Dar instrucciones de respiración al paciente antes de cada adquisición36,37. Para una exploración inspiratoria, diga "Tome en una respiración profunda ... y déjelo salir. Respira hondo... y déjalo salir. Respira hondo y aguanta la respiración. Sigue conteniendo la respiración"37.
- Obtener escaneos inspiratorios a plena inspiración35,36.
- Utilice la colimación más delgada, el tiempo de rotación más corto y el tono más alto para asegurarse de que las imágenes sin movimiento se obtienen36. Los parámetros de escaneado sugeridos se detallan en la Tabla 137.
- Para una calidad óptima de escaneos volumétricos, obtenga imágenes de sección delgada (<2 mm) con reconstrucción de alta resolución espacial35,,36.
- Revise los escaneos inmediatamente después de la adquisición y repita si cualquier artefacto de movimiento está presente o se ha producido una inspiración inadecuada37.
2. Informes
- Prepare un informe interpretativo.
- Comparta el informe y las imágenes de HRCT con el equipo de atención del paciente y agréguelas a los registros médicos del paciente.
Diagnóstico
Las características clave de SSc-ILD en HRCT suelen incluir un patrón de neumonía intersticial no específica (NSIP) con opacidades periféricas de vidrio molido y bronquiectasia de tracción extensa(Figura 1 y Figura 2). Las opacidades de vidrio molido tienen una etiología amplia y a menudo no son específicas40,,41,,42. El predominio central o la distribución periférica con moderación subpleural es altamente sugerente de NSIP (Figura 3).
Típicamente, los patrones de ILD en imágenes HRCT incluyen reticulaciones con distorsión arquitectónica que resulta en bronquiectasia de tracción/bronquiolectasis (consistente con una forma fibrosa de NSIP). De hecho, la bronquiectasia de tracción y la bronquiolectasis de tracción son a menudo las características predominantes de SSc-ILD (Figura 4)43. Otros hallazgos pueden incluir panal de abeja(Figura 5; más común en formas limitadas de SSc), engrosamiento interlobular del tabique y líneas intralobulares, y micronódulos40,,44. El panal se refiere a los espacios aéreos quísticos agrupados de diámetro típicamente consistente (3-10 mm) con paredes gruesas y bien definidas31. El panal y la bronquiectasia de tracción son características clave de la neumonía intersticial habitual (UIP) en HRCT. Aunque este patrón se asocia más comúnmente con fibrosis pulmonar idiopática (IPF), el prototipo de ILD fibrosa con un fenotipo progresivo, a veces se puede observar en pacientes con SSc-ILD10. Recientemente, se han identificado varios signos en pacientes con ILD relacionada con la enfermedad del tejido conectivo (incluyendo SSc-ILD) y el patrón UIP en HRCT, pero no en aquellos con IPF. Estos son el signo del borde recto (es decir, el aislamiento de la fibrosis a las bases pulmonares con demarcación aguda en el plano craneocaudal sin extensión sustancial a lo largo de los márgenes laterales de los pulmones en imágenes coronales), el signo predominante (o exuberante) de panal (>70% de las porciones fibromáticas del pulmón) y el signo del lóbulo superior superior (es decir, concentración de fibrosis dentro del aspecto anterior de los lóbulos superiores, con relativa moderación de los otros aspectos de los lóbulos superiores, y afectación concomitante del lóbulo inferior)45. El signo de borde recto también se ha asociado con la patología NSIP46,que es el patrón de TC principal en SSc-ILD10.
El esófago lleno de aire dilatado se observa con frecuencia en pacientes con SSc (Figura 6)47,48,49 y en pacientes con SSc-ILD47,48. Si bien no se acepta un límite de edad superior aceptado cuando un esófago dilatado ya no puede ayudar a diferenciar la SSc-ILD y la IPF, un esófago dilatado puede ser más difícil de interpretar en pacientes mayores de 65 años debido al aumento de la incidencia de trastornos de la motilidad esofágica. La linfadenopatía mediastinal (generalmente reactiva), en la que el eje corto del ganglio linfático supera los 10 mm, también se observa a menudo en pacientes con SSc-ILD47,,50. El tamaño de la arteria pulmonar mayor que la aorta ascendente adyacente sugiere hipertensión pulmonar coexistente (Figura 6), incluso en pacientes sin enfermedad pulmonar fibrosa51,52,53. Las áreas de consolidación sugieren infección superpuesta, aspiración, organización de neumonía, hemorragia o neoplasia maligna. Los nódulos deben ser monitoreados debido al mayor riesgo de cáncer de pulmón en SSc-ILD7; el cáncer primario más común que ha surgido en pacientes con SSc-ILD es el adenocarcinoma7,,54.
SSc-ILD comparte una serie de similitudes clínicas, mecanicistas y patológicas con IPF15,,55. Sin embargo, algunas características radiológicas permiten la diferenciación de estos dos ILD15,45. En SSc-ILD, en comparación con el IPF, hay una mayor proporción de opacidad del vidrio molido y la fibrosis es menos gruesa. En los casos de UIP en SSc, el panal se puede observar en más del 70% del tejido fibrotico-pulmón, el signo de panal exuberante56,57. Además, el signo de cuatro esquinas (también conocido como el signo del lóbulo superior anterior) es significativamente más común en SSc-ILD que en IPF; este es un patrón de inflamación y/o fibrosis de forma focal o desproporcionada que afecta a los lóbulos superiores anterolaterales bilaterales y a los lóbulos inferiores posterosuperior58.
Las radiografías de tórax pueden detectar inicialmente la ILD; sin embargo, no ofrecen suficiente resolución de contraste para un diagnóstico confiable. En radiografías de tórax de pacientes con SSc-ILD, el patrón más frecuente es la reticulación predominante basal59. Otras características pueden incluir bronquiectasia visible, pérdida de volumen y panal. Al igual que con HRCT, la presencia de un esófago lleno de aire dilatado puede ser útil para apoyar el diagnóstico de SSc-ILD47.
Pronóstico
Se ha demostrado que varios hallazgos de imágenes diferentes están asociados con el pronóstico en SSc-ILD. Se ha notificado que el riesgo de mortalidad es mayor en pacientes con una extensión de la enfermedad de al menos el 20% en el HRCT (la supervivencia de 10 años fue del 43% frente al 67%, respectivamente, en pacientes con una extensión de la enfermedad superior frente a menos del umbral del 20%)60. Del mismo modo, se ha asociado una puntuación alta de fibrosis en HRCT (basada en el grado de reticulación y panal) con un aumento de la mortalidad61. Los grandes diámetros del esófago se asocian con una mayor gravedad de la ILD y una disminución del DLCO48. La densidad pulmonar y el diámetro de la arteria pulmonar se pueden utilizar potencialmente para predecir el riesgo de hipertensión pulmonar62. También se podrían aprovechar los parámetros cuantitativos computarizados de tc para identificar el riesgo de disminución o mortalidad de la función pulmonar de los pacientes. Un estudio sugirió que el alcance de la ILD, cuantificado a partir de HRCT, podría utilizarse para predecir la disminución de la CVF durante 12meses 63. En otro estudio, los parámetros cuantitativos de la TC torácica proporcionaron resultados de riesgo de mortalidad que fueron consistentes con los modelos de predicción clínica64. A pesar de su potencial aparente, los biomarcadores basados en imágenes se consideran actualmente mejor a nivel de población, ya que no se ha establecido su utilidad clínica en pacientes individuales.
Respuesta al tratamiento
La ciclofosfamida y el micofenolato mofettil proporcionan un beneficio modesto en pacientes con SSc-ILD. En el histórico estudio pulmonar de la esclerodermia I, el tratamiento con ciclofosfamida condujo a una progresión más lenta de la fibrosis en comparación con placebo65. Más recientemente, el Estudio pulmonar de la escleroderma II informó de una eficacia similar y una mayor tolerabilidad con micofenolato mofettil en comparación con la ciclofosfamida66. Sin embargo, sigue siendo necesario mejorar las opciones de tratamiento para los pacientes con SSc-ILD. Las terapias que se están investigando actualmente incluyen anticuerpos monoclonales (por ejemplo, rituximab, abituzumab), agentes antifibroticos (por ejemplo, nintedanib, pirfenidona), el inhibidor directo de la trombina dabigatrán, el inhibidor del proteasoma bortezomib y el trasplante de células madre hematopoyéticas19,,67.
Exploraciones seriales de HRCT que muestran la progresión de la enfermedad en un paciente con SSc-ILD
Las evaluaciones de HRCT realizadas en diferentes puntos de tiempo se pueden utilizar para investigar la progresión de la enfermedad. La Figura 7 muestra dos conjuntos de imágenes de HRCT en el pecho axial y coronal tomadas con 10 años de diferencia en un paciente con SSc-ILD. Las imágenes axiales y coronales iniciales(Figura 7A,B)del tórax HRCT muestran la opacidad y reticulación predominantes de vidrio molido basilar con bronquiectasia de tracción suave y moderación subpleural consistente con NSIP en este paciente con SSc. Este último conjunto de imágenes(Figura 7C, D) tomadas 10 años más tarde, muestran un aumento de la reticulación y la bronquiolectasis de tracción en las bases pulmonares con disminución de la opacidad del vidrio molido en las imágenes axiales y coronales(Figura 7C,D) de la TC torácica consistente con un empeoramiento leve de la fibrosis pulmonar. Las exploraciones seriales HRCT también se pueden utilizar para monitorear la respuesta del tratamiento68,69,70; esto se demostró en el Estudio Pulmonar de escleroderma II, en el que se utilizaron puntuaciones de diagnóstico asistidas por ordenador basadas en exploraciones HRCT para comparar la eficacia de la ciclofosfamida con micofenolato mofettil en pacientes con SSc-ILD68.
| Fase | Detector Colimación | Tensión (kV) | Corriente (mAs) | escanear Intervalo | alquitrán | Rotación | Corriente del tubo Modulación |
| Inspiración supina | Helicoidal 1,2 mm | 120 (se puede reducir) | 230 (se puede reducir) | N/A | 1,0 | 0,5 segundos o más rápido | En |
| Espiratorio supinoso | Axial 2 x 1,0 mm | 120 | 150 | 20 mm | N/A | 1.0 segundos | En |
| Inspiratorio propenso | Axial 2 x 1,0 mm | 120 | 150 | 20 mm | N/A | 1.0 segundos | En |
Tabla 1: Parámetros de adquisición de tomografía computarizada37. N/A no aplicable.
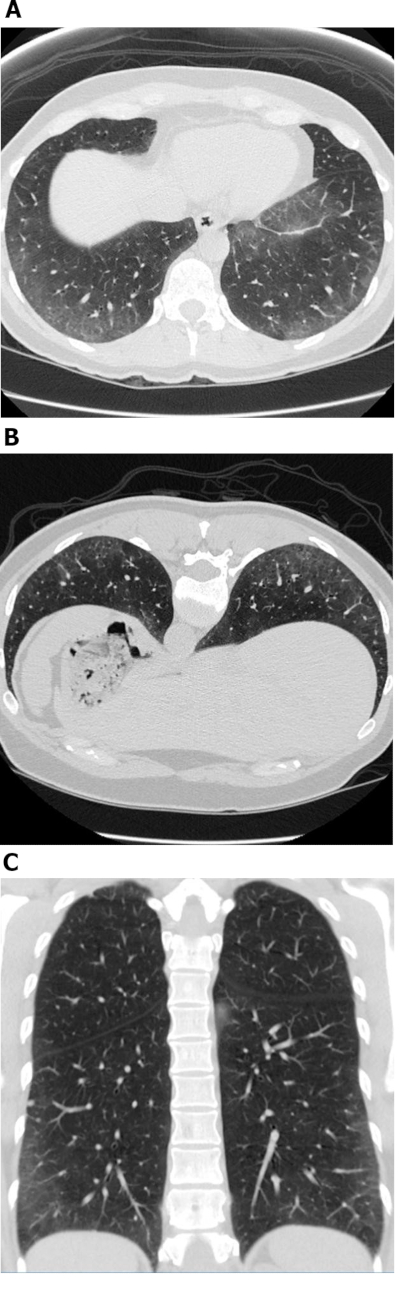
Figura 1: Esclerosis sistémica con un patrón celular de NSIP de enfermedad. Las imágenes de tomografía computarizada de alta resolución axial (A), propensas (B) y coronales (C), muestran extensas opacidades de vidrio molido predominantes periféricos y basales; estas son observaciones típicas con NSIP. La falta de bronquiectasia de tracción sugiere un patrón celular de NSIP de enfermedad. NSIP : neumonía intersticial no específica. Haga clic aquí para ver una versión más grande de esta figura.
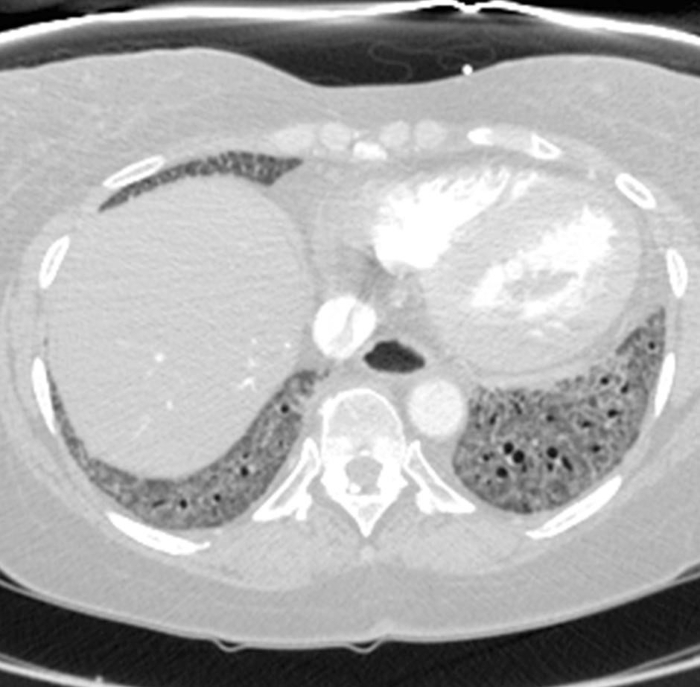
Figura 2: Esclerosis sistémica con un patrón de neumonía intersticial no específica fibrosa de la enfermedad. La imagen de tomografía computarizada axial muestra extensas opacidades de vidrio molido predominantes basalmente con bronquiectasia de tracción asociada. En particular, el esófago muestra una dilatación marcada; esto es típico de la esclerodermia. Haga clic aquí para ver una versión más grande de esta figura.
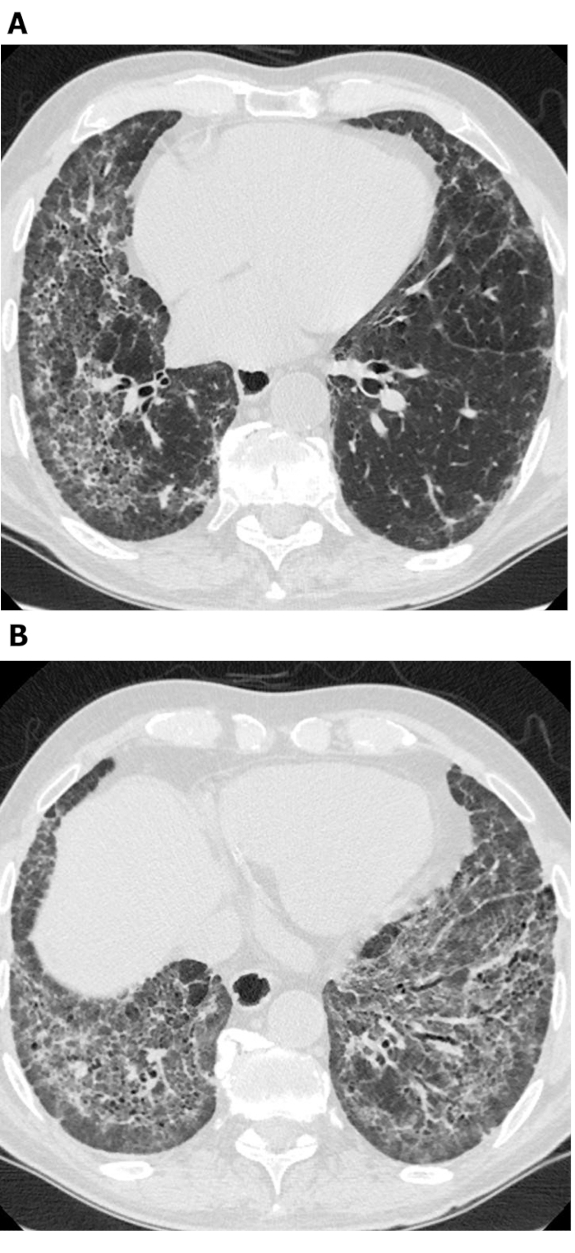
Figura 3: Esclerosis sistémica con un patrón NSIP fibroso. Las imágenes axiales de tomografía computarizada de alta resolución (A y B) muestran extensas opacidades de vidrio molido, reticulación, distorsión arquitectónica y bronquiectasia de tracción. En particular, el escaymos subpleurales es evidente; esto es típico de NSIP y se ve en aproximadamente el 50% de todos los casos. NSIP : neumonía intersticial no específica. Haga clic aquí para ver una versión más grande de esta figura.
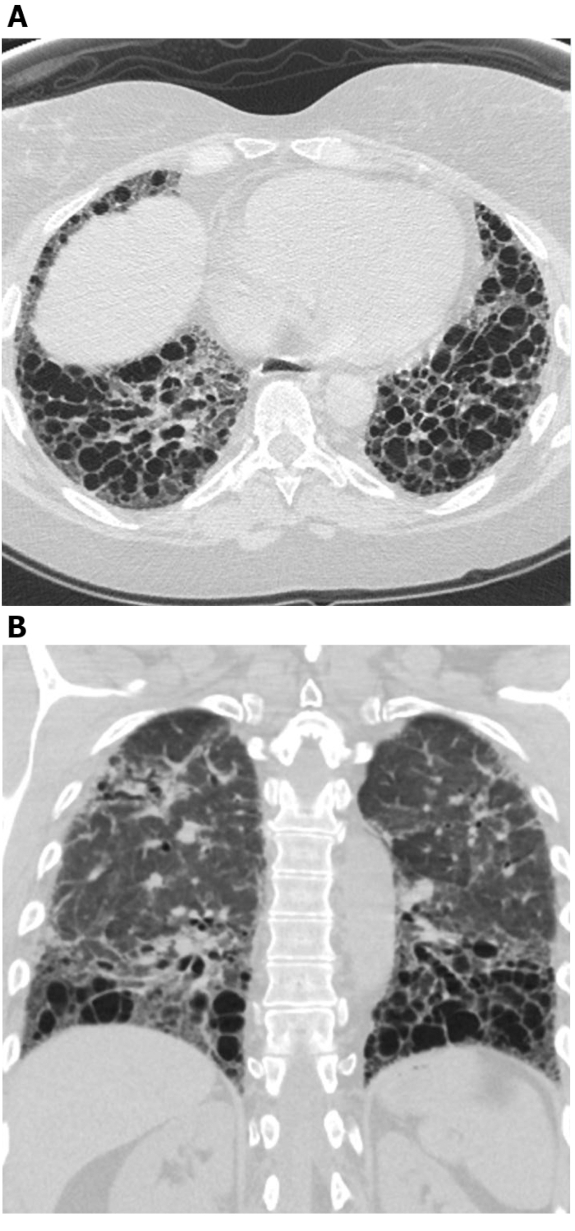
Figura 4: Esclerosis sistémica con bronquiectasia de tracción exuberante. Las imágenes de tomografía computarizada de alta resolución axial (A) y coronal (B) muestran una bronquiectasia de tracción predominante en la zona pulmonar media e inferior extensa. Si bien esto puede confundirse con panal, las áreas quísticas se conectan entre sí y evitan el pulmón subpleural inmediato; esto es típico de la bronquiectasia. Haga clic aquí para ver una versión más grande de esta figura.
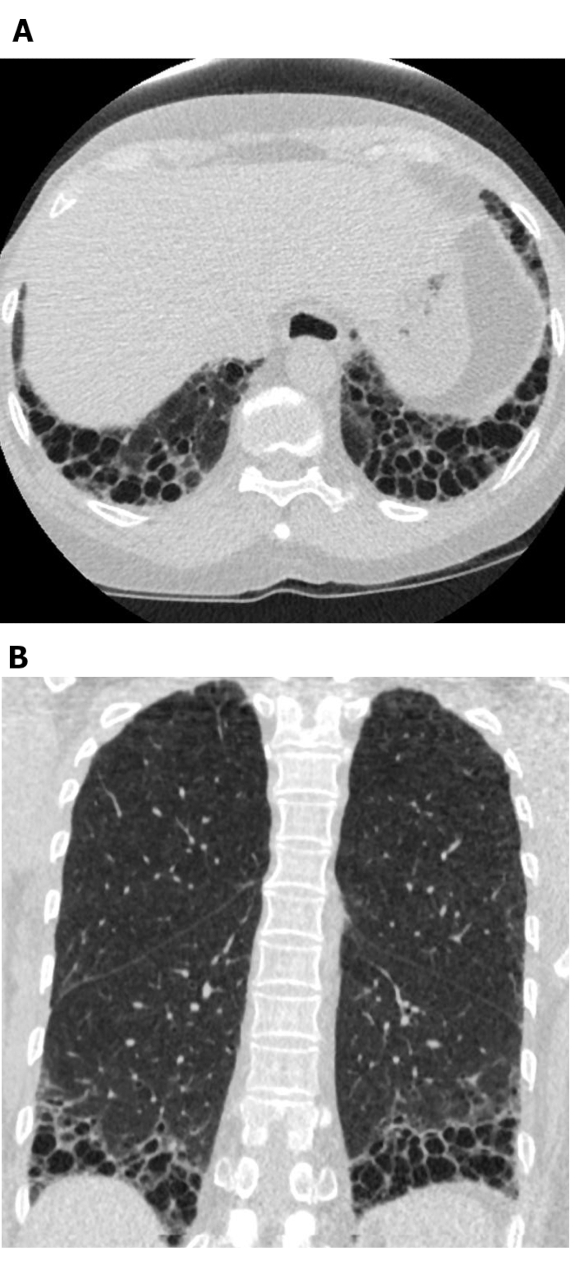
Figura 5: Esclerosis sistémica con un patrón UIP de fibrosis pulmonar. Las imágenes de tomografía computarizada axial (A) y coronal (B) muestran panal de abeja y tracción predominantes periféricos y basales de acuerdo con el patrón típico UIP de fibrosis pulmonar. Tenga en cuenta el esófago dilatado (atribuible a la esclerodermia) y el panal "exuberante" (sugerente de la EPI relacionada con la enfermedad del tejido conectivo en lugar de la fibrosis pulmonar idiopática). UIP: neumonía intersticial habitual. Haga clic aquí para ver una versión más grande de esta figura.
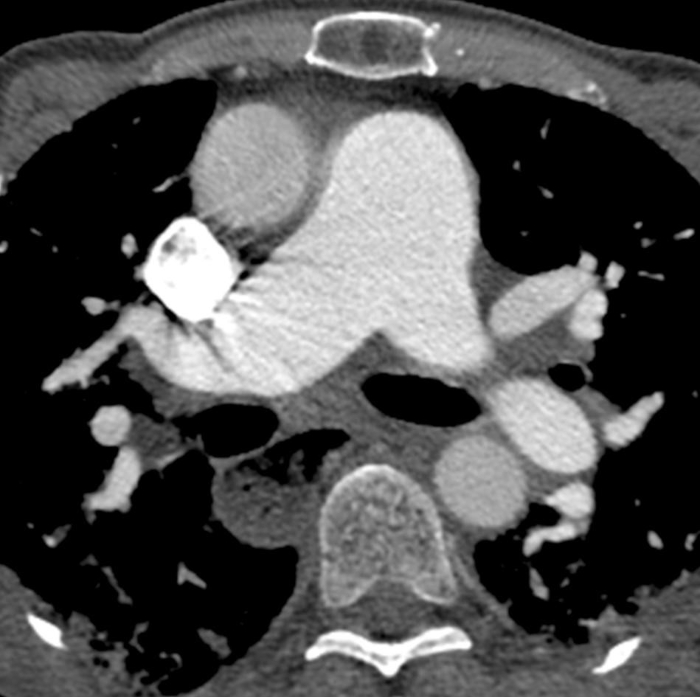
Figura 6: Esclerosis sistémica con hipertensión pulmonar y esófago dilatado. La tomografía computarizada del pecho mejorada por contraste muestra un marcado agrandamiento del tronco pulmonar, con una medida mayor que la aorta ascendente adyacente que sugiere hipertensión pulmonar subyacente. El esófago está marcadamente dilatado; esto es atribuible a la esclerodermia. Haga clic aquí para ver una versión más grande de esta figura.
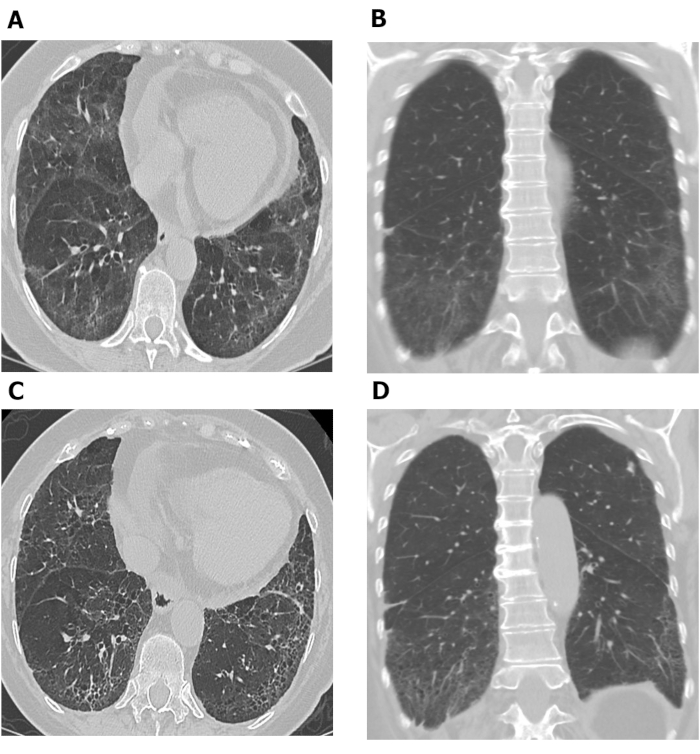
Figura 7: Imágenes de HRCT en el tórax serie que muestran la progresión de la fibrosis pulmonar en pacientes con SSc-ILD. Las imágenes axiales (A) y coronales (B) del tórax HRCT muestran una opacidad y reticulación de vidrio molido predominante basilar con bronquiectasia de tracción suave y moderación subpleural consistente con neumonía intersticial no específica en este paciente con SSc. Después de 10 años, se observa un aumento de la reticulación y la bronquiolectasis de tracción en las bases pulmonares con disminución de la opacidad del vidrio molido en imágenes de HRCT axial(C)y coronal(D),consistentes con un empeoramiento leve de la fibrosis pulmonar. HRCT : tomografía computarizada de alta resolución; SSc-ILD : enfermedad pulmonar intersticial asociada a la esclerodermia sistémica. Haga clic aquí para ver una versión más grande de esta figura.
Si bien el HRCT es actualmente el método de diagnóstico por imágenes definitivo para diagnosticar y evaluar la SSc-ILD, utiliza radiación ionizante y es relativamente costoso. En su lugar, se pueden llevar a cabo radiografías de tórax, aunque no facilitan el diagnóstico diferencial en la misma medida que el HRCT, y una radiografía de tórax normal no elimina la posibilidad de ILD. Tal vez el mejor uso de las radiografías de tórax es monitorear la enfermedad progresiva entre las exploraciones de HRCT y la exclusión de enfermedades que complican, como la neumonía infecciosa, en el entorno de empeoramiento agudo de los síntomas.
Una limitación percibida del HRCT es la exposición a la radiación. Como se describió anteriormente, los nuevos métodos para realizar tomografías computarizadas pueden permitir reducir la exposición a la radiación31y, además, los escáneres de TC actuales proporcionan una serie de técnicas avanzadas que ofrecen la posibilidad en el futuro de reducir la exposición a la radiación a niveles de radiografía casi tórax. Alternativamente, los métodos de diagnóstico por imágenes como la RMN o el ultrasonido pulmonar podrían utilizarse potencialmente para evitar exponer al paciente a la radiación en el futuro32,,71,,72,,73. Creemos que, si bien existen consideraciones de riesgo-beneficio asociadas con la utilización de imágenes, las ventajas de la tomografía computarizada en el diagnóstico y el manejo del paciente superan con creces los riesgos potenciales.
Los datos de imágenes, en particular el HRCT, proporcionan sin duda la información más importante para permitir el diagnóstico de SSc-ILD. La consideración detallada de los patrones y características de las exploraciones HRCT suele ser suficiente para distinguir la SSc-ILD de otras enfermedades pulmonares, con el beneficio de evitar la necesidad de un procedimiento de biopsia invasiva.
La evaluación visual de los escaneos HRCT introduce un grado de subjetividad y la posibilidad de variabilidad entre observadores. Los métodos informáticos de interpretación de escaneo HRCT han sido investigados como un posible enfoque para mejorar la precisión63,,74. Por ejemplo, se pueden utilizar enfoques cuantitativos para la evaluación de la fibrosis pulmonar o el alcance de la enfermedad para evaluar la respuesta al tratamiento68,70,75. Sin embargo, estos métodos no son ampliamente utilizados en la práctica clínica diaria en este momento.
Esperamos que la información presentada en este manuscrito sirva como una guía práctica para ayudar a los médicos en el uso de escaneos de HRCT para diagnosticar SSc-ILD y determinar el pronóstico. Los métodos mejorados para obtener imágenes y para interpretar las exploraciones tienen el potencial de reducir la exposición de los pacientes a la radiación y mejorar la precisión diagnóstica/pronostica.
Jonathan Chung no declara posibles conflictos de intereses con respecto a la investigación, autoría y/o publicación descrita de este artículo; Christopher Walker declara la recepción de regalías de Amirsys (Elsevier); y Stephen Hobbs declara la recepción de regalías de Elsevier y Wolters Kluwer Health. La publicación de Open Access de este artículo está patrocinada por Boehringer Ingelheim Pharmaceuticals, Inc.
Los autores cumplen los criterios de autoría recomendados por el Comité Internacional de Editores de Revistas Médicas (ICMJE). Los autores no recibieron ninguna compensación directa relacionada con el desarrollo del video. El soporte de escritura médica para el video guion fue proporcionado por Leon Newman, PhD, de GeoMed, una compañía de Ashfield, que fue contratada y financiada por Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI). Se dio a BIPI la oportunidad de revisar el vídeo final para la exactitud médica y científica, así como las consideraciones de propiedad intelectual.
Los autores cumplen con los criterios de autoría recomendados por el Comité Internacional de Editores de Revistas Médicas (ICMJE). Los autores no recibieron ninguna compensación directa relacionada con el desarrollo del manuscrito. La asistencia para escribir fue proporcionada por Ken Sutor, BSc, de GeoMed, una compañía de Ashfield, parte de UDG Healthcare plc, que fue contratada y financiada por Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI). SE dio a BIPI la oportunidad de revisar el manuscrito para la exactitud médica y científica, así como consideraciones de propiedad intelectual.
| Name | Company | Catalog Number | Comments |
| CT scanners | Philips | NA | Multiple |
- Denton, C. P., Khanna, D. Systemic sclerosis. Lancet. 390 (10103), 1685-1699 (2017).
- Arnett, F. C., et al. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis & Rheumatism. 44 (6), 1359-1362 (2001).
- Barnes, J., Mayes, M. D. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Current Opinion in Rheumatology. 24 (2), 165-170 (2012).
- Marie, I., Gehanno, J. F. Environmental risk factors of systemic sclerosis. Seminars in Immunopathology. 37 (5), 463-473 (2015).
- Silman, A. J. Epidemiology of scleroderma. Annals of the Rheumatic Diseases. 50, 846-853 (1991).
- Scholand, M. B., et al. Interstitial lung disease in systemic sclerosis: diagnosis and management. Rheumatology. 1, 008 (2012).
- Solomon, J. J., et al. Scleroderma lung disease. European Respiratory Review. 22 (127), 6-19 (2013).
- Walker, U. A., et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Annals of the Rheumatic Diseases. 66 (6), 754-763 (2007).
- Hinchcliff, M., Fischer, A., Schiopu, E., Steen, V. D., Investigators, P. Pulmonary hypertension assessment and recognition of outcomes in scleroderma (PHAROS): baseline characteristics and description of study population. The Journal of Rheumatology. 38 (10), 2172-2179 (2011).
- Giacomelli, R., et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatology International. 37 (6), 853-863 (2017).
- Sanchez-Cano, D., et al. Interstitial lung disease in systemic sclerosis: data from the spanish scleroderma study group. Rheumatology International. 38 (3), 363-374 (2018).
- Silver, K. C., Silver, R. M. Management of systemic-sclerosis-associated interstitial lung disease. Rheumatic Diseases Clinics of North America. 41 (3), 439-457 (2015).
- Fischer, A., Kong, A. M., Swigris, J. J., Cole, A. L., Raimundo, K. All-cause healthcare costs and mortality in patients with systemic sclerosis with lung involvement. The Journal of Rheumatology. 45 (2), 235-241 (2018).
- Tyndall, A. J., et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Annals of the Rheumatic Diseases. 69 (10), 1809-1815 (2010).
- Herzog, E. L., et al. Review: interstitial lung disease associated with systemic sclerosis and idiopathic pulmonary fibrosis: how similar and distinct. Arthritis & Rheumatology. 66 (8), 1967-1978 (2014).
- Rubio-Rivas, M., Royo, C., Simeon, C. P., Corbella, X., Fonollosa, V. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Seminars in Arthritis and Rheumatism. 44 (2), 208-219 (2014).
- Stock, C., et al. Serum KL-6 as a marker of disease progression in SSc-ILD. European Respiratory Journal. 52, (2018).
- Winstone, T. A., et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: a systematic review. Chest. 146 (2), 422-436 (2014).
- Khanna, D., et al. Ongoing clinical trials and treatment options for patients with systemic sclerosis-associated interstitial lung disease. Rheumatology (Oxford). 58 (4), 567-579 (2019).
- Behr, J., Furst, D. E. Pulmonary function tests. Rheumatology (Oxford). 47, 65-67 (2008).
- Hax, V., et al. Clinical algorithms for the diagnosis and prognosis of interstitial lung disease in systemic sclerosis. Seminars in Arthritis and Rheumatism. 47 (2), 228-234 (2017).
- Molberg, O., Hoffmann-Vold, A. M. Interstitial lung disease in systemic sclerosis: progress in screening and early diagnosis. Current Opinion in Rheumatology. 28 (6), 613-618 (2016).
- Raghu, G., Goldman, L., Schafer, A. I. Interstital lung disease. Goldman-Cecil Medicine. , 575-588 (2016).
- Showalter, K., et al. Performance of forced vital capacity and lung diffusion cutpoints for associated radiographic interstitial lung disease in systemic sclerosis. The Journal of Rheumatology. 45 (11), 1572-1576 (2018).
- Suliman, Y. A., et al. Brief report: pulmonary function tests: high rate of false-negative results in the early detection and screening of scleroderma-related interstitial lung disease. Arthritis & Rheumatology. 67 (12), 3256-3261 (2015).
- Roth, M. D., et al. Predicting treatment outcomes and responder subsets in scleroderma-related interstitial lung disease. Arthritis & Rheumatology. 63 (9), 2797-2808 (2011).
- Kalra, M. K., Maher, M. M., Rizzo, S., Kanarek, D., Shepard, J. A. Radiation exposure from chest CT: issues and strategies. Journal of Korean Medical Science. 19 (2), 159-166 (2004).
- Siegel, J. A., Pennington, C. W., Sacks, B., Welsh, J. S. The birth of the illegitimate linear no-threshold model: an invalid paradigm for estimating risk following low-dose radiation exposure. American Journal of Clinical Oncology. 41 (2), 173-177 (2018).
- Frauenfelder, T., et al. Screening for interstitial lung disease in systemic sclerosis: performance of high-resolution CT with limited number of slices: a prospective study. Annals of the Rheumatic Diseases. 73 (12), 2069-2073 (2014).
- Kubo, T., et al. Radiation dose reduction in chest CT: a review. American Journal of Roentgenology. 190 (2), 335-343 (2008).
- Nguyen-Kim, T. D. L., et al. The impact of slice-reduced computed tomography on histogram-based densitometry assessment of lung fibrosis in patients with systemic sclerosis. Journal of Thoracic Disease. 10 (4), 2142-2152 (2018).
- Muller, C. S., Warszawiak, D., Paiva, E. D. S., Escuissato, D. L. Pulmonary magnetic resonance imaging is similar to chest tomography in detecting inflammation in patients with systemic sclerosis. Revista Brasileira de Reumatologia English Edition. 57 (5), 419-424 (2017).
- Pinal-Fernandez, I., et al. Fast 1.5 T chest MRI for the assessment of interstitial lung disease extent secondary to systemic sclerosis. Clinical Rheumatology. 35 (9), 2339-2345 (2016).
- Sverzellati, N. Highlights of HRCT imaging in IPF. Respiratory Research. 14, 3 (2013).
- Lynch, D. A., et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. The Lancet Respiratory Medicine. 6 (2), 138-153 (2018).
- Raghu, G., et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. American Journal of Respiratory and Critical Care Medicine. 198 (5), 44-68 (2018).
- . Exam: CT thorax for interstitial lung disease Available from: https://www.pulmonaryfibrosis.org/docs/default-source/medical-community-documents/ct-chest-for-ild-protocol.pdf?sfvrsn=0 (2019)
- Doyle, T. J., Hunninghake, G. M., Rosas, I. O. Subclinical interstitial lung disease: why you should care. American Journal of Respiratory and Critical Care Medicine. 185 (11), 1147-1153 (2012).
- Peroni, D. G., Boner, A. L. Atelectasis: mechanisms, diagnosis and management. Paediatric Respiratory Reviews. 1 (3), 274-278 (2000).
- Branley, H. M. Pulmonary fibrosis in systemic sclerosis: diagnosis and management. Respiratory Medicine CME. 3, 10-14 (2010).
- Engeler, C. E., Tashjian, J. H., Trenkner, S. W., Walsh, J. W. Ground-glass opacity of the lung parenchyma: a guide to analysis with high-resolution CT. American Journal of Roentgenology. 160 (2), 249-251 (1993).
- Goldin, J. G., et al. High-resolution CT scan findings in patients with symptomatic scleroderma-related interstitial lung disease. Chest. 134 (2), 358-367 (2008).
- Strollo, D., Goldin, J. Imaging lung disease in systemic sclerosis. Current Rheumatology Reports. 12 (2), 156-161 (2010).
- Pandey, A. K., et al. Significance of various pulmonary and extrapulmonary abnormalities on HRCT of the chest in scleroderma lung. Indian Journal of Radiology and Imaging. 23 (4), 304-307 (2013).
- Chung, J. H., et al. CT Features of the usual interstitial pneumonia pattern: differentiating connective tissue disease-associated interstitial lung disease from idiopathic pulmonary fibrosis. American Journal of Roentgenology. 210 (2), 307-313 (2018).
- Zhan, X., et al. Differentiating usual interstitial pneumonia from nonspecific interstitial pneumonia using high-resolution computed tomography: the "Straight-edge Sign". Journal of Thoracic Imaging. 33 (4), 266-270 (2018).
- Farrokh, D., Abbasi, B., Fallah-Rastegar, Y., Mirfeizi, Z. The extrapulmonary manifestations of systemic sclerosis on chest high resolution computed tomography. Tanaffos. 14 (3), 193-200 (2015).
- Salaffi, F., et al. Relationship between interstitial lung disease and oesophageal dilatation on chest high-resolution computed tomography in patients with systemic sclerosis: a cross-sectional study. La Radiologia Medica. 123 (9), 655-663 (2018).
- Vonk, M. C., et al. Oesophageal dilatation on high-resolution computed tomography scan of the lungs as a sign of scleroderma. Annals of the Rheumatic Diseases. 67 (9), 1317-1321 (2008).
- Chowaniec, M., Skoczynska, M., Sokolik, R., Wiland, P. Interstitial lung disease in systemic sclerosis: challenges in early diagnosis and management. Reumatologia. 56 (4), 249-254 (2018).
- McCall, R. K., Ravenel, J. G., Nietert, P. J., Granath, A., Silver, R. M. Relationship of main pulmonary artery diameter to pulmonary arterial pressure in scleroderma patients with and without interstitial fibrosis. Journal of Computer Assisted Tomography. 38 (2), 163-168 (2014).
- Pandey, A. K., et al. Predictors of pulmonary hypertension on high-resolution computed tomography of the chest in systemic sclerosis: a retrospective analysis. Canadian Association of Radiologists Journal. 61 (5), 291-296 (2010).
- Raymond, T. E., Khabbaza, J. E., Yadav, R., Tonelli, A. R. Significance of main pulmonary artery dilation on imaging studies. Annals of the American Thoracic Society. 11 (10), 1623-1632 (2014).
- Colaci, M., et al. Lung cancer in scleroderma: results from an Italian rheumatologic center and review of the literature. Autoimmunity Reviews. 12 (3), 374-379 (2013).
- Distler, O., et al. Design of a randomised, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease (SENSCIS). Clinical and Experimental Rheumatology. 35 (4), 75-81 (2017).
- Desai, S. R., et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology. 232 (2), 560-567 (2004).
- Mira-Avendano, I., et al. Interstitial lung disease and other pulmonary manifestations in connective tissue diseases. Mayo Clinic Proceedings. 94 (2), 309-325 (2019).
- Walkoff, L., White, D. B., Chung, J. H., Asante, D., Cox, C. W. The four corners sign: a specific imaging feature in differentiating systemic sclerosis-related interstitial lung disease from idiopathic pulmonary fibrosis. Journal of Thoracic Imaging. 33 (3), 197-203 (2018).
- Kotnur, M. R., Suresh, P., Reddy, V. S., Sharma, T., Salim, N. A. Systemic sclerosis with multiple pulmonary manifestations. Journal of Clinical & Diagnostic Research. 10 (6), 16-17 (2016).
- Goh, N. S., et al. Interstitial lung disease in systemic sclerosis: a simple staging system. American Journal of Respiratory and Critical Care Medicine. 177 (11), 1248-1254 (2008).
- Takei, R., et al. Radiographic fibrosis score predicts survival in systemic sclerosis-associated interstitial lung disease. Respirology. 23 (4), 385-391 (2018).
- Bakker, M. E., et al. Lung density and pulmonary artery diameter are predictors of pulmonary hypertension in systemic sclerosis. Journal of Thoracic Imaging. 32 (6), 391-397 (2017).
- Khanna, D., et al. Predictors of lung function decline in scleroderma-related interstitial lung disease based on high-resolution computed tomography: implications for cohort enrichment in systemic sclerosis-associated interstitial lung disease trials. Arthritis Research & Therapy. 17, 372 (2015).
- Ariani, A., et al. Quantitative chest computed tomography is associated with two prediction models of mortality in interstitial lung disease related to systemic sclerosis. Rheumatology (Oxford). 56 (6), 922-927 (2017).
- Goldin, J., et al. Treatment of scleroderma-interstitial lung disease with cyclophosphamide is associated with less progressive fibrosis on serial thoracic high-resolution CT scan than placebo: findings from the scleroderma lung study. Chest. 136 (5), 1333-1340 (2009).
- Tashkin, D. P., et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. The Lancet Respiratory Medicine. 4 (9), 708-719 (2016).
- Distler, O., et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. New England Journal of Medicine. 380, 2518-2528 (2019).
- Cappelli, S., et al. Interstitial lung disease in systemic sclerosis: where do we stand. European Respiratory Review. 24 (137), 411-419 (2015).
- Goldin, J. G., et al. Longitudinal changes in quantitative interstitial lung disease on CT after immunosuppression in the Scleroderma Lung Study II. Annals of the American Thoracic Society. 5 (11), 1286-1295 (2018).
- Wangkaew, S., Euathrongchit, J., Wattanawittawas, P., Kasitanon, N. Correlation of delta high-resolution computed tomography (HRCT) score with delta clinical variables in early systemic sclerosis (SSc) patients. Quantitative Imaging in Medicine and Surgery. 6 (4), 381-390 (2016).
- Kim, H. J., et al. Transitions to different patterns of interstitial lung disease in scleroderma with and without treatment. Annals of the Rheumatic Diseases. 75 (7), 1367-1371 (2016).
- Tardella, M., et al. Ultrasound B-lines in the evaluation of interstitial lung disease in patients with systemic sclerosis: cut-off point definition for the presence of significant pulmonary fibrosis. Medicine (Baltimore). 97 (18), 0566 (2018).
- Hassan, R. I., et al. Lung ultrasound as a screening method for interstitial lung disease in patients with systemic sclerosis. Journal of Clinical Rheumatology. , (2018).
- Wang, Y., Gargani, L., Barskova, T., Furst, D. E., Cerinic, M. M. Usefulness of lung ultrasound B-lines in connective tissue disease-associated interstitial lung disease: a literature review. Arthritis Research & Therapy. 19 (1), 206 (2017).
- Ariani, A., et al. Quantitative CT indexes are significantly associated with exercise oxygen desaturation in interstitial lung disease related to systemic sclerosis. The Clinical Respiratory Journal. 11 (6), 983-989 (2017).
- Kim, H. J., et al. Quantitative texture-based assessment of one-year changes in fibrotic reticular patterns on HRCT in scleroderma lung disease treated with oral cyclophosphamide. European Radiology. 21 (12), 2455-2465 (2011).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved