Near Simultaneous Laser Scanning Confocal and Atomic Force Microscopy (Conpokal) on Live Cells
In This Article
Summary
Presented here is the protocol of the Conpokal technique, which combines confocal and atomic force microscopy into a single instrument platform. Conpokal provides same cell, same region, near simultaneous confocal imaging and mechanical characterization of live biological samples.
Abstract
Techniques available for micro- and nano-scale mechanical characterization have exploded in the last few decades. From further development of the scanning and transmission electron microscope, to the invention of atomic force microscopy, and advances in fluorescent imaging, there have been substantial gains in technologies that enable the study of small materials. Conpokal is a portmanteau that combines confocal microscopy with atomic force microscopy (AFM), where a probe “pokes” the surface. Although each technique is extremely effective for the qualitative and/or quantitative image collection on their own, Conpokal provides the capability to test with blended fluorescence imaging and mechanical characterization. Designed for near simultaneous confocal imaging and atomic force probing, Conpokal facilitates experimentation on live microbiological samples. The added insight from paired instrumentation provides co-localization of measured mechanical properties (e.g., elastic modulus, adhesion, surface roughness) by AFM with subcellular components or activity observable through confocal microscopy. This work provides a step by step protocol for the operation of laser scanning confocal and atomic force microscopy, simultaneously, to achieve same cell, same region, confocal imaging, and mechanical characterization.
Introduction
Micro- or nano-scale mechanical evaluation tools are often employed to uncover the deformation characteristics on the single cell or microorganism level, while separate high resolution microscopy tools are employed to visualize subcellular processes and structural features. Conpokal, is the combination of laser scanning confocal microscopy and atomic force microscopy (AFM) into a single instrumental platform. The Conpokal technique was first implemented in a non-biological polymer system, where the goal was to determine the adhesion, elasticity, and surface tension of hard surfaces in contact with soft surfaces. Confocal images provided a visual rendering of how the polymer deforms, adheres, and releases from the hard probe1,2. From polymers to biological samples, this technique provides the opportunity for near simultaneous live cell or microorganism confocal imaging and AFM.
Changes in cell elasticity have been implicated in the pathogenesis of many human diseases3 including vascular disorders4, malaria5,6, sickle cell anemia7, arthritis8, asthma9, and cancer6,10,11,12,13,14,15. Common techniques to measure the mechanics of cells include the use of magnetic beads16,17, optical tweezers18,19, micropipette aspiration8,20,21,22, and AFM11,23,24,25,26. Since the application of AFM to living cells, it has readily been adapted to characterize cell topography27,28 as well as mechanical properties11,24,29,30,31 with nanoscale precision. AFM has been further adapted for microrheology32,33, frequency modulation34,35, and creep25,36,37 experiments to study the viscoelastic properties of various cell lines. The use of an appropriate nanocontact model is critical to the extraction of quantitative elasticity values from AFM-produced force indentation measurements1,2. AFM studies that investigate the influence of cytoskeletal drugs on cell elasticity have shown the elastic modulus to be highly affected38. Based on the elastic modulus measurements, many researchers have shown that cancerous cells are softer than their non-transformed counterparts11,38,39. Increased deformability likely plays a prominent role in the ability of cancer cells to metastasize and infiltrate tissues40. Such behavior is regulated by modifications in the cytoskeletal organization of cells38,41,42. In addition to cancer, another class of mechanical-dependent diseases are respiratory illnesses. For example, acute respiratory stress syndrome affects more and more humans each year. It has been shown that when patients are put on mechanical ventilation, with high concentrations of oxygen, their condition can worsen43. In a series of studies observing alveolar epithelial macrophages with AFM, scientists found that the addition of an oxygen-rich environment increased the stiffness of cells due to actin formation; a prime example of the impact that AFM has on our detailed understanding of atmospheric environmental influence on respiratory function at the cellular level and, ultimately, human healing and health44,45,46. Epithelial cell stiffness during migration toward a wound can also be assessed via AFM and that a variation in elastic modulus occurs to signal future cell spreading47,48. Therefore, there is a practical need to measure cell mechanics quantitatively to understand how diseased cells differ from, and interact with, healthy ones.
AFM is also used to study adhesive properties and surface structure. An atomic force microscope has a variety of modes to collect information about a sample using contact mechanics. For living biological samples, two of the primary objectives are to obtain quantitative mechanical property values (e.g., elastic moduli, adhesive forces) as well as map surface height. These modes include tapping mode (also known as intermittent contact), which maintains a constant cantilever amplitude intermittently contacting the sample surface and contact mode, which raises or lowers the cantilever to maintain a constant deflection49. Adhesion maps can be collected using force mapping on the AFM system. The cantilever indents the sample to a certain force and is then retracted. The force resisting retraction is measured by the detector to generate a force-displacement graph. The adhesion force is the maximum force measured during retraction. Surface morphology provides a detailed view of the sample on the micro- or nano-level. The cantilever completes a raster scan across the sample based on the indentation and deflection captured by the motion of the cantilever at each pixel, revealing micro- and nano-sized features. With AFM, the size of small features such as peptidoglycan structure50, polymer chain alignment51, flagella52, lamellipodia53, and filipodia54 can be measured. While AFM’s power lies in force-displacement curves and non-optical topological images, confocal microscopy offers detailed imaging of fluorescently labeled samples.
Confocal laser scanning microscopy (CLSM) is a dominant technique for imaging live cells, fixed cells, and tissues55,56,57. CLSM has been employed for biological imaging since 1955 i.e., since its inception58,59. While the working principle of confocal microscopes (i.e., the rejection of out of focus light through a pinhole) has remained largely the same, the technical implementation has become very diverse56,57,58,60. Confocal imaging can be implemented via multipoint scanning, as in a spinning disc system61, point scanning of a single laser beam, as in line scanning62, or imaging the point spread function with subsequent pixel reassignments via a multi-element detector57. Recent advances that expand the utility of confocal imaging include laser scanning capability, high speed resonance scanning, and high sensitivity detectors63,64,65. The advantage that all confocal systems have over widefield microscopes is the ability to perform optical sectioning in the z axis with greater detail, especially in thick samples. Widefield microscopes using camera-based detection collect light that is emitted from the focal plane and, simultaneously, light emitted from everywhere in the illumination path. By closing the pinhole, at the cost of reducing the signal, both the axial and lateral resolution can be increased66. In CLSM, images are built up pixel by pixel through emission signal collection as an excitation laser is scanned across an x-y field of view. Collection of several x-y planes along the z axis of the sample can then be used to reconstruct the 3-dimensional architecture of the sample57,67. Confocal microscopy in conjunction with the introduction of fluorescent proteins, has revolutionized our understanding of cell biology68. For example, it has become an essential tool in neuroscience69. The CLSM is regularly used to observe cleared tissue, and to obtain comprehensive images of immune stained brain and neuronal network architecture70. This application has resulted in new insights into synaptic contacts and communication among neurons and glial cells in the brain71. From brain cells to vesicles, fluorescent staining protocols support inspection and capture of cell functions via confocal microscopy for advancements in therapeutic techniques72,73.
Although they are impressive and versatile tools individually, the combination of confocal and atomic force microscopy (Conpokal) allows researchers to uniquely correlate topographical and micromechanical cellular/tissue properties with the observation of various cellular organelles and their respective dynamics. Paired instrumentation allows users to, essentially, “see” what they are probing; a huge advantage over one technique on its own. With Conpokal, force mapping through AFM is overlaid on confocal images to correlate cell stiffness, adhesion, or morphology to the cytoskeletal structure within the sample, for example. The overall goal of the Conpokal method is to provide a platform for researchers to study living samples in a concise, effective manner, providing both image and material property data in a single platform. In this work, we demonstrate the operation of Conpokal including proper sample selection and preparation, instrument setup, cantilever calibration, and provide guidelines to successful troubleshooting. After the protocol, we provide representative results that include successful bacteria and cell AFM height mapping, bacteria and cell AFM modulus mapping, and confocal imaging of multi-labeled cells, through same-cell confocal and AFM. We conclude with a discussion of the Conpokal procedure critical steps, troubleshooting recommendations, technique limitations, significance, and future outlook for the method.
Protocol
1. Preparation of instruments, culture media, dishes
- Turn on the power sources for both the confocal microscope and the atomic force microscope, as well as any other associated instruments or devices utilized during Conpokal. Allow enough time for the instruments to thermally equilibrate and stabilize before beginning measurements. Typically, 1 h is sufficient.
- Prepare one clean, system-approved, glass-bottomed Petri dish and fill half full, but no more than two-thirds full, with phosphate buffered saline (PBS), or similar clear liquid. This dish (referred to as the “calibration dish”) is separate from the sample dishes and will be used to locate and calibrate the atomic force microscope (AFM) cantilever.
NOTE: It is important that the liquid is clear and has low autofluorescence to prevent any interference with the fluorescence spectra. PBS is recommended because it mimics the biological environment. - Prepare experimental sample dishes such that the associated liquid has filled the dish at least half full. If the sample is fluorescent, ensure it is covered or in a dark place until needed. Clean the bottom of all glass dishes using lens cleaner and lens wipes.
- Create data storage folders for the confocal and AFM on the computer(s) hard drive(s).
2. Selection of cells or microorganisms
- To create bacteria stock solution, inoculate Streptococcus mutans bacteria, using an inoculation loop, in 15 mL of Todd Hewitt Yeast (THY) overnight (for exponential phase) in a 5% CO2 incubator.
- 1 h before the overnight incubation is complete, coat experimental sample dishes with 1 mL of poly-l-lysine solution or enough to cover the glass portion of the glass-bottomed dish and leave in the biosafety cabinet for 1 h. After setting, aspirate poly-l-lysine solution and rinse the dishes 3x with PBS.
- After 18 – 24 h of bacterial incubation, inoculate THY with 1 mL of bacterial suspension until an optical density of 0.6 – 0.7 is achieved (typical values for inoculation are 3 mL of THY with 1 mL of bacterial suspension per dish). Add 1 mL of new bacteria solution to each poly-l-lysine-coated dish and incubate for 1 h. During the last 10 min, add 1 mL of green fluorescent membrane stain (concentration of 1 µM) to each dish.
- Rinse each dish 3x with PBS. Gently fix the bacteria with about 1 mL of 4% paraformaldehyde solution for 15 min at room temperature inside a biosafety cabinet. Rinse each dish 3x with PBS after fixation.
- Store the Human Embryonic Kidney (HEK) 293 cells in liquid nitrogen. Perform fast thawing at 37 ˚C water bath before plating.
- Add 1 mL of basement membrane matrix solution in cell culture media to each cell sample dish and incubate (5% CO2) at 37 ˚C for 1 h. Aspirate the solution and wash the dishes with cell culture media. Plate the required number of HEK 293 cells (e.g., 100,000 cells per 35 mm dish) and add 2 mL of cell culture media. Then, incubate (5% CO2) the cells at 37 ˚C for 48 h.
- After 48 h, add 2 µL of a fluorescent microtubule cytoskeleton dye (concentration of 1x) to each cell sample dish and incubate for 30 min. Aspirate the solution containing the dye, wash the cells 3x with PBS, and add 2 mL of cell culture media.
- Add 1 µL of plasma membrane dye (concentration of 10 µM) to the sample dishes and incubate for 30 minutes. Wash the solution containing the dye 3x with PBS and add 2 mL of cell culture media.
- Counterstain the nucleus by adding 1 µL of a nucleic acid dye (concentration of 10 µM) solution to the sample dishes and incubate for 30 min. Wash the solution containing the dye 3x with PBS and add 2 mL of culture media.
3. AFM procedure
- Open the atomic force microscope (AFM) operating software by clicking on the software icon on the computer.
- Rotate to or insert a low magnification air objective (recommended 10x or 20x). A long working distance of 5 mm or more is beneficial during tip lowering and calibration.
- Mount the chosen cell sample stage if not already installed. For live samples, use a dish heater to keep the sample at desired temperature.
- Load the clean, system-approved Petri dish filled half full of PBS (prepared in step 1.2) or similar clear liquid (calibration dish). If sample stage has built-in clasps, use these, otherwise, use vacuum grease to stabilize the sample.
- Select an appropriate AFM cantilever for the desired data collection and install it following the steps below.
- Using gloves, mount the AFM chip into the glass block by using the AFM chip mounting stage, tweezers, and a small screwdriver. Carefully place the AFM chip onto the glass block and orient it such that it is centered. The cantilever, plus a very small portion of the AFM chip, should be in the visible, non-opaque portion of the mounting block.
- Secure the chip with the screwdriver by tightening the screw until the chip is snug against the glass block. Check that the AFM cantilever is oriented correctly using a stereo microscope or a handheld spyglass. Adjust as needed. Once properly oriented, place the glass block into the AFM head in the proper orientation and lock it in place.
NOTE: The orientation of the AFM chip within the glass block is important so that the system lasers aim at the backside of the cantilever and reflect onto the photodetector correctly. Be careful not to overtighten the screws during the placement as this procedure could cause the AFM chip, cantilever, or tip to break.
- Using the brightfield option of the confocal microscope, locate the bottom of the calibration dish by using the focus knob. Take note of this z height as the value where the bottom of the dish is located with respect to the microscope objective.
- Using the AFM system’s motor control panel on the computer that operates the z stepper motors, move the AFM cantilever away from the sample upwards 2000 µm.
- Using both hands, gently place the AFM head with the AFM chip onto the sample stage making sure that each of the vertical points on the AFM head are firmly set in their designated grooves on the AFM stage. Once the AFM head is in place, visually see how close the cantilever holder is to the dish. If it appears to be too close, such that the AFM head is in contact or within 1 mm of the top of the sample dish, back it out using the z stepper motors before lowering the AFM tip towards the sample.
- Using brightfield illumination, slowly lower the AFM chip to the bottom of the glass dish using the z stepper motor control panel on the AFM software. Locate the manual micrometers that control x-y or in-plane motion of the AFM head on the instrument platform. Adjust the position of the AFM cantilever within the field of view by correcting the AFM head as the cantilever comes into view.
- Since the location with respect to the bottom of the dish is unknown at this time, lower with steps of 100 – 200 µm to avoid crashing the AFM tip into the Petri dish. Typically, the tip needs to be lowered 800 – 2000 µm. As the tip is lowered, watch for a shadow to appear on the microscope software view, which indicates the AFM tip is getting closer to the bottom of the dish.
- Decrease increments to 20 – 50 µm. Be sure to adjust the look up tables (LUTs) of the camera image as the tip lowers into the dish by using the LUTs panel on the confocal software. Continue to lower the AFM tip using small steps until it is mostly in-focus.
- Ensure that the tip is dark enough so that the general shape of it can be seen and centered in the field of view. Also ensure that the tip appears blurry, which confirms that it has not encounter the bottom of the dish, yet.
- Adjust the microscope focus such that the tip is now in focus, keeping in mind the working distance of the objective. Before moving to a higher magnification objective, use the microscope measurement tool to collect the width and length of the AFM cantilever by accessing the measurement tools panel on the confocal software. Take note of these measurements.
NOTE: It is important not to crash the objective into the bottom of the sample dish which may also cause the sample dish to come into contact with the AFM tip, potentially damaging it. - If present, remove the laser light filter, and ensure the AFM laser is on, so that the laser light becomes visible in the optics.
- Using the laser alignment dials, move the laser into the field of view and near the AFM tip. Once the laser is at this location, replace the laser light filter.
- Using the laser alignment dials, place the laser on the backside of where the AFM tip is located on the cantilever. At this point, the laser alignment panel should be reading a sum signal greater than 0.0 V. If no sum signal is obtained, adjust the mirror by using the manually controlled mirror knob until a sum signal greater than 0.0 V reads on the screen.
- Move the laser in small amounts in all directions on the AFM cantilever until the maximum sum signal is achieved but the position is at the AFM tip. Once the laser position is set, zero the vertical and lateral deflection by using the manually controlled deflection dials.
- Observe the laser alignment panel on the AFM software and by using the vertical and lateral deflection knobs on the AFM head, align the detector so that the target is centered (red dot in center of crosshairs) and there is no vertical or lateral deflection.
- Open the calibration window in the AFM software. Input all experiment specific information. Before calibrating, turn off the confocal microscope light source and close the AFM enclosure, if applicable, to dampen any potential noise coming from the room light or room vibrations. Then press the calibration button to automatically allow the system to calibrate the tip. After calibration is complete, the stiffness of the cantilever (N/m) and its sensitivity (nm/V) will be provided within the calibration panel. Note these for later analysis.
- Retract the AFM cantilever at least 2000 µm with the stepper motors. Take the AFM head off the stage and remove the calibration dish. Rotate to or insert the desired objective. Retract the objective 10% of the working distance if the system is programmed to maintain the same focal plane between objectives. If this is an oil objective, place one drop of immersion oil on the eye of the objective.
NOTE: The retraction of the objective reduces the likelihood of the objective contacting the sample dish during placement. - To securely hold the sample dish in place, use a cotton swab to apply small dabs of vacuum grease around the edge of the sample ring within the sample stage, or employ sample clips. Carefully load the sample dish into the sample stage.
- Once the sample is placed, rotate it a few times to ensure a good seal of the dish to the sample holder via the grease. Raise the objective to the bottom of the dish until the oil just wicks over the eye of the objective.
- Using the microscope, without the AFM head on the stage, focus the imaging system so that the sample is in focus. Note this z height for reference.
- Carefully replace the AFM head onto the platform.
- Using the z stepper motors, lower the tip towards the sample. As the AFM tip is lowered, adjust the look up tables (LUTs) in the brightfield image as needed. Lower the tip until almost sharp. Using the manually operated AFM tip position micrometers, move the AFM head so that the AFM tip is in the center or left centered in the field of view.
NOTE: Since the z height of the AFM was noted before in step 3.6, larger steps may be taken to lower the AFM tip, however, grease has been added to the bottom of the dish and potentially oil to the objective, so this value is just for reference. It is recommended to take 50 – 100 µm steps and adjusting smaller as the tip comes into focus - Readjust the laser position by using the manual micrometers on the AFM head. If needed, zero the vertical and lateral deflections by adjusting the respective knobs and recalibrate the AFM cantilever in the sample dish.
NOTE: If the cantilever was calibrated in a medium different from the sample, it must be recalibrated in the scanning medium to account for a potential change in refractive index. Additionally, the laser may drift from the backside of the cantilever due to noise or temperature changes. Only readjust the laser and recalibrate if necessary and in the sample dish above the glass substrate if using non-contact calibration. If using contact calibration, recalibrate inside of the calibration dish using the sample medium. - To achieve high quality confocal microscope images, replace the current attenuating light filter with the filter that blocks all AFM laser light. This light filter ensures no interference from the bright light of the AFM laser.
- Using the automated approach command button, typically denoted by a downward facing arrow, lower the tip to the bottom of the sample dish. Set the size/area of the scan, the resolution, the setpoint, the z length, and the pixel time (or use the calibrated/propagated values) on the imaging control panel and begin scanning.
- After a scan is complete, lift the AFM chip 50 – 100 µm up before navigating to a new measurement position in the sample dish. Once a new location is found, approach the glass, and start to scan again following the steps 3.21 and 3.22.
- Select the Autosave option, or manually save each generated file from each individual scan by clicking on Save.
4. Confocal procedure
- Open the confocal operating software. Ensure all associated peripherals, light sources, and controllers are switched on and functioning normally.
- Rotate or insert an objective of low magnification (10x or 20x) to view sample. Ensure the objective is at a safe working distance from where the sample dish will be and if needed, back-out the objective to avoid any potential collision of the objective with the sample. Using a cotton swab, dab vacuum grease around the edge of the sample dish ring. If using an oil objective, place a single drop of immersion oil onto the front lens of the objective
- Place desired sample into the sample stage. Spin the sample around a few times to ensure that vacuum grease has generated a tight bond to the sample stage insert or employ sample clips. If using an oil objective, raise the objective to the bottom of the dish so the oil just wicks over the glass bottom. To avoid excessive bleaching, minimize ambient lighting.
- Using brightfield or epifluorescence modes via the camera or eyepieces, locate the sample and using the focus knob, focus on an area of interest containing multiple cells or a single cell. Often, the field of view within the optical microscope will be larger than the x-y scanning window on the AFM system (100 x 100 μm). Make adjustments of the AFM position within the confocal field of view by using the motor control panel in the AFM software so that the AFM scan and confocal optics are imaging the same features.
NOTE: Within the field of view, there are typically only a few cells, so it is easier to determine which cell is desired to probe. As AFM is conducted, the cell becomes visible on the AFM software and, from there, the user can pinpoint specific regions of interest to probe. - If desired, capture images in bright-field or epifluorescence using a camera or CLSM modes based on the label of the sample, focus knobs, and capture buttons on the microscope software.
- Using the microscope software, select the option or open the panel to enable confocal capabilities (e.g., lasers and subsequent parameters). This option is typically noted by the laser line wavelength values.
- Select the laser lines appropriate for the dyes that are used for staining the samples. Turn on one or multiple laser lines to excite and image those features in the sample.
- Set the gain to a value that optimizes the samples fluorescence but limits the amount of noise. A typical starting value is a gain of 70.
- Adjust the laser power to avoid saturated pixels but maximize the dynamic range. A typical starting range for the laser power is 1 – 3 %.
- Set the pinhole size to 1 Airy unit to maximize the resolution for the optical sectioning. If the sample is dim and the laser power is already high, open up the pinhole to increase the signal at the cost of decreasing the z axis resolution.
- Set the pixel dwell time (i.e., the scanning speed). Start off with a 2 µs dwell time and if needed, adjust to reflect the sample brightness. Choose the pixel size/scan size for the selected objective by letting the instrument calculate it via the Nyquist option button and the chosen number of pixels in the image.
NOTE: Longer dwell times in thicker samples can lead to excessive bleaching when z stacks are collected. - Select the Scan option on the instrument’s software and begin data collection.
NOTE: If the instrument has multiple lasers, each can be optimized independently, and then ran simultaneously for multi-fluorescent images. - Use the focus knob to zoom in and out and locate the optimal field of view in the sample.
- Capture images by using the system’s Capture button and save all necessary file formats of the sample to a desired location/folder on the computer’s hard drive, or local external hard drive.
- Continue to adjust gain, laser power, pinhole size, sampling rate, and other values accordingly to optimize the image. The pixel saturation index can be activated to check if pixels are over-saturated. If this over-saturation is present, gain and laser power can be readjusted to eliminate saturated pixels. Use the LUTs as a guide to make specific adjustments.
- If possible, use the system’s Nyquist sampling rate, which is displayed in the software as a panel or button, to ensure images are collected at the maximum obtainable resolution.
- Activate the confocal system’s z stack or image volume collection tool. Using a bottom-to-top option, with only one laser line, specifically the laser line that illuminates a feature in the sample the clearest, set the start and finish planes for the volume to be measured.
- Use the suggested spacing between planes in the z stack, which is calculated to fulfill the Nyquist sampling criteria for the best obtainable axial resolution afforded by the instrument and used laser line.
- When multiple laser lines are used, use the shortest wavelength for calculation of the spacing. When the acquisition finishes, save the file to the appropriate folder.
- Alternatively, if a priori knowledge of the sample’s thickness exists, define the volume by selecting the middle plane, the one that appears the brightest and sharpest. In this situation, set the top to half the sample thickness above the middle plane and the bottom to half the thickness below the middle plane.
- After sample collection in this field of view is complete, or the sample has bleached, use the motorized or manually controlled stage to find another place of interest on the sample and repeat the steps for image collection.
5. Clean up procedure
- Ensure all data files, both from the CLSM and AFM, are saved to the proper locations.
- Retract the AFM chip using the z stepper motors at least 2000 µm or the distance that was traveled to get to the sample surface. Remove the AFM head from the stage and place it carefully in its resting place.
- Retract the microscope objective so that it is far away from the sample. If an oil objective was used, the objective should be retracted far enough such that there is no longer contact between the objective and the sample substrate.
- Using gloves, remove the sample and clean off the grease, and oil if applicable, from the bottom of the sample dish. Acquire a new, clean, empty Petri dish and fill it with 70% ethanol solution one-half to three-quarters full. Place it into the sample holder and replace the AFM head to allow the AFM chip to soak in the ethanol solution for 5 min.
- While the AFM chip is soaking, it is recommended to begin copying the experiment data onto an external hard drive. After immersion of the AFM chip in ethanol solution for at least 5 minutes, remove the AFM head and put it in its resting place.
- Using gloves, remove the AFM chip holder and place it into the mounting station. Carefully remove the AFM chip using tweezers with rubber grips. Place the used tip back into the location of the box it came from oriented off axis to denote that it has been used. For future reference, note which tip was used in the experiment.
- Take the Petri dish filled with the ethanol solution out of the system and dispose of the liquid and the dish. Using gloves, lens cleaner, and lens wipes, gently clean the oil from the microscope objective following standard protocols. Clean the sample holder by removing any grease. Dispose of all waste materials according to biosafety protocols and return tools to their known locations.
- Finish data collection from the computer(s) and shut them down. Power off all instruments, accessories, peripherals, or other devices utilized for the experiment.
Representative Results
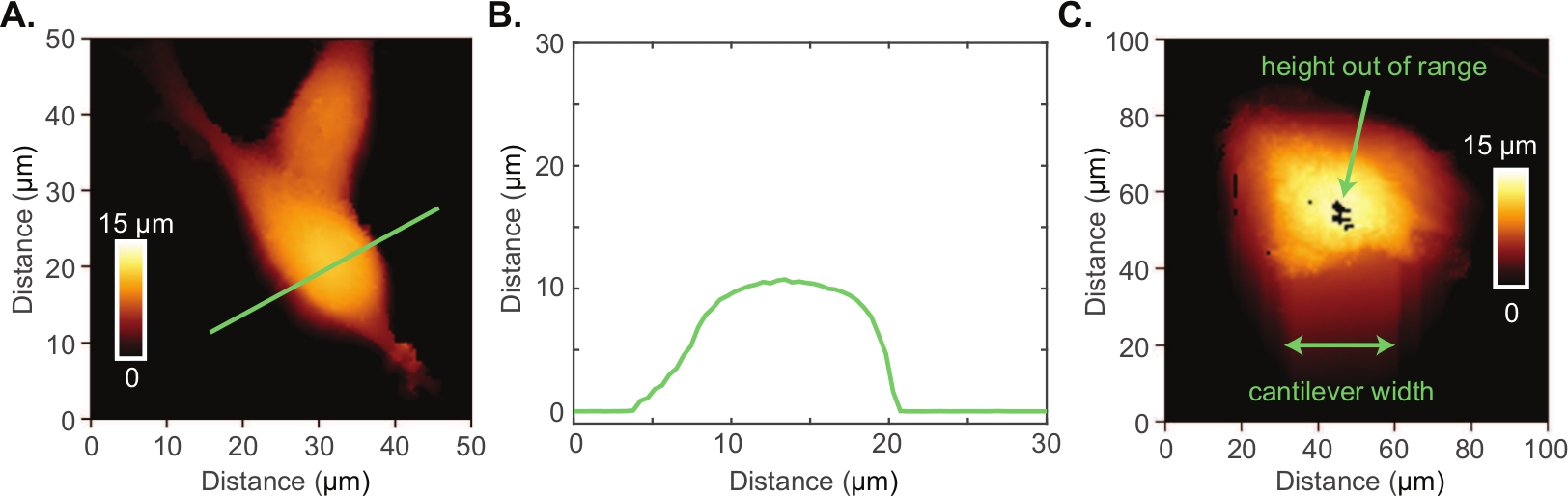
The Conpokal technique is schematically represented in Figure 1A and an image of the setup is shown in Figure 1B. In order to accurately co-locate biological structures via confocal microscopy with the same structures in AFM, the alignment of the two systems during installation is critical. Select reputable confocal and AFM manufacturers that are familiar with each other’s spatial requirements. Additionally, successful implementation of the technique relies upon good staining procedures, immobilization of the biological structure, and the appropriate choice of AFM tip. The height of living cells often presents a challenge for AFM imaging. An AFM scan over a living HEK cell is shown in Figure 2A. The height for this particular HEK cell was around 10 μm, demonstrated by the line scan in green (Figure 2B). An AFM tip with either zero offset (tip is at the very end of the cantilever) or the tip height greater than the cell height (e.g., tip height ≥ 10 μm) will produce a highly resolved image of individual cells. An example of a poor scan due to improper AFM tip choice is included in Figure 2C. In this image, black pixels appeared at the apex of the cell indicating that the AFM piezo is out of range due to a large cell height. The end of the AFM cantilever appeared in the image (a rounded square) because of tip offset combined with insufficient tip height compared to cell height. These artifacts in the AFM image indicate a different AFM tip should be chosen to image the cell.
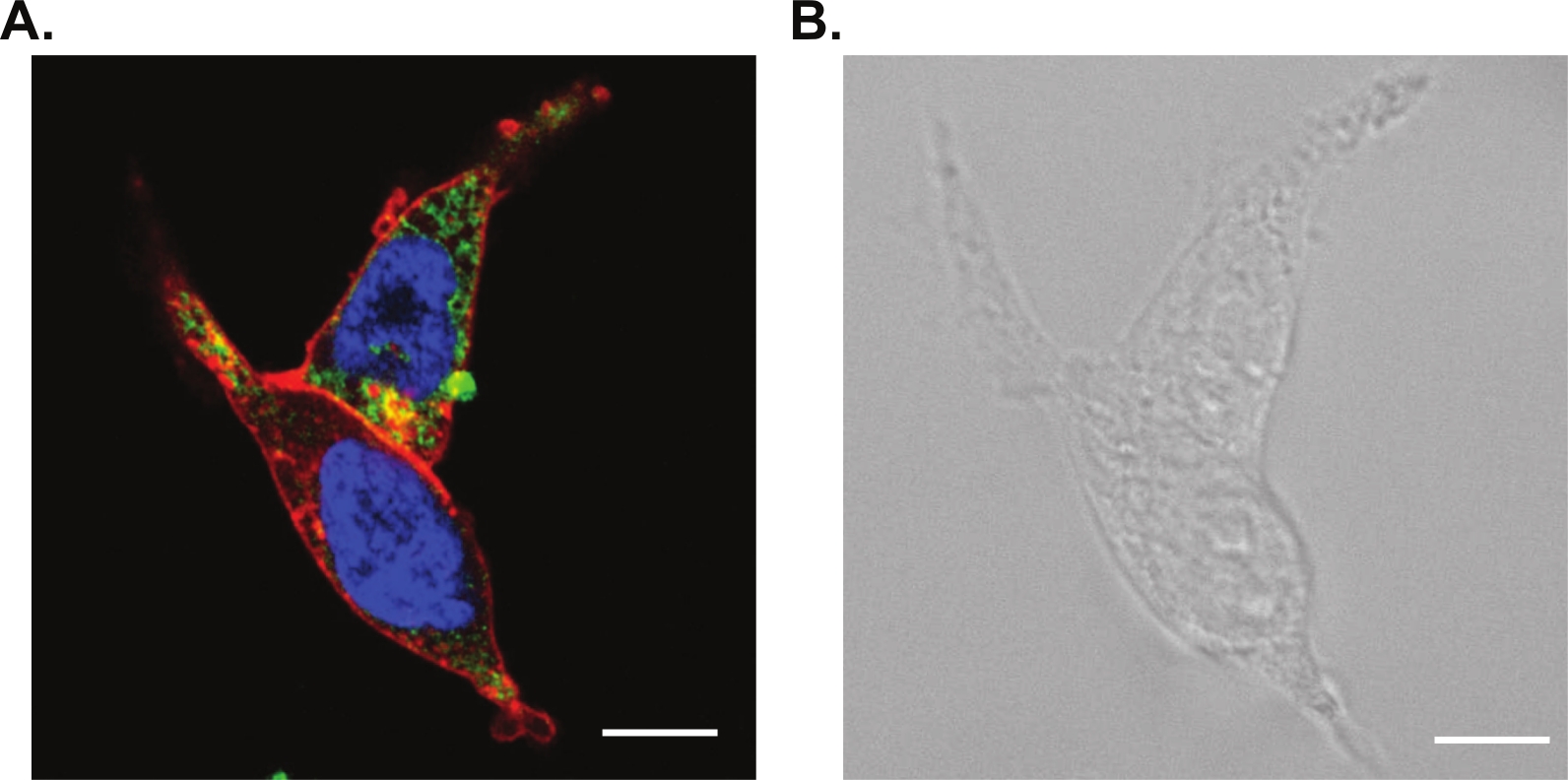
Efficient labeling in fluorescence staining along with the correct probe for live cell imaging are required for this method. We performed a three-color confocal image shown in Figure 3A with a nuclear stain (fluorescing blue), a microtubule stain (fluorescing green), and a lipophilic membrane stain (fluorescing red). These live cell probes were chosen because of their limited spectral overlap and their compatibility with standard filter cubes. We have also included the brightfield optical image in Figure 3B.
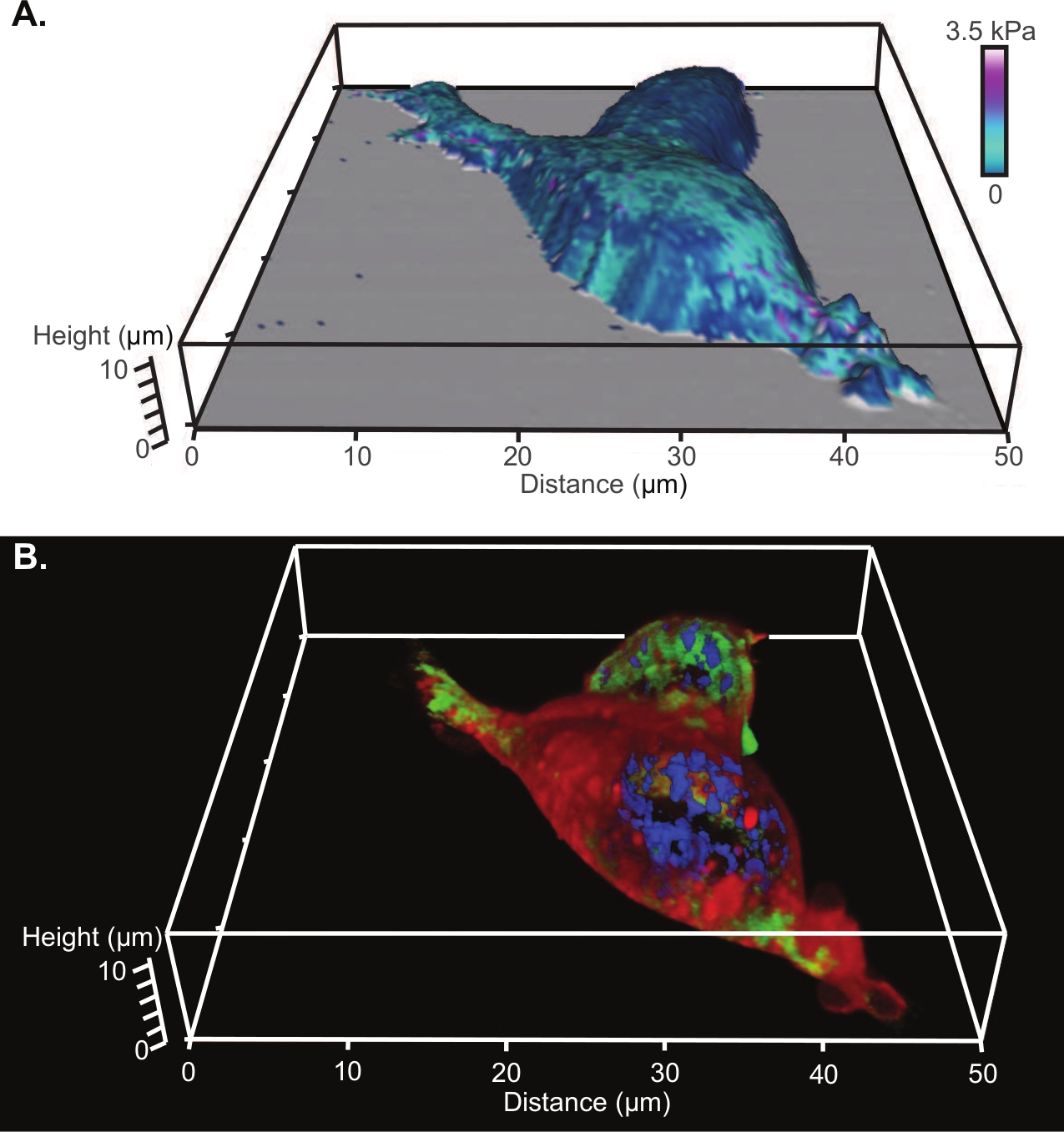
From AFM, analysis of the tip indentations along with a nanomechanical model, produces a modulus map of the surface. An example of a modulus map constructed using a Hertz model fit and a parabolic profile (radius of 8 nm) for the tip shape is shown overlaid onto the 3D reconstruction of the cell shape in Figure 4A (which is the same two cells shown in Figure 3). The corresponding 3D projection of the laser confocal z stack is shown in Figure 4B.
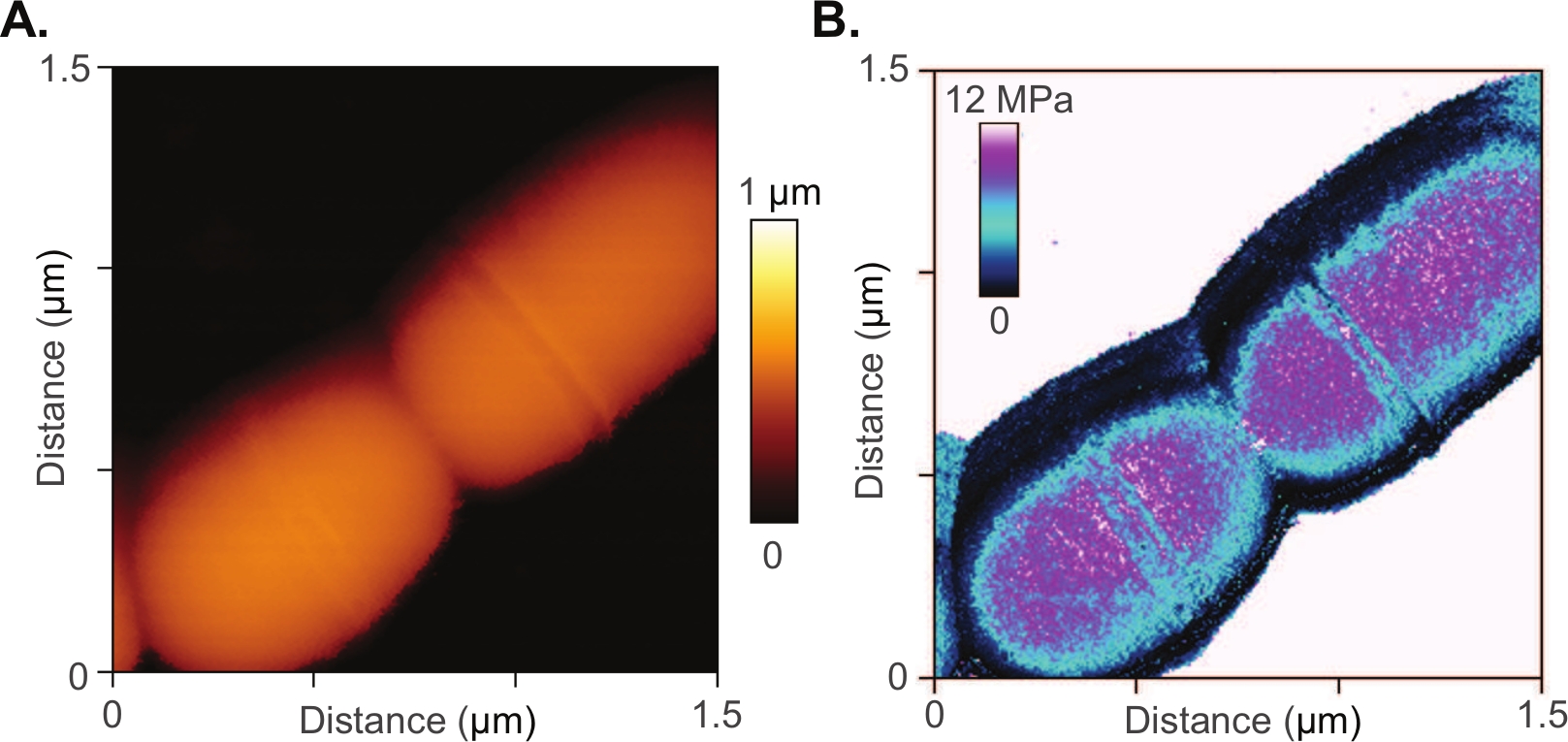
The method is also appropriate for smaller biological structures, including single bacteria whose micro and nanoscopic features are difficult to resolve using light microscopy. An on-going study utilizes the Conpokal technique to explore mechanical mechanisms within the cell wall that influence antibiotic resistance in Streptococcus mutans.74 Manipulation of the cell wall components resulted in changes in morphology and antibiotic resistance. Conpokal facilitates live, in-solution, data collection (e.g., surface morphology, elastic modulus, adhesion strength, and surface roughness) on same-cell samples to couple mechanical properties with the presence or absence of cell wall components. Figure 5A,B include an AFM scan and measured modulus map of a Streptococcus mutans bacterium. Better resolution was achieved at this scale with AFM than with traditional confocal microscopy.
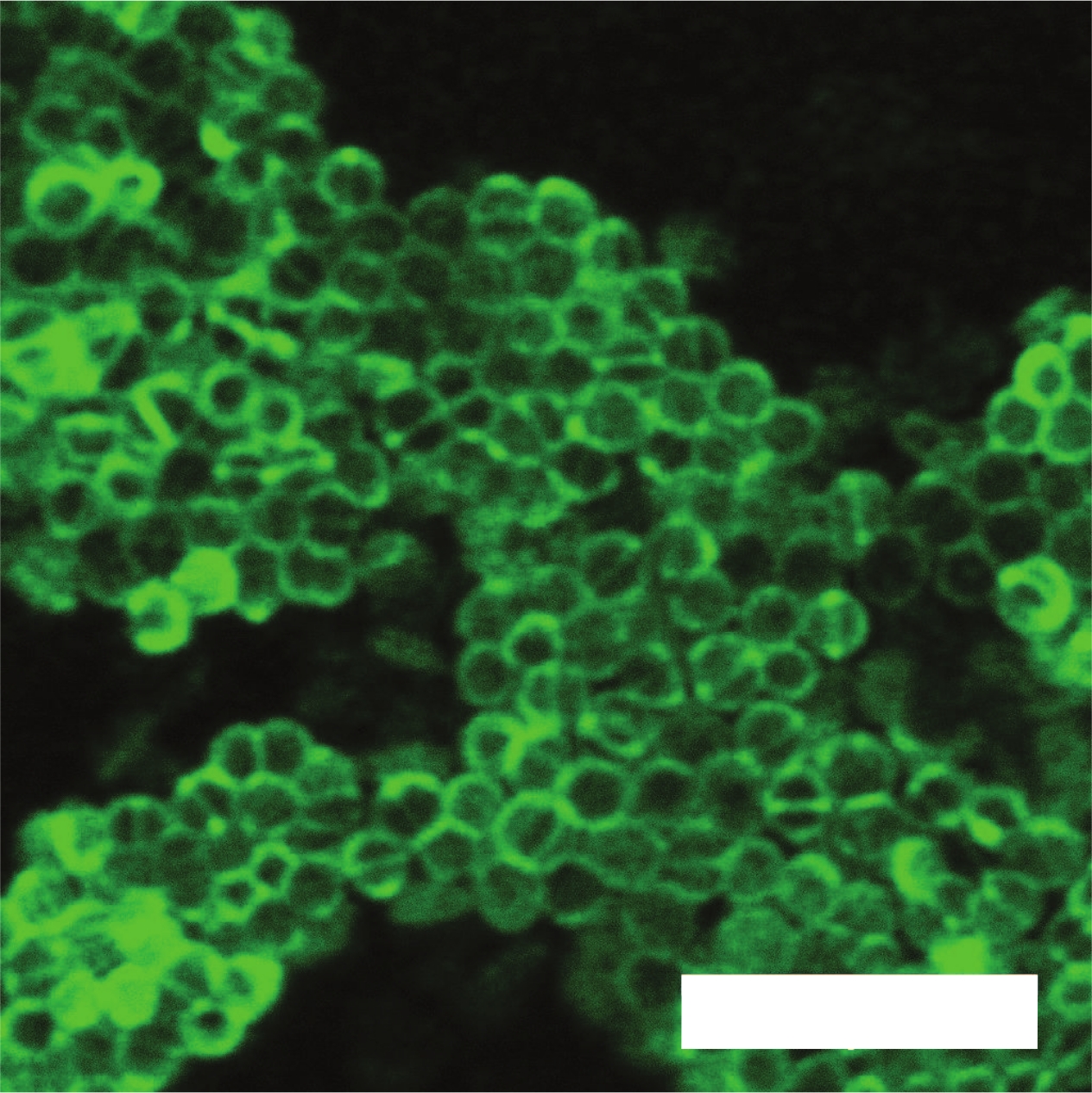
Figure 6 contains a confocal image of a colony of Enterococcus faecalis stained with a green fluorescent cell structure stain, which attaches to the cell wall. Figure 5 and Figure 6 demonstrate the applicability of the Conpokal technique to microbes in addition to eukaryotic cells.

Figure 1: The Conpokal setup.
(A) Schematic of Conpokal technique. (B) Image of the instrument setup. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Atomic force microscopy of living cells.
(A) AFM image of two HEK cells. (B) Height profile (green line in A) of an HEK cell indicating the cell height is quite large at 10 μm. (C) Poor AFM image of an astrocyte cell where the AFM piezo is out range at the apex of the cell and there is evidence of reverse imaging of the cantilever end. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Confocal and brightfield microscopy of living cells.
(A) Laser scanning confocal microscopy of living HEK cells stained with a nuclear stain (fluorescing blue), a microtubule stain (fluorescing green) and a lipophilic membrane stain (fluorescing red). (B) Corresponding brightfield optical image. Scale bars are 10 μm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Comparison of 3D renderings of AFM and confocal microscopy of living HEK cells.
(A) Modulus map constructed using a Hertz model fit and a parabolic profile (radius of 8 nm) for the tip shape overlaid onto the 3D reconstruction of the cell from AFM. (B) The corresponding 3D projection of the laser confocal z stack with a nuclear stain (fluorescing blue), a microtubule stain (fluorescing green) and a lipophilic membrane stain (fluorescing red). Please click here to view a larger version of this figure.
{kind=link}

Figure 5: AFM of bacteria sample.
(A) AFM scan and (B) measured modulus map of a Streptococcus mutans bacterium. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Confocal microscopy of bacteria sample.
Confocal image of a colony of Enterococcus faecalis with green fluorescent membrane stain. Scale bar is 5 μm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Conpokal is an advanced, effective technique for collecting high-quality images and in situ mechanical properties of live biomaterials in a liquid environment. The ability to collect exceptional morphology and topography images combined with live-sample mechanical properties extends past typical electron and light microscopy techniques. Separately, a confocal microscope provides brightfield, epifluorescence, and laser scanning confocal capabilities to achieve high-quality, detailed fluorescent images of samples. An AFM provides mechanical characterization, but without auxiliary optics it becomes difficult to navigate the sample. With the two systems combined, a user can collect both images and mechanical properties of the exact same cell during the experiment, which is a great advantage over two separate instruments. The purpose of this manuscript is to inform and guide users on the extensive capabilities of the combined Conpokal system for the future of biology, engineering, and health. The protocol involves preparation of instruments, culture media, dishes, selection of microorganisms, AFM procedure, confocal procedure, and clean up. The most critical steps for a successful live, in-solution, Conpokal session are sample immobilization, judicious tip selection, and live stain execution. The discussion section for this manuscript will elaborate on culture recommendations, troubleshooting tips and operational guidelines, and future work for the Conpokal technique. Culture recommendations will cover guidance on sample culture, immobilization, and staining. AFM, confocal, and Conpokal tips and guidelines will discuss tip selection and calibration, resolution, limitations, and near simultaneous operation. Future work includes the outlook and potential for prospective research pathways.
The protocol covers culture, probing, and imaging of both live cell and fixed bacteria samples. One challenge present when conducting AFM in liquid stems from cantilever movement obstruction due to sample hindrance and hydrodynamic drag. If the sample is not well adhered to the substrate, there is potential for fouling of the cantilever by sections of the sample floating in the liquid. In that case, measurements are compromised since they are a supposition of elastic adherence and micromechanical properties of the sample. The HEK cells were not treated with fixative, however, S. mutans and E. faecalis require additional immobilization, similar to other prokaryotic cells. The bacteria chosen showed active movement which hindered cell probing and imaging, therefore, the samples were gently, chemically fixed to promote immobilization. There are alternatives to chemical fixation, such as filter membranes that physically trap individual cells in pores75.
Tip selection is also a critical component to the setup and operation of the AFM. When mechanical properties based on deformation of the biomaterial are sought, one must consider cantilever stiffness and material stiffness compatibility, which is a non-trivial task. The main goal is to best match the stiffness of the material with the stiffness of the selected cantilever. If the cantilever is much softer than the sample, it will be subject to too much deflection. If the cantilever is stiffer than the sample, then the AFM detector may be unable to capture such small deflections. It is recommended that when selecting a suitable AFM cantilever, to choose based on experimental application. To collect detailed images in intermittent contact mode, AFM tips within a range of 0.1 - 0.3 N/m for stiffness and tip radius within 5 nm – 100 nm are effective for live cell imaging. Small conical tips are recommended. Sharp tips provide a small contact area and ability to indent further aiding in collecting small details and features in sample morphology76. However, sharp tips can also be problematic as they are prone to puncturing samples when the settings (e.g., set point, pixel time, approach velocity) require too much indentation or the indentation is too fast. To avoid high-strain rate effects, local strain hardening near a sharp tip, or simply to control the contact area and approach speed, many choose to use force spectroscopy with a colloidal tip to measure an elastic modulus.
AFM tips within a range of 0.01 - 1.0 N/m for stiffness and tip radius within 1 – 5 µm are recommended to measure the elastic moduli of living cells. Large tip sizes, typically glass colloids, provide a known contact area and are unlikely to puncture the cell while in contact. Rectangular cantilevers are preferred over triangular because during calibration, the contact-free method can be used for rectangular geometry as compared to contact-based calibration for triangular geometries. Also, due to the delicate nature of AFM tips, it is recommended that the operator implements tweezers with rubber tips to reduce the potential for damaging the delicate AFM chip or mounting block. Other parameters to keep in mind include the approach velocity of the AFM tip and the amount of indentation into the sample. A good guideline is to keep indentation to about 10% of the thickness (or height) of the sample and choose a velocity that precludes potential hydrodynamic resistance experienced by the cantilever38.
Intricate experimental instruments typically come with roadblocks that require troubleshooting in the instrument set up, calibration, and operation. Crashing the AFM tip or cantilever into the sample or sample substrate is a common mistake for new users. To avoid this issue, the protocol suggests backing the cantilever out 2000 µm. This step ensures the tip will not come into contact with the bottom of the dish if the previous user forgot to back out the tip when cleaning up after experimentation. However, the 2000 µm distance was chosen for the selected dish holder used in this protocol. A different range, larger or smaller, may need to be selected depending on the dish holder style being used. During alignment of the laser and detector, the protocol mentions adjustment of a mirror knob to maximize the sum signal. A mirror adjustment knob may not be present on all AFMs. If present, however, one way of manipulating the instrument to troubleshoot a low sum signal is to adjust the mirror knob, used to account for the medium in which scanning will take place; either liquid (e.g., water, media, PBS) or air. Due to the difference in index of refraction for light through air and through liquid, the mirror knob may need to be adjusted. The maximum bending sensitivity of the AFM cantilever will be at the location of the AFM tip, therefore, the laser light, which returns the bending position through its placement on a photodiode, must be located at the location of the AFM tip. Depending on the AFM tip chosen, the sum signal value could vary from 0.3 – 3.0 V. A backside coating on the AFM cantilever such as Cr-Au or Al will increase the sum signal and the sensitivity of the measurement.
Tip geometry is pertinent when completing tip calibration during AFM operation. Good agreement has been observed between non-contact and contact calibration. If the chosen AFM cantilever is not rectangular, contact calibration will need to be performed. Keep in mind that the medium in which the tip is calibrated must be the same as the sample medium. If those fluids differ, the user must recalibrate. When using the AFM tips in liquid, the frequency measured by the system should be one-fourth to one-third of that of the natural resonance frequency denoted by the manufacturer. A good way to check that the system calibrated the tip correctly is to verify values within the thermal noise file that is generated. Ensure this file saves to the appropriate folder. If the system has trouble calibrating or non-probable values come out, recalibrate, or adjust the laser position slightly then recalibrate.
Another cause of poor sum signal may be due to AFM cantilever alignment. When the AFM chip is mounted into the glass block, it is essential that the tip of the cantilever remains within the small laser window (smooth glass area). If the chip is too far forward, the angle at which the cantilever naturally rests may cause an issue with the reflection off the cantilever, missing the photodetector and resulting in poor sum signal. If the chip is too far back, the laser will be unable to reflect off the back of the cantilever, causing poor sum signal. For these reasons, the mounted AFM chip may need to be adjusted. Additionally, another issue that may be encountered occurs with the height of the sample. The AFM instrument used in this protocol has a maximum piezo z range (height) of 15 µm. If a user finds that the software is unable to collect the height data and force maps in a particular pixel, a black box will appear indicating that the system is out of range (Figure 2C). One way to trouble shoot this issue is to set the piezo height to a lower value, such as 2 or 3 µm so most of the 15 µm range is committed to mapping the anticipated height of the cell. This technique should, in most cell- or bacteria-related experiments, fix the issue associated with the z range.
Experimentalists that require an extended z range for tall samples with heights greater than 10 – 15 µm may need to pursue an additional module on the AFM. AFM manufacturers have this option available at additional costs for most systems. By extending the z range, the experimentalist has the availability to scan samples that are considered tall at the micro-scale with little issues for out of range values or AFM piezo motor modification. Although these modules cost extra, some, depending on the manufacturer, can offer additional height, up to 100 µm in the z direction. Confocal is still possible with taller samples if the user has a long working distance, high magnification objective or is willing to use an air objective, perhaps a 20x or 40x. By lowering microscope objective magnification, the working distance increases, gaining distance to view the apex of a taller sample. This modification to a lower magnification objective will sacrifice resolution. In the Conpokal setup referred to in this manuscript, the 60x TIRF (total internal reflection fluorescence) objective has a working distance of near 100 µm past the coverslip of the glass-bottomed sample dish.
In regard to the confocal microscope referred to in this manuscript, a few important stipulations are discussed. The confocal system used for the production of the figures in this manuscript implemented a 60x TIRF oil objective with a numerical aperture of 1.49. Laser lines at 405 nm, 488 nm and 561 nm excitation wavelengths were used for live cell sample imaging, shown in Figure 3 and Figure 4. The diffraction limit of the confocal microscope can be determined by using the Abbe Resolution equation, Abbe Resolution(x,y) = λ/2NA, where λ is the excitation wavelength for Alexa 488, at 488 nm, and NA is the numerical aperture for the confocal condenser, which is 0.3. Therefore, an axial resolution of 272 nm is determined. For epifluorescence imaging, two cases are considered to determine the resolution where the pinhole is set to one airy unit (AU) and 0.5 AU. In the latter case, the pinhole is closed down such that significant light loss occurs, but the resolution increases. The confocal software calculates lateral native and axial native resolutions at 170 nm and 290 nm for the pinhole at 0.5 AU, and 200 nm and 370 nm for the pinhole at 1 AU, respectively. Spherical aberrations introduced in the system can be accounted for through a deconvolution process to increase contrast and resolution in microscope images. Due to the diffraction limitations that are inherent with confocal microscopes, the confocal image of the bacteria colony in Figure 6 lacks the matching resolution for the detail seen in the AFM scan of bacteria in Figure 5. AFM provides access to nanoscale features and detail that is difficult to capture with a confocal microscope. However, depending on the required fluorescence resolution needed, Figure 5 and Figure 6 demonstrate the applicability of the Conpokal technique to microbes in addition to eukaryotic cells.
An advantage of using a confocal microscope allows the operator to collect 3D images of specific regions in a sample with sharpened detail. These images correlate with the AFM by viewing the surface that was probed via the AFM image and a confocal scan of the same region. Since the microscope is inverted, an epifluorescence image collects light information from the opposite side of the sample that was probed. The pinhole within the confocal system helps limit to a single plane from a certain distance while filtering out light that comes from the rest of the sample or even the room. Essentially, the pinhole helps isolate the light coming back from the single plane of interest in the sample. Typically, this plane of interest must contain strong fluorophore markers because, for inverted confocal systems, single molecule detection would be limited to higher-resolution confocal microscopes. Epifluorescence illumination is less desirable due to the fact that in epifluorescence imaging mode, any light from the sample that is reflected into the objective is collected and used to generate the image, therefore, impossible to isolate a single plane. Confocal techniques provide a more isolated single plane image of the sample feature in question because of the pinhole77. If, for example, the apex of a eukaryotic cell is probed by AFM, that same surface can be isolated with the laser scanning confocal capabilities of the microscope, rather than with epifluorescence imaging mode. It is recommended to monitor the health/shape of the cells during data acquisition via imaging simultaneously in differential interference contrast mode with the aid of the transmitted light detector and the 488 nm laser line. When capturing a z stack of the sample, for the procedure detailed above, only the gain of the detector is adjusted. Any morphological change in the cells during the measurement, which is not necessarily visible in the fluorescent channels, indicates that artifacts are introduced into the measurement.
Ideal spacing between planes can be obtained by following the software recommendation for the shortest wavelength used in the imaging technique. The native resolution and the signal to noise ratio in the image volumes can be effectively improved by employing deconvolution algorithms available in the image processing module of the acquisition software. However, performing fluorescent microscopy, selection of specific stains and dyes is vital to avoid early on-set bleaching or crosstalk from overlapping excitation/emission spectra. On occasion, a user may experience a malfunction in confocal light generation. If a user experiences a lack of light emission or malfunctioning laser lines, one way to troubleshoot is to reset the system, typically done by restarting the operating software. If the issue persists, the transmitted light detector may have failed to move into or out of place within the optical path of the light microscope. Resetting the position of the transmitter detector may help to alleviate light collection or laser imaging issues.
The focus of the Conpokal instrument is to provide users with the ability to collect optical and force-based information on live biomaterials in a liquid environment, simultaneously and on the same cell or feature. This work explicitly describes how to perform these experiments in liquid, a natural home for many biomaterials, although, dry experiments can still be performed using the instrumentation. With samples prepared in Petri dishes, the dish height is a limitation. Due to the configuration of the glass block that holds the AFM cantilever, the side walls of the dishes must be under 10 mm in height; if the dish is too tall, the instrument will not be able to lower the AFM tip to the surface of the sample or the substrate.
Although there is a limitation for sample size, there is no limitation with the instrument or software capabilities with regard to a time delay. Simultaneous confocal and AFM is possible with the right factors in place. The limitation that contributes to its near simultaneous capability refers to the noise that is generated when performing certain confocal microscopy functions and atomic force microscopy functions simultaneously. The vibrations from the motors that move the microscope objective during collection of a z stack will be added to the signal from the AFM probe tip during its motion. The noise will be enhanced by an oil objective, as the motors move up and down in the z direction to illuminate sequential planes in the sample. Therefore, the recommended protocol includes sequential collection of AFM scans and confocal z stack images. Simultaneous CLSM and AFM would require stationary imaging with the confocal, however, with the current technique, the delay time for the two instruments could be as short as tens of seconds. The actual time to switch from AFM operation to confocal imaging was around 2 – 4 minutes for the images collected in Figure 2A and Figure 4B. This value was determined by subtracting the two image collection time periods from the total time of 33 minutes, which includes the time to begin and complete the AFM scan, switch instrument modes to activate confocal imaging, and begin and complete the confocal image z stack.
The future of Conpokal aims to explore new structure-function relationships in addition to keen insight of single cell processes. For example, experimentation of in situ drug treatments on cell or bacterial samples to determine the effects of cell elasticity would be an advancement to the fields of biomaterials, biology, and biomechanics. Treatment therapy into the sample dish while imaging and probing would provide knowledge of how the sample responds to therapeutics in a live and carefully controlled environment, over time. Incorporating a new drug or environmental challenge would broaden the understanding of how the cytoskeleton or organelle location affects locomotion, topology, stiffness, etc. Another potential advancement of Conpokal is the ability to have complete environmental control of the system. The current Conpokal mentioned in this protocol is housed inside of an acoustic enclosure designed to reduce noise from inside of the laboratory. Advancement of this housing would provide the ability to test within, perhaps, one or a combination of factors, not limited to those such as a sterile environment, temperature-controlled, or even variable-gravity. As it stands, the Conpokal method provides an effective and useful approach for characterizing live, in-liquid biomaterials, but the future of the technique will only advance these capabilities further.
Acknowledgements
We acknowledge NIH COBRE funding under number P20GM130456. We thank the UK Light Microscopy Core, which is supported by the Vice President for Research, for assistance in this work. Strains of S. mutans and E. faecalis were provided by Dr. Natalia Korotkova. The first mention of the play-on-words, Conpokal, to describe the combination of confocal microscopy and atomic force microscopy is attributed to Dr. Brad Berron, Chemical and Materials Engineering, at the University of Kentucky, in discussion with Dr. Martha Grady (corresponding author) in anticipation of the arrival of a seminar speaker Dr. Jonathan Pham, who joined the faculty in Chemical and Materials Engineering at the University of Kentucky in the Fall of 2017.
Materials
| Name | Company | Catalog Number | Comments |
| Acoustic Enclosure | JPK-Bruker | Acoustic Enclosure | Chamber used to mitigate noise |
| BioTracker 488 Green Microtubule Cytoskeleton Dye | Millipore Sigma | SCT142 | Used to label microtubules in mammalian cells |
| CP-qp-CONT-BSG AFM tips | NanoAndMore | CP-qp-CONT-BSG-B | Collodial AFM tips good for modulus measurements on biological samples |
| Dil Stain (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindocarbocyanine Perchlorate ('DiI'; DiIC18(3))) | ThermoFischer Scientific | D282 | Used to label plasma membrane of mammalian cells |
| Dulbecco's Modified Eagle Medium | VWR | VWRL0100-0500 | Component of cell culture media |
| Dulbecco's Phosphate-Buffered Saline | ThermoFisher Scientific | A1285601 | Used to simulate biological environment |
| Fetal Bovine Serum | Fischer Scientific | 45000-736 | Component of cell culture media |
| Fisher BioReagents Microbiology Media Additives: Yeast Extract | Fischer Scientific | BP1422-500 | Component of bacterial culture medium |
| FluoroDish Cell Culture Dish | World Precision Instruments (WPI) | FD35-100 | Glass-bottomed dishes whose side walls are less than 10 mm, necessary for full function of Nanowizard 4a AFM |
| Hoechst 33342 nucleic acid stain | ThermoFischer Scientific | 62249 | Used to label nucleus of mammalian cells, a good nuclear marker for live cells |
| Human embryonic kidney 293 cells | ATCC Global Bioresource Center | CRL-1573 | Mammalian cells used in protocol |
| Matrigel | VWR | 47743-716 | Coating in cell culture dish |
| Model: PFQNM-LC-A-CAL AFM tips | Bruker | PFQNM-LC-A-CAL | AFM tips that work well for samples that have large heights (such as eukaryotic cells) |
| NanoWizard 4a | JPK-Bruker | NanoWizard 4a | AFM |
| Nikon A1 Laser Scanning Confocal Microscope | Nikon | A1 HD25 | Microscope used for brightfield, epi-fluorescence, and confocal imaging |
| PENICILLIN/STREPTOMYCIN | VWR | 16777-164 | Component of cell culture media |
| PetriDishHeater | JPK-Bruker | PetriDishHeater | Maintains desired Petri dish sample temperature |
| qp-BioAC-Cl AFM tips | Nanosensors | qp-BioAC-Cl-10 | Three different AFM cantilevers of varying stiffness |
| Replaceable Carbon Fiber Tip Tweezers | Ted Pella Inc. | 5051 | Used to carefully place and remove delicate AFM tips into the glass mounting block |
| Todd Hewitt Broth Bacto | BD DIFCO Bacterius Ltd | Bacto-249240 | Component of bacterial culture medium |
| Vancomycin, BODIPY FL Conjugate | Thermo Fisher Scientific | V34850 | Used to fluorescently label bacterial cell wall |
References
- Butt, H. J., Pham, J. T., Kappl, M. Forces between a stiff and a soft surface. Current Opinion in Colloid & Interface Science. 27, 82-90 (2017).
- Pham, J. T., Schellenberger, F., Kappl, M., Butt, H. J. From elasticity to capillarity in soft materials indentation. Physical Review Materials. 1 (1), 015602 (2017).
- Lee, G. Y. H., Lim, C. T. Biomechanics approaches to studying human diseases. Trends Biotechnology. 25 (3), 111-118 (2007).
- Qiu, H., et al. Short Communication: Vascular Smooth Muscle Cell Stiffness As a Mechanism for Increased Aortic Stiffness With Aging. Circulation Research. 107 (5), 615-619 (2010).
- Glenister, F. K., Coppel, R. L., Cowman, A. F., Mohandas, N., Cooke, B. M. Contribution of parasite proteins to altered mechanical properties of malaria-infected red blood cells. Blood. 99 (3), 1060-1063 (2002).
- Suresh, S., et al. Connections between single-cell biomechanics and human disease states: gastrointestinal cancer and malaria. Acta Biomaterialia. 1 (1), 15-30 (2005).
- Nash, G. B., Johnson, C. S., Meiselman, H. J. Mechanical properties of oxygenated red blood cells in sickle cell (HbSS) disease. Blood. 63 (1), 73-82 (1984).
- Trickey, W. R., Lee, G. M., Guilak, F. Viscoelastic properties of chondrocytes from normal and osteoarthritic human cartilage. Journal of Orthopedic Research. 18 (6), 891-898 (2000).
- An, S. S., Fabry, B., Trepat, X., Wang, N., Fredberg, J. J. Do Biophysical Properties of the Airway Smooth Muscle in Culture Predict Airway Hyperresponsiveness. American Journal of Respiratory Cell and Molecuar Biology. 35 (1), 55-64 (2006).
- Suresh, S. Biomechanics and biophysics of cancer cells. Acta Materialia. 55 (12), 3989-4014 (2007).
- Rebelo, L. M., de Sousa, J. S., Mendes, J., Radmacher, M. Comparison of the viscoelastic properties of cells from different kidney cancer phenotypes measured with atomic force microscopy. Nanotechnology. 24 (5), 055102 (2013).
- Cross, S. E., Jin, Y. S., Rao, J., Gimzewski, J. K. Nanomechanical analysis of cells from cancer patients. Nature Nanotechnology. 2 (12), 780-783 (2007).
- Remmerbach, T. W., et al. Oral Cancer Diagnosis by Mechanical Phenotyping. Cancer Research. 69 (5), 1728-1732 (2009).
- Wirtz, D., Konstantopoulos, K., Searson, P. C. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nature Reviews Cancer. 11 (7), 512-522 (2011).
- Yallapu, M. M., et al. The Roles of Cellular Nanomechanics in Cancer. Medical Research Reviews. 35 (1), 198-223 (2015).
- Fabry, B., et al. Scaling the Microrheology of Living Cells. Physical Review Letters. 87 (14), 148102 (2001).
- Ingber, D. E., Prusty, D., Sun, Z., Betensky, H., Wang, N. Cell shape, cytoskeletal mechanics, and cell cycle control in angiogenesis. Jouranl of Biomechica. 28 (12), 1471-1484 (1995).
- Svoboda, K., Block, S. M. Biological Applications of Optical Forces. Annual Reviews of Biophysics and Biomolecular Structures. 23 (1), 247-285 (1994).
- Dao, M., Lim, C. T., Suresh, S. Mechanics of the human red blood cell deformed by optical tweezers. Journal of the Mechicanics and Physics of Solids. 51 (11-12), 2259-2280 (2003).
- Thoumine, O., Cardoso, O., Meister, J. J. Changes in the mechanical properties of fibroblasts during spreading: a micromanipulation study. European Biophys with Biophyics Letters. 28 (3), 222-234 (1999).
- Reynolds, N. H., et al. On the role of the actin cytoskeleton and nucleus in the biomechanical response of spread cells. Biomaterials. 35 (13), 4015-4025 (2014).
- Jones, W. R., et al. Alterations in the Young’s modulus and volumetric properties of chondrocytes isolated from normal and osteoarthritic human cartilage. Journal of Biomechics. 32 (2), 119-127 (1999).
- Radmacher, M., Fritz, M., Kacher, C. M., Cleveland, J. P., Hansma, P. K. Measuring the viscoelastic properties of human platelets with the atomic force microscope. Biophysics Journals. 70 (1), 556-567 (1996).
- Ketene, A. N., Roberts, P. C., Shea, A. A., Schmelz, E. M., Agah, M. Actin filaments play a primary role for structural integrity and viscoelastic response in cells. Integrative Biology. 4 (5), 540-549 (2012).
- Ketene, A. N., Schmelz, E. M., Roberts, P. C., Agah, M. The effects of cancer progression on the viscoelasticity of ovarian cell cytoskeleton structures. Nanomedicine-Nanotechnology. 8 (1), 93-102 (2012).
- Darling, E. M., Topel, M., Zauscher, S., Vail, T. P., Guilak, F. Viscoelastic properties of human mesenchymally-derived stem cells and primary osteoblasts, chondrocytes, and adipocytes. Journal of Biomechics. 41 (2), 454-464 (2008).
- Henderson, E., Haydon, P., Sakaguchi, D. Actin filament dynamics in living glial cells imaged by atomic force microscopy. Science. 257 (5078), 1944-1946 (1992).
- Rotsch, C., Radmacher, M. Drug-Induced Changes of Cytoskeletal Structure and Mechanics in Fibroblasts: An Atomic Force Microscopy Study. Biophysics Jouranl. 78 (1), 520-535 (2000).
- Darling, E. M., Zauscher, S., Guilak, F. Viscoelastic properties of zonal articular chondrocytes measured by atomic force microscopy. Osteoarthritis Cartilage. 14 (6), 571-579 (2006).
- Lekka, M., et al. Local elastic properties of cells studied by SFM. Applied Surface Science. 141 (3-4), 345-349 (1999).
- Li, Q. S., Lee, G. Y. H., Ong, C. N., Lim, C. T. AFM indentation study of breast cancer cells. Biochemical and Biophysical Research Communication. 374 (4), 609-613 (2008).
- Rother, J., Noding, H., Mey, I., Janshoff, A. Atomic force microscopy-based microrheology reveals significant differences in the viscoelastic response between malign and benign cell lines. Open Biology. 4 (5), 140046 (2014).
- Nalam, P. C., Gosvami, N. N., Caporizzo, M. A., Composto, R. J., Carpick, R. W. Nano-rheology of hydrogels using direct drive force modulation atomic force microscopy. Soft Matter. 11 (41), 8165-8178 (2015).
- Raman, A., et al. Mapping nanomechanical properties of live cells using multi-harmonic atomic force microscopy. Nature Nanotechnology. 6 (12), 809 (2011).
- Caporizzo, M. A., et al. Strain-rate dependence of elastic modulus reveals silver nanoparticle induced cytotoxicity. Nanobiomedicine. 2, 9 (2015).
- Corbin, E. A., Kong, F., Lim, C. T., King, W. P., Bashir, R. Biophysical Properties of Human Breast Cancer Cells Measured Using Silicon MEMS Resonators and Atomic Force Microscopy. Lab Chip. 15 (3), 839-847 (2014).
- Moreno-Flores, S., Benitez, R., Vivanco, M. D., Toca-Herrera, J. L. Stress relaxation and creep on living cells with the atomic force microscope: a means to calculate elastic moduli and viscosities of cell components. Nanotechnology. 21 (44), 445101 (2010).
- Grady, M. E., Composto, R. J., Eckmann, D. M. Cell elasticity with altered cytoskeletal architectures across multiple cell types. Journal of the Mechanical Behavior of Biomedical Materials. 61, 197-207 (2016).
- Park, S., Koch, D., Cardenas, R., Kas, J., Shih, C. K. Cell motility and local viscoelasticity of fibroblasts. Biophysical Journal. 89 (6), 4330-4342 (2005).
- Ribeiro, A. S., Khanna, P., Sukumar, A., Dong, C., Dahl, K. Nuclear Stiffening Inhibits Migration of Invasive Melanoma Cells. Cellular and Molecular Bioengineering. 7 (4), 1-8 (2014).
- Buda, A., Pignatelli, M. Cytoskeletal network in colon cancer: from genes to clinical application. The International Journal of Biochemistry & Cell Biology. 36 (5), 759-765 (2004).
- Lindberg, U., Karlsson, R., Lassing, I., Schutt, C. E., Höglund, A. S. The microfilament system and malignancy. Seminars in Cancer Biology. 18 (1), 2-11 (2008).
- Altemeier, W. A., Sinclair, S. E. Hyperoxia in the intensive care unit: why more is not always better. Current Opinion in Critical Care. 13 (1), 73-78 (2007).
- Roan, E., Waters, C. M., Teng, B., Ghosh, M., Schwingshackl, A. The 2-pore domain potassium channel TREK-1 regulates stretch-induced detachment of alveolar epithelial cells. PLoS One. 9 (2), 89429 (2014).
- Roan, E., et al. Hyperoxia alters the mechanical properties of alveolar epithelial cells. American Journal of Physiology. 302 (12), 1235-1241 (2012).
- Wilhelm, K. R., Roan, E., Ghosh, M. C., Parthasarathi, K., Waters, C. M. Hyperoxia increases the elastic modulus of alveolar epithelial cells through Rho kinase. The FEBS Journal. 281 (3), 957-969 (2013).
- Roan, E., Wilhelm, K. R., Waters, C. M. Kymographic Imaging of the Elastic Modulus of Epithelial Cells during the Onset of Migration. Biophysical Journal. 109 (10), 2051-2057 (2015).
- Wagh, A. A., et al. Localized elasticity measured in epithelial cells migrating at a wound edge using atomic force microscopy. American Journal of Physiology. 295 (1), 54-60 (2008).
- Liu, S., Wang, Y. Application of AFM in microbiology: a review. Scanning. 32 (2), 61-73 (2010).
- Hayhurst, E. J., Kailas, L., Hobbs, J. K., Foster, S. J. Cell wall peptidoglycan architecture in Bacillus subtilis. Proceedings of the National Academy of Sciences U.S.A. 105 (38), 14603-14608 (2008).
- Dover, R. S., Bitler, A., Shimoni, E., Trieu-Cuot, P., Shai, Y. Multiparametric AFM reveals turgor-responsive net-like peptidoglycan architecture in live streptococci. Nature Communications. 6 (1), 1-10 (2015).
- Gillis, A., Dupres, V., Delestrait, G., Mahillon, J., Dufrêne, Y. F. Nanoscale imaging of Bacillus thuringiensis flagella using atomic force microscopy. Nanoscale. 4 (5), 1585-1591 (2012).
- Laurent, V. M., et al. Gradient of rigidity in the lamellipodia of migrating cells revealed by atomic force microscopy. Biophysical Journal. 89 (1), 667-675 (2005).
- Collart-Dutilleul, P. Y., et al. Initial stem cell adhesion on porous silicon surface: molecular architecture of actin cytoskeleton and filopodial growth. Nanoscale Research Letters. 9 (1), 564 (2014).
- Ettinger, A., Wittmann, T. Fluorescence live cell imaging. Methods Cell Biology. 123, 77-94 (2014).
- Ilie, M. A., et al. Current and future applications of confocal laser scanning microscopy imaging in skin oncology. Oncology Letters. 17 (5), 4102-4111 (2019).
- Jonkman, J., Brown, C. M. Any way you slice it-a comparison of confocal microscopy techniques. Journal of Biomolecular Techniques. 26 (2), 54 (2015).
- Minsky, M. Memoir on inventing the confocal scanning microscope. Scanning. 10 (4), 128-138 (1988).
- Paddock, S. Over the rainbow: 25 years of confocal imaging. Biotechniques. 44 (5), 643-648 (2008).
- Amos, W., White, J. How the confocal laser scanning microscope entered biological research. Biology of the Cell. 95 (6), 335-342 (2003).
- Oreopoulos, J., Berman, R., Browne, M. Spinning-disk confocal microscopy: present technology and future trends. Methods in Cell Biology. 123, 153-175 (2014).
- Wolleschensky, R., Zimmermann, B., Kempe, M. High-speed confocal fluorescence imaging with a novel line scanning microscope. Journal of Biomedical Optics. 11 (6), 064011 (2006).
- Brakenhoff, G., Van der Voort, H., Van Spronsen, E., Nanninga, N. Three-dimensional imaging in fluorescence by confocal scanning microscopy. Journal of Microscopy. 153 (2), 151-159 (1989).
- Jerome, W. G., Price, R. L. . Basic Confocal Microscopy. , (2018).
- Webb, R. H. Confocal optical microscopy. Reports on Progress in Physics. 59 (3), 427 (1996).
- Pawley, J. . Handbook of biological confocal microscopy. , 236 (2010).
- Webb, D. J., Brown, C. M. Epi-fluorescence microscopy. Cell Imaging Techniques. , 29-59 (2012).
- Day, R. N., Davidson, M. W., Press, C. . The fluorescent protein revolution. , (2014).
- Fine, A., Amos, W., Durbin, R., McNaughton, P. Confocal microscopy: applications in neurobiology. Trends in Neurosciences. 11 (8), 346-351 (1988).
- Livet, J., et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 450 (7166), 56-62 (2007).
- Pan, Y. A., Livet, J., Sanes, J. R., Lichtman, J. W., Schier, A. F. Multicolor Brainbow imaging in zebrafish. Cold Spring Harbor Protocols. 2011 (1), 5546 (2011).
- Fu, X., et al. Brain Region Specific Single-Molecule Fluorescence Imaging. Anals in Chemistry. 91 (15), 10125-10131 (2019).
- Snell, A. A., et al. Cell-Derived Vesicles for in Vitro and in Vivo Targeted Therapeutic Delivery. ACS Omega. 4 (7), 12657-12664 (2019).
- Edgar, R. J., et al. Discovery of glycerol phosphate modification on streptococcal rhamnose polysaccharides. Nature Chemical Biology. 15 (5), 463-471 (2019).
- Kailas, L., et al. Immobilizing live bacteria for AFM imaging of cellular processes. Ultramicroscopy. 109 (7), 775-780 (2009).
- Gavara, N. A beginner's guide to atomic force microscopy probing for cell mechanics. Microscopy Research and Technique. 80 (1), 75-84 (2017).
- Wilson, T. The role of the pinhole in confocal imaging system. Handbook of Biological Confocal Microscopy. , 167-182 (1995).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved