Isolating Human Peripheral Blood Mononuclear Cells and CD4+ T cells from Sézary Syndrome Patients for Transcriptomic Profiling
In This Article
Summary
We present a simple protocol for the isolation of peripheral blood mononuclear cells from whole blood obtained from patients diagnosed with Sézary Syndrome, followed by selection of CD4+ T cells, their stimulation with phorbol12-myristate13-acetate and A23187 ionophore, and preparation of RNA for transcriptomic profiling.
Abstract
Cutaneous T-cell lymphomas (CTCL) are derived from the transformation and uncontrolled proliferation of mature skin-homing T cells, and mycosis fungoides (MF) and Sézary syndrome (SS) represent the most common subtypes. Despite a number of studies on characterizing gene expression, genetic alterations, and epigenetic abnormalities of CTCL, the molecular pathogenesis of MF/SS remains unclear. MF refers to the more common CTCL with a skin-predominance, and is usually limited to skin, whereas SS is an aggressive leukemic variant of CTCL with widespread skin involvement and is characterized by neoplastic distribution mainly involving blood, skin, and lymph node. Focusing on clinical practice, the identification of gene expression biomarkers has enormous potential to improve diagnosis and treatment of MF/SS. Indeed, recent transcriptomic studies have identified potential diagnostic biomarkers from differences in gene expression between normal and malignant T cells, which may improve our understanding of SS biology, and reveal potential therapeutic targets. This manuscript describes a detailed reproducible protocol for the isolation of peripheral blood mononuclear cells from fresh whole blood from patients diagnosed with SS, selection of CD4+ memory T cells (CD4+CD45RO+ T cells), chemical stimulation, and preparation of RNA suitable for transcriptomic profiling to discover novel prognostic molecular markers to gain additional insight in disease etiology. The stimulation using chemical agonist to activate nuclear regulation provides more specific assessment for pathways important in the dynamic transcription regulation and gene expression and eliminates confounding defects that may arise from upstream signaling defects arising from TCR antigen loss at the cell membrane. The data obtained from comparison of transcriptome of unstimulated to stimulated SS T cells unmasks functional regulatory gene expression defects not evident from analysis of quiescent unstimulated cells. Furthermore, the method outlined from this approach can be adapted for studying T cell gene expression defects in other T cell immune diseases.
Introduction
Cutaneous T-cell lymphoma (CTCL), including the most common subtypes mycosis fungoides (MF) and Sézary syndrome (SS), is a heterogeneous group of diseases derived from transformation and uncontrolled proliferation of mature skin-homing T cells1,2. The neoplastic T cells have a mature CD4+CD45RO+, memory phenotype3, and express skin homing adhesion markers, increasing epidermotropism4, which manifests as a rash particularly in early disease. The clinical course of MF is often indolent when under routine managed care, but a subset of patients can progress to more advanced disease. In these MF cases, skin lesions grow and thicken into large tumors, and neoplastic T cells may disseminate to lymph nodes and visceral organs. In contrast, SS is a more aggressive, leukemic variant of CTCL5, characterized by a triad of symptoms: generalized erythroderma (defined as affecting >80% of total body surface area), lymphadenopathy, and presence of more than 1000/mm3 circulating clonal atypical T cells with cerebriform nuclei, so called Sézary cells6,7. The prognosis for SS patients is significantly worse than MF. SS is rare with an incidence rate of 0.1/100,000, and represents approximately 3% of total CTCL cases8,9. CTCL typically presents in older adults with a median age of about 60 years10. The incidence rate for CTCL had been increasing and while the cause is unclear, the rate has stabilized since 199811,12.
The molecular pathogenesis of SS remains unclear. Genetic, epigenetic, and gene expression studies have produced a wealth of novel data, however there remain inconsistent findings, primarily due to the small patient cohorts studied2, as well as differences in experimental design and control populations13,14. Improved genomic and transcriptomic characterization may shed light on both disease mechanisms and previously unexplored therapeutic targets. Therefore, more studies from a larger patient population are needed to better understand this heterogenous malignancy. Biomarker panels that are highly sensitive and specific in one SS cohort have performed less uniformly in other cohorts15, which represents a serious obstacle in the development of reliable diagnostic and prognostic biomarkers for SS16. Ideal diagnostic biomarkers will be consistently and highly over-expressed in malignant T cells, while absent or nearly absent in normal T cells17. The discovery of disease-specific biomarkers is important for the advancement of diagnostic and therapeutic protocols for SS.
High quality transcriptomic data for both malignant and normal T cells requires an efficient and reliable approach to sample preparation. Here, we will discuss a detailed yet simple strategy to obtain RNA samples from T cell populations relevant to SS. We will discuss the isolation of peripheral blood mononuclear cells (PBMC) from whole blood, negative magnetic selection of disease-relevant CD4+CD45RO+ T cell populations, chemical activation to reveal differences in functional responses, and preparation of RNA for transcriptomic profiling. In the current protocol, chemical activation has been performed using phorbol myristate acetate (PMA) and calcium ionophore (A23187)18,19, because previous studies have shown defective T-cell receptor signaling in CTCL, and stimulation with PMA/A23187 bypasses the T-cell receptor20,21. Also, PMA/A23187 permits a more direct proximal activation of nuclear signals needed for cytokine gene activation. Finally, stimulation of T cells provides an additional level of insight into the regulation of gene expression that could not be gained from resting T cells where dynamic change is absent.
Protocol
Human cells are potentially infectious. Therefore, the experiments are performed strictly in accordance with the required precautions and procedures discussed as occupational safety and health administration (OSHA) and personal protection equipment (PPE).
1. Isolation of PBMCs from whole blood
- Collect all the materials needed from Table 1 and bring them to room temperature (RT). Warm RP10F to 37 °C. Adjust centrifuge to RT. Except for centrifugations and cell counting, perform all the steps using viable cells in a biological safety cabinet.
- Obtain blood in five 10 mL tubes (desired amount) containing anticoagulant. Store the whole blood at ambient temperature (18‒24 °C). Label 50 mL separation tubes with the human research subject number for the blood sample to be processed.
- Transfer 10‒15 mL of blood into each separation tube(s) with the matching subject number. Dilute the blood at least 2 fold with Hank’s balanced salt solution (HBSS). Do not exceed 35 mL of diluted blood per tube.
- Carefully and slowly underlay the blood with ~13 mL of density medium. Watch through the transparent density medium at the bottom of the tube, and stop pipetting when the pipette is almost empty (to prevent bubble release). Carefully remove the pipette to avoid mixing the blood and density medium layers.
- Carefully transfer the filled separation tubes to the centrifuge without disturbing the layers.
- Centrifuge at 500 x g for 30 min with the centrifuge brake off (deceleration set to zero).
NOTE: If the centrifuge only displays rpm, consult rotor specifications to estimate the rpm equivalent for 500 x g. - Carefully remove separation tubes from the centrifuge without disturbing the layers. Observe the buffy coat, which has been formed in between the density medium and plasma layers.
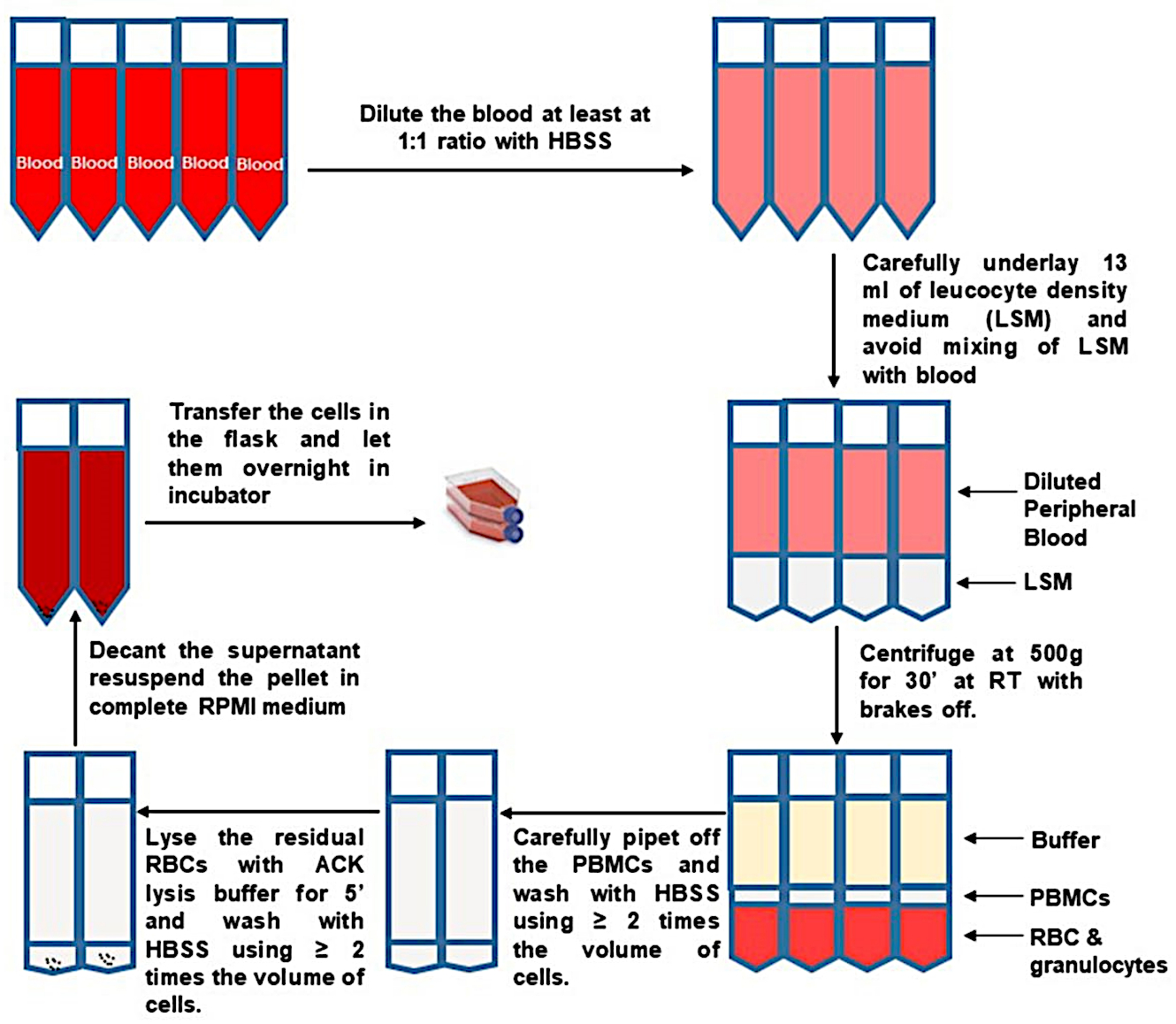
- Pipette from the top to remove and discard most of the upper plasma fraction, so that 10 mL remains above the buffy coat. Carefully and slowly collect the buffy coat. Transfer the buffy coats from two separation tubes into a new pre-labeled and sterile 50 mL tube, as shown in Figure 1.
- Dilute the PBMCs at least 2 fold with HBSS, bringing the volume in each new tube up to 50 mL. Remember to switch the centrifuge brake to full. Pellet PBMCs by centrifugation at 400 x g for 10 min. Remove the supernatant as much as possible and tap the bottom of the tube to loosen the pellet.
- To lyse residual red blood cells (RBC), resuspend each cell pellet in 1‒2 mL of ammonium-chloride-potassium (ACK) lysis buffer per 10 mL original blood volume. Incubate for exactly 5 min. Promptly stop lysis with an equal or greater volume of HBSS and adjust volume to 50 mL. Centrifuge at 400 x g for 10 min.
- Remove the supernatant and tap the bottom of tube to loosen the cell pellet. Pool cells from the same donor. Bring volume up to 50 mL with HBSS. Centrifuge at 400 x g for 10 min.
- Remove the supernatant and tap the bottom of tube to loosen the cell pellet. Resuspend cells in 10 mL of warm RP10F medium, and take an aliquot for viable cell counting using trypan blue.
- Calculate the total cell number in each sample using a hemocytometer.
2. Purification of CD4+CD45RO+ T cells from PBMCs
NOTE: Purification of CD4+CD45RO+ T cells from PBMCs is done by using commercially available magnetic separation (see Table of materials) with minor modifications. It is preferred to follow the kit manual for incubation time as each commercial kit has their own instructions.
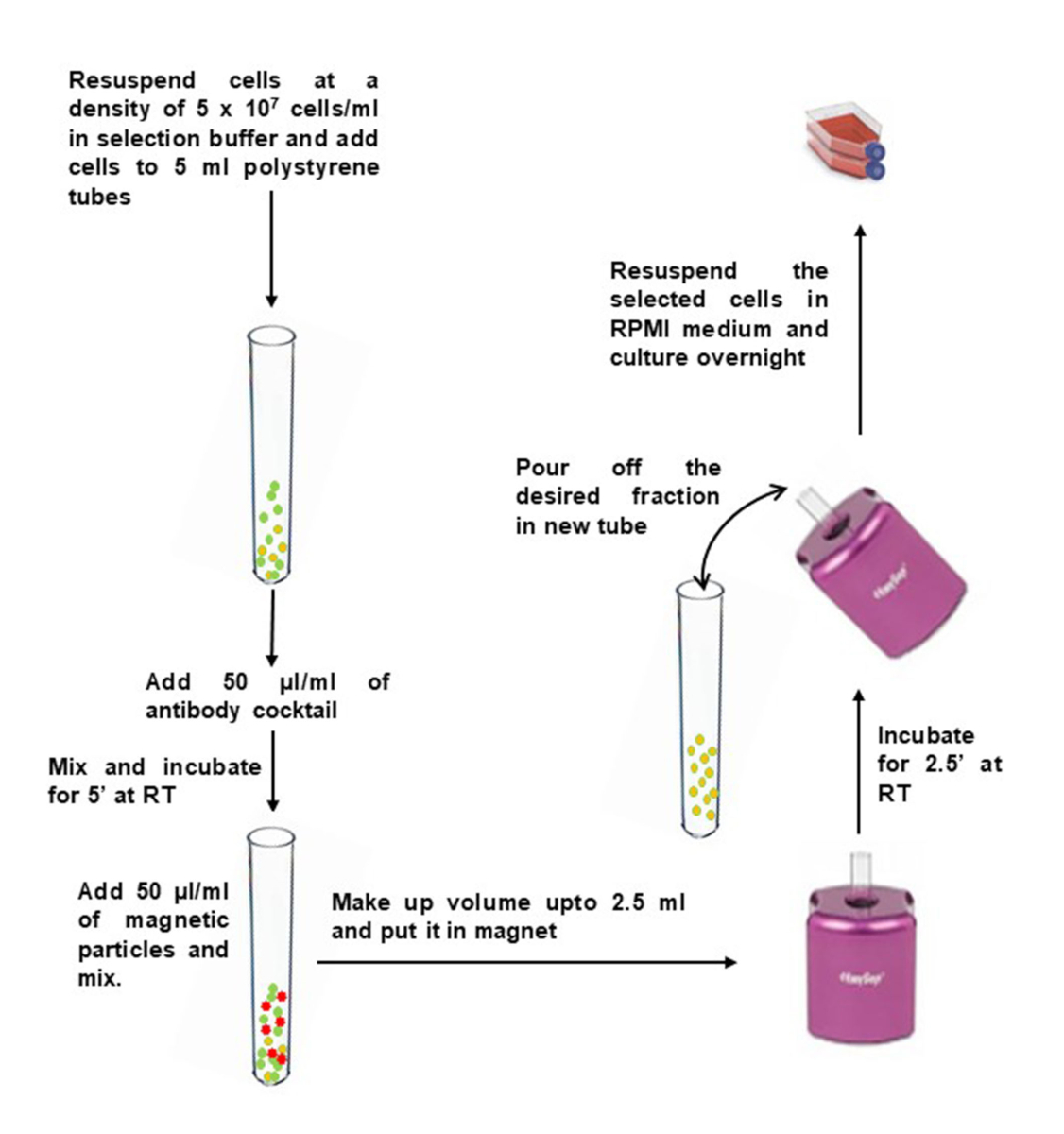
- Wash the desired quantity of PBMCs in 10 mL of selection buffer. Centrifuge at 400 x g for 10 min. Remove the supernatant and tap the bottom of the tube to loosen the pellet.
- Dilute PBMCs to 5 x 107 cells/mL in selection buffer and transfer them to a 5 mL of polystyrene round-bottom tube (12 x 75 mm).
- Add 50 µL of antibody cocktail per 1 mL of sample, and mix gently. Incubate at room temperature for 5 min.
- Immediately before use, vortex magnetic particles for 30 sec on high speed. Add 50 µL of magnetic particles per 1 mL of sample to the tube containing PBMCs, and mix gently.
- Bring the volume up to 2.5 mL with selection buffer and mix gently. Place the tube (without lid) into the magnet, and incubate at RT for 2.5 min.
- Pick up the magnet, and in a continuous motion, invert the magnet and tube to pour the enriched cell suspension into a new sterile tube.
- To increase recovery, add 2.5 mL of selection buffer to the tube remaining in the magnet, without disturbing the immobilized beads. Keep in the magnet for another 2.5 min, and repeat step 2.6 to recover additional cells.
- Take an aliquot for viable cell counting using trypan blue. Calculate the total cell number using a hemocytometer or cell counter.
- Confirm the purity by flow cytometry (Figure 3).
3. Chemical activation
- Adjust CD4+CD45RO+ T cells to 5 x 106 cells/mL with warm RP10F medium, and distribute cells into culture dishes of the desired size. Rest cells in a humidified 37 °C 5% CO2 incubator overnight.
- Pellet rested cells by centrifugation at 400 x g for 10 min. Remove the supernatant and tap the bottom of tube to loosen the cell pellet.
- Adjust cell concentration to 5 x 106 cells/mL with warm RP10F medium, and distribute 0.5‒1 x 107 cells into each of three sterile, screw-cap tubes.
NOTE: If sufficient cells are available, duplicate stimulations or additional time points may be included in the experimental design. - Stimulate cells in tubes 2 and 3 with PMA and A23187. Add PMA to 25 ng/mL and A23187 to 500 ng/mL and mix gently. Add an equal volume of dimethyl sulfoxide (DMSO) to the cells in tube 1 that will serve as a vehicle (control). For instance, if using 1 µL of PMA and 1 µL of A23187, then add 2 µL of DMSO to the vehicle tube. Keep the final concentration of DMSO below 0.5% in all tubes.
- Loosen the caps on the tubes, and return cells to the 37 °C 5% CO2 incubator for 2 h (tube 2) and 6 h (tubes 1 and 3). At the indicated time, centrifuge cells at 500 x g for 10 min.
- Prior to lysis, discard as much of the supernatant as possible, without disturbing the cell pellet.
- Promptly lyse the cells, as directed by instructions from the commercially available RNA isolation kit (see Table of Materials). Proceed with RNA isolation, or freeze the lysate at -80 °C to process later with additional samples.
NOTE: RNA purity and integrity may be verified using microcapillary electrophoresis. - Optional: To completely remove all traces of DNA from the purified RNA sample, use RNA clean up kit (see Table of Materials), according to manufacturer’s instructions.
Representative Results
This protocol includes procedures for the isolation of PBMCs from SS blood, purification of CD4+CD45RO+ T cells by negative selection, stimulation of purified T cells, and isolation of total RNA for transcriptomic profiling. Figure 1 describes the process of PBMC isolation from whole blood. Please note that the total yield of SS PBMCs will vary with starting blood volume and circulating tumor burden of each patient. In our laboratory, the average yield of SS PBMCs was 4.6 × 106 cells/mL of whole blood (1.85 × 106 – 3.25 x 107 cells/mL for 7 SS). The mean viability of isolated PBMCs was 95‒99%. Figure 2 shows high purity and viability of selected CD4+CD45RO+ memory T cells. The average yield of CD4+CD45RO+ T cells from SS PBMCs was 75% (75.6% – 84%), compared to 15.9% (3% – 30%) from normal donors (ND) PBMCs obtained from leukoreduction system (LRS) chambers. The viability and purity of CD4+CD45RO+ T cells obtained by this negative selection protocol has been consistently high (Figure 3).
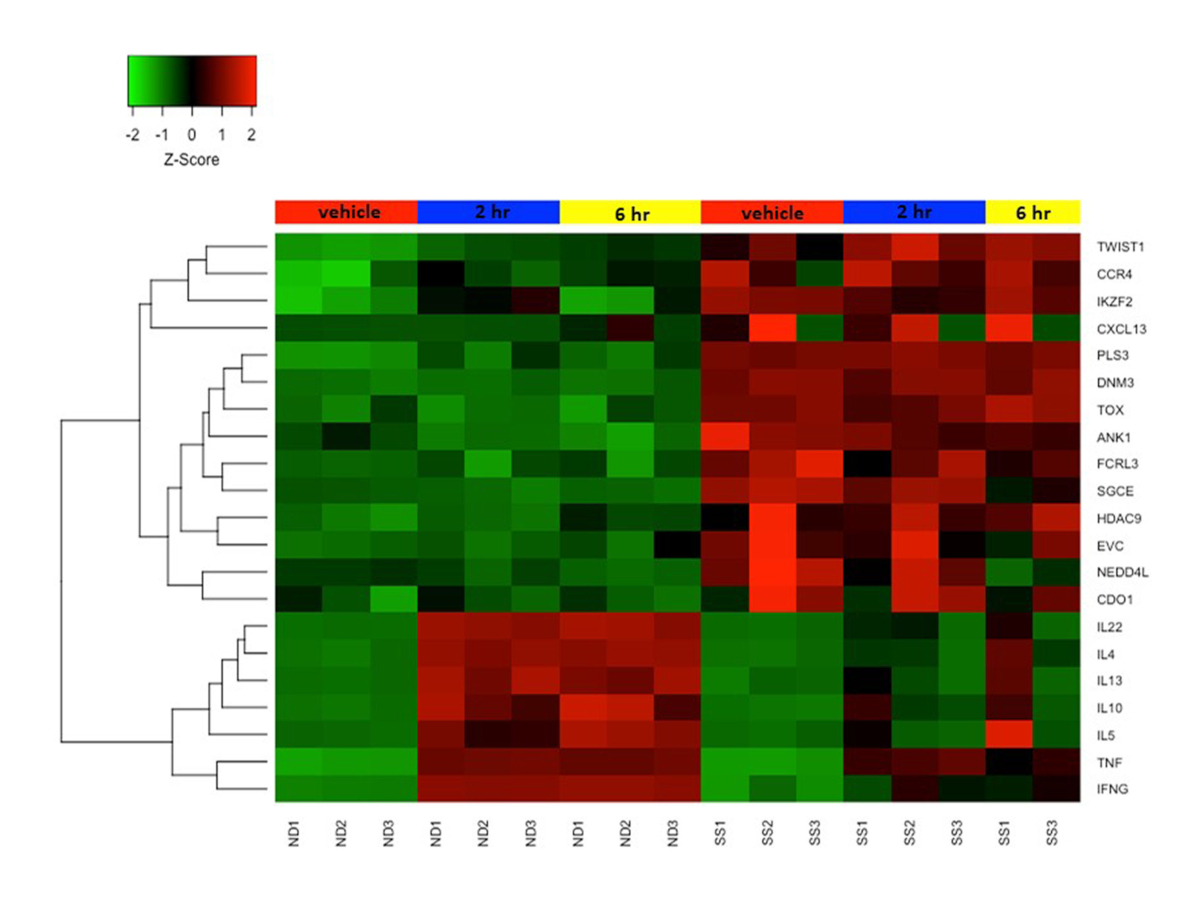
We previously combined the activation protocol above with microarrays to study the functional changes in the transcriptomes of both SS and ND T cells, and have demonstrated that SS memory T cells and SS PBMCs poorly express cytokine and other immune response genes compared to ND T cells and PBMCs19,22,23. Figure 4 shows the robust activation of several cytokine genes including IL4, IL 10, IL13 and IL22 in ND T cells, but not in SS T cells. This defect in functional gene expression in SS T cells have since been confirmed by other groups24. In addition, many genes not normally expressed in ND T cells are highly expressed in SS T cells, both at rest and following stimulation (Figure 4). These include the previously described SS biomarker genes DNM3, PLS3, TOX and TWIST125,26,27, as well as ANK1 and SGCE, which were first reported by our group. These positive biomarkers are highly expressed in SS, but not ND, and avoid technical pitfalls associated with negative biomarkers.

Figure 1: PBMC isolation from whole blood. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Negative selection of CD4+ memory T cells from isolated PBMCs. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Purity of CD4+CD45RO+ T cells was confirmed by flow cytometry. Lymphocytes were gated by light scatter (A), live lymphocytes excluded eFluor780 viability dye (B), and (C) represents non-selected normal donors (ND). Negative selection resulted in nearly pure populations of CD45RO+ T cells in ND (D) and SS patients (E). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Differential gene expression in resting and activated CD4+CD45RO+ memory T cells from SS and ND. Gene expression z-score is represented by a color scale from red (high expression) to green (low expression). Colored bars at the top of the heat map represents cell treatments: mock/vehicle treated (red), 2 h stimulated (blue), and 6 h stimulated (yellow). Several SS biomarker genes are highly expressed and cytokine genes are poorly expressed in SS T cells compared to ND T cells. Please click here to view a larger version of this figure.
{kind=link}
| Reagents | |
| Density Medium | Lymphocyte Separation Medium, Ficoll-Hypaque, or equivalent density medium with density = 1.077-1.080g/ml at 20oC. |
| HBSS | 1x Hank’s balanced salt solution, 4.2 mM NaHCO3, 10 mM HEPES, pH 7.2 |
| RP10F | RPMI 1640 medium, 10 % heat inactivated fetal bovine serum (FBS), 1x penicillin-streptomycin solution, pH 7.2 |
| ACK lysis buffer | 155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, No need to adjust pH. It should be ~ 7.3. |
| Selection buffer | 1x HBSS, 2% FBS, 2 mM EDTA |
| phorbol12-myristate13-acetate (PMA) | 50 µg/ml in DMSO |
| A23187 ionophore | 500 µg/ml in DMSO |
Table 1: Reagents.
Discussion
Several ways of isolating PBMCs have been developed, and each has their own advantages and limitations28. We routinely collect up to 50 mL of blood in five 10 mL tubes containing anticoagulant. The volume of the blood for PBMC isolation depends on several factors such as health and age of the research subject and also on phlebotomist expertise. A critical procedural step in the protocol is the formation of the step gradient. Poor layering may result in partial or complete failure of PBMCs to sediment at the interface. We prefer the under-layering method described here, as it is easy to start the bottom layer. To completely dispense all of the density medium below the blood, it is critical to use a pipet aid with no air leaks. Contamination of the PBMC fraction by undesired cell types can be minimized by careful and consistent collection of the buffy coat, which should be performed in the same way for each isolation. If PBMCs will not be further fractionated, collecting different amounts of the density gradient and plasma layers between isolations should be avoided. RBC lysis is performed to minimize the potential impact of contaminating RBC- and reticulocyte-derived RNA on downstream gene expression analyses. Hypotonic lysis will be inhibited by excess isotonic buffer.
Further isolation of T cell subset is important for molecular studies. Here we described subsequent CD4+CD45RO+ T cells selection by negative selection to remove undesired cell types. Negative selection relies on antibodies recognizing specific cell surface markers for all undesired cells. Antibody coated cells are then removed by magnetic beads. This selection protocol removes unwanted cells while allowing untouched and unstimulated target cells remain free floating, which is essential in studying gene activation. However, care must be taken to avoid cell clumps, which reduce the final purity of selected CD4+ CD45RO+ T cells. Ethylenediaminetetraacetic acid (EDTA) present in the selection buffer minimizes cell clumping. Yield of T cells depends on factors such as initial volume of the blood, patient variables such as the treatment being administered to the patient and disease stage at the time of sample collection. Treatment given to the patients may also affect the cell viability. In addition, sample collection before any procedure such as photopheresis also have positive impact on CD4+CD45RO+ T cells purity. We have observed that sample collection after photopheresis treatment procedure has negative impact on CD4+CD45RO+ T cells yield.
Neoplastic T cell clones from SS patients most frequently express surface markers consistent with a mature, memory CD4 T cell phenotype29,30. However, phenotypic plasticity has been occasionally observed with respect to surface markers including CD4, CD45RO, CD45RA, CD7 and/or CD2631. Previous studies have also shown the heterogeneity in CD45RO and CD45RA expression among SS patients29, whereas the majority of SS cases are still CD45RO+. Roelens et al.31 also showed that SS may exhibit interindividual and intraindividual heterogeneity with mixed population of naïve (TN), central memory (TCM), transitional memory (TTM), effector memory (TEM), and terminal effector memory (TEMRA) subsets. However, their results clearly show that majority of the SS cells has TCM phenotype. We focused our study on the CD45RO+ surface immunophenotype most common in SS patients, and confirmed phenotype by flow cytometry. In planning studies of T cell subsets in patients, it is important to consider phenotypic heterogeneity of the disease being studied, and purification strategy may therefore be adjusted as needed to obtain the desired T cell population for analysis.
There are several ways to stimulate T cells and PBMCs to examine functional gene expression. We prefer chemical activation (PMA + A23187 ionophore), since we are interested in gene regulation in the nucleus. Chemical activation is a best option for this purpose because it acts as a broad activator and is more uniform compared to antigen specific stimulation. PMA is a small organic compound that diffuses through the cell membrane into the cytoplasm, and directly activates protein kinase C. A23187 allows calcium to pass through membranes. These compounds bypass surface receptors, and together mimic the effects of T cell receptor ligation with co-stimulation mediated by CD28. The chemicals activate several intracellular signaling pathways, resulting in nuclear transcription factor activation and upregulation of cytokine genes that are accessible to transcription activation. Although chemical activation and CD3CD28 ligation produce strikingly similar global gene expression profiles in normal cells32, chemical activation with PMA + A23187 is a good choice since SS T cells can lose expression of surface receptors including TCR components33. Chong et al.22 compared the activation of cytokine genes between PMA/A23187 to anti-CD3 and anti-CD28 antibodies in PBMCs from normal, early MF/CTCL, and late MF/CTCL patients. They reported that PMA/A23187 caused more rapid and intense activation of the IL-2 gene as compared to anti-CD3/CD28 stimulation. Additionally, they showed that the slower activation kinetics with anti-CD3/CD28 antibodies is potentially from cross-linking and membrane signaling necessary for stimulation. Furthermore, trends in expression of cytokines among the different cell populations studied were preserved with PMA/A23187. Since, we are interested in gene expression activation, chemical stimulation is an ideal approach because it acts as a broad activator and is more consistent compared to antigen specific stimulation. CD3/CD28 ligation is ideal to investigate pathways important in membrane based signal transduction. In addition, chemical activation is less expensive and does not require special equipment. In the current study, PMA + A23187 significantly activated cytokine genes in ND but not SS T cells, suggesting that SS T cells have functional deficiencies downstream of the TCR.
In summary, this protocol provides phenotypically pure T cells from precious patient-derived blood, and a method for assessing genome-wide changes in functional gene expression. We demonstrate that transcriptomic profiling of SS T cells compared to normal CD45RO+ T cells reveal profound differences in gene activation in fresh human T cells from patients with CTCL. These studies will aid the development of diagnostic biomarkers and therapeutic strategies targeting novel markers in CTCL. In addition, this strategy and protocol in studying primary human T cells may be valuable in adapting to studies of other T cell mediated diseases.
Disclosures
The authors have nothing to disclose.
Ethical Disclosure:
This research protocol was approved by the Institutional Review Board (IRB) of the University of Arkansas for Medical Sciences (UAMS, Little Rock, AR) The microarray data presented in this study was done on the samples recruited under a research protocol approved by the IRB of Henry Ford Hospital (Detroit, MI).
Acknowledgements
We thank the patients and volunteers who participated in our research.
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 ml microcentrifuge tubes | Fisher Scientific | 02-681-320 | |
| 10 ml Disposable Plastic Pipette | Thermo Scientific | 170356 | |
| 1000 ul Pipet tips | VWR | 10017-038 | |
| 15 ml Conical Tubes | Corning | 352196 | |
| 25 ml Disposable Plastic Pipette | Thermo Scientific | 170357 | |
| 5 ml Disposable Plastic Pipette | Thermo Scientific | 170355 | |
| 50 ml Conical Tubes | Thermo Scientific | 339652 | |
| A23187 ionophore | Fisher Scientific | BP595 | |
| Centrifuge | Thermo Scientific | 75004381 | |
| DMSO | Sigma | D2650-5x5ml | |
| EasySep Human Memory CD4+ T cell Enrichment Kit | StemCell | 19157 | |
| FBS | GIBCO | 16140-071 | |
| HBSS | GIBCO | 14185-052 | |
| HEPES | Fisher Bioreagents | BP310-500 | |
| KHCO3 | Fisher Bioreagents | P184-500 | |
| Lymphocyte Separation Medium | Corning | 25-072-CV | |
| Na2EDTA | ACROS | 10378-23-1 | |
| NaHCO3 | Fisher Bioreagents | S233-500 | |
| NH4Cl | Fisher Bioreagents | A661-500 | |
| Penicillin-streptomycin solution | GIBCO | 15140122 | |
| phorbol12-myristate13-acetate (PMA) | Sigma | P-8139 | |
| Pipet-Lite LTS Pipette L-1000XLS+ | RAININ | 17014382 | |
| Pipet-Lite LTS Pipette L-10XLS+ | RAININ | 17014388 | |
| Pipet-Lite LTS Pipette L-200XLS+ | RAININ | 17014391 | |
| Pipet-Lite LTS Pipette L-20XLS+ | RAININ | 17014392 | |
| RNA Clean & Concentrator-5 | Zymo Research | R1013 | |
| Rneasy Plus Mini Kit | Qiagen | 74136 | |
| RPMI 1640 | GIBCO | 31800-022 | |
| T-25 Flask | Thermo Scientific | 2024-10 |
References
- Kim, E. J., et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. Journal of Clinical Investigation. 115, 798-812 (2005).
- Wong, H. K., et al. Evolving Insights in the Pathogenesis and Therapy of Cutaneous T-cell lymphoma (Mycosis Fungoides and Sezary Syndrome). British Journal of Haematology. 155 (2), 150-166 (2011).
- Whittaker, S. Biological insights into the pathogenesis of cutaneous T-cell lymphomas (CTCL). Seminars in oncology. 33 (3), 3-6 (2006).
- Kallinich, T., et al. Chemokine receptor expression on neoplastic and reactive T cells in the skin at different stages of Mucosis Fungoides. Journal of Investigative Dermatology. 121 (5), 1045-1052 (2003).
- Rodd, A. L., et al. Current and Emerging Therapeutics for Cutaneous T-Cell Lymphoma: Histone Deacetylase Inhibitors. Lymphoma. 2012, 1-10 (2012).
- Olsen, E., et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 110, 1713-1722 (2007).
- Hameetman, L., et al. EPHA4 is overexpressed but not functionally active in Sézary syndrome. Oncotarget. 6 (31), 31868-31876 (2015).
- Willemze, R., et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 105, 3768-3785 (2005).
- Bradford, P. T., et al. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 113, 5064-5073 (2009).
- Wilson, L. D., et al. Age, race, sex, stage, and incidence of cutaneous lymphoma. Clinical Lymphoma Myeloma and Leukemia. 12, 291-296 (2012).
- Li, Y., et al. Management of cutaneous T cell lymphoma: new and emerging targets and treatment options. Cancer Management and Research. 4, 75-89 (2012).
- Korgavkar, K., et al. Changing incidence trends of cutaneous T-cell lymphoma. Jama Dertmatology. 149 (11), 1295-1299 (2013).
- Dulmage, B. O., Geskin, L. J. Lessons learned from gene expression profiling of cutaneous T-cell lymphoma. British Journal of Dermatology. 169 (6), 1188-1197 (2013).
- Boonk, S. E., et al. Evaluation of Immunophenotypic and Molecular Biomarkers for Sézary Syndrome Using Standard Operating Procedures: A Multicenter Study of 59 Patients. Journal of Investigative Dermatology. 136 (7), 1364-1372 (2016).
- Scarisbrick, J. J., et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. Journal of Clinical Oncology. 10 (33), 3766-3773 (2015).
- Benoit, B. M., et al. CD164 identifies CD4+ T cells highly expressing genes associated with malignancy in Sézary syndrome: the Sézary signature genes, FCRL3, Tox, and miR-214. Archives of Dermatological Research. 309, 11-19 (2017).
- Dulmage, B., et al. The biomarker landscape in mycosis fungoides and Sézary syndrome. Experimental Dermatology. 26 (8), 668-676 (2017).
- Pick, E., et al. Intracellular Mediation of Lymphokine Action: Mimicry of Migration Inhibitory Factor (MIF) Action by Phorbol Myristate Acetate (PMA) and the Ionophore A23187. Annals of the New York Academy of Sciences. 94 (332), 378-394 (1979).
- Chong, B. F., et al. Induced Sézary syndrome PBMCs poorly express immune response genes up-regulated in stimulated memory T cells. Journal of Dermatological Science. 60 (1), 8-20 (2010).
- Fargnoli, M. C., et al. Diminished TCR signaling in cutaneous T cell lymphoma is associated with decreased activities of Zap70, Syk and membrane-associated Csk. Leukemia. 11, 1338-1346 (1997).
- Hansen, E. R., et al. Leukemic T cells from patients with cutaneous T-cell lymphoma demonstrate enhanced activation through CDw60, CD2, and CD28 relative to activation through the T-cell antigen receptor complex. Journal of Investigative Dermatology. , 667-673 (1993).
- Chong, B. F., et al. Immune Function Abnormalities in Peripheral Blood Mononuclear Cell Cytokine Expression Differentiates Stages of Cutaneous T-Cell Lymphoma/Mycosis Fungoides. Clinical Cancer Research. 14 (3), 646-653 (2008).
- Moerman-Herzog, A. M., et al. Transcriptome analysis of Sézary syndrome and lymphocytic-variant hypereosinophilic syndrome T cells reveals common and divergent genes. Oncotarget. 10, 5052-5069 (2019).
- Fanok, M. H., et al. Role of Dysregulated Cytokine Signaling and Bacterial Triggers in the Pathogenesis of Cutaneous T-Cell Lymphoma. Journal of Investigative Dermatology. 138, 1116-1125 (2018).
- Su, M. W., et al. Aberrant expression of T-plastin in Sezary cells. Cancer Research. 63, 7122-7127 (2003).
- van Doorn, R., et al. Aberrant expression of the tyrosine kinase receptor EphA4 and the transcription factor twist in Sezary syndrome identified by gene expression analysis. Cancer Research. 64, 5578-5586 (2004).
- Booken, N., et al. Sezary syndrome is a unique cutaneous T-cell lymphoma as identified by an expanded gene signature including diagnostic marker molecules CDO1 and DNM3. Leukemia. 22, 393-399 (2008).
- Dagur, P. K., McCoy, J. P. Collection , storage, and preparation of human blood cells. Current Protocol Cytometry. 73, 1-16 (2016).
- Fierro, M. T., et al. Heterogeneity of Circulating CD4+ Memory T-cell Subsets in Erythrodermic Patients: CD27 Analysis Can Help to Distinguish Cutaneous T-cell Lymphomas From Inflammatory Erythroderma. Dermatology. 216 (3), 213-221 (2008).
- Campbell, J. J., et al. Sézary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 116 (5), 767-771 (2010).
- Roelens, M., et al. Circulating and skin-derived Sezary cells: clonal but with phenotypic plasticity. Blood. 130 (12), 1468-1471 (2017).
- Diehn, M., et al. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proceedings of the National Academy of Sciences of the United States of America. 99 (18), 11796-11801 (2002).
- Fuji, K. New therapies and immunological findings in cutaneous T-cell lymphoma. Frontiers in Oncology. 8, 12-28 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved