Fluorescence In Situ Hybridization on DNA Halo Preparations to Reveal Whole Chromosomes, Telomeres and Gene Loci
In This Article
Summary
Combining DNA halo preparations with fluorescence in situ hybridization enables high-resolution analysis of genomic interactions with the nucleoskeleton. Attached genome leads to hybridized fluorescent signals located within the residual extracted nuclei, whereas non-attached genome is in the halo of DNA surrounding the residual nuclei.
Abstract
The genome is associated with several structures inside cell nuclei, in order to regulate its activity and anchor it in specific locations. These structures are collectively known as the nucleoskeleton and include the nuclear lamina, the nucleoli, and nuclear bodies. Although many variants of fluorescence in situ hybridization (FISH) exist to study the genome and its organization, these are often limited by resolution and provide insufficient information on the genome's association with nuclear structures. The DNA halo method uses high salt concentrations and nonionic detergents to generate DNA loops that remain anchored to structures within nuclei through attachment regions within the genome. Here, soluble nuclear proteins, such as histones, lipids, and DNA not tightly bound to the nuclear matrix, are extracted. This leads to the formation of a halo of unattached DNA surrounding a residual nucleus which itself contains DNA closely associated with internal nuclear structures and extraction-resistant proteins. These extended DNA strands enable increased resolution and can facilitate physical mapping. In combination with FISH, this method has the added advantage of studying genomic interactions with all the structures that the genome is anchored by. This technique, termed HALO-FISH, is highly versatile whereby DNA halos can be coupled with nucleic acid probes to reveal gene loci, whole chromosomes, alpha satellite, telomeres and even RNA. This technique provides an insight into nuclear organization and function in normal cells and in disease progression such as with cancer.
Introduction
The "nuclear matrix" was first described by Berezney and Coffey in 19741. After performing extractions with high salt molarities and nuclease treatment on rat liver nuclei, they identified a proteinaceous structural framework. The DNA halo procedure was subsequently adapted from this method and involves the removal of soluble proteins so that only the nuclear matrix (NM) and NM-associated proteins and chromosomes persist. DNA attachment regions are located at the base of DNA loops and are called matrix attached regions (MARs) or scaffold attachment regions (SARs), which are resistant to extraction with high salt concentrations and ionic detergent lithium-3,5-diiodosalicylate (LIS) respectively. In DNA halos, DNA associated with MARs/SARs are bound within the residual nucleus whereas the DNA loops extend away from these sites and form the DNA halo. We know now that the genome is anchored via lamina associated domains (LADs) to the nuclear lamina and through nucleolar associated regions (NADs) and possibly through other nuclear structures such as specific nuclear bodies.
The DNA halo method can be used for physical mapping of DNA, genes, and chromosomal regions as the extended DNA and chromatin provides a greater resolution because the chromatin is stripped of histones and the DNA is stretched out2,3,4,5,6. However, there are some limitations when using DNA halos for this application. For instance, DNA tightly associated with residual nuclei of DNA halos can be inaccessible to probes thus precluding it from analysis and physical mapping6. Other techniques such as fiber-FISH2,4,5,7 and molecular combing8 also enable physical mapping and have the advantage of being relatively quick and easy to perform. Both are preferentially used for DNA mapping of genes over DNA halos. These methods extract chromatin fibers via the use of solvent or salt extractions from the nucleus, however, molecular combing tends to have better reproducibility8,9.
There is an increasing evidence that the nucleoskeleton has a role in supporting key nuclear processes, such as attachment sites for DNA, chromatin remodeling, DNA transcription, DNA repair and DNA replication11,12. As such, the DNA halo technique was developed to investigate the interactions between the nucleoskeleton and genome during these cellular activities and has been routinely used and reported in research. This technique has also been used to investigate interactions between the genome and nucleoskeleton in relation to disease progression with malignancy-associated changes in nuclear structure being identified11.
The DNA halo technique has also been used to investigate the relationship between the genome and nucleoskeleton during development and differentiation12. A number of studies have used a variation of the DNA halo technique known as halosperm13 or SpermHalo-FISH if coupled with FISH14. Spermatozoa chromatin is tightly bound to proteins known as protamines and this technique was developed to improve access to the sperm DNA. Halosperm has been used to investigate the integrity of spermatozoa DNA and determine if DNA damage is present. Spermatozoa with less DNA damage correlate to a larger DNA halo size, whereas spermatozoa with increased levels of fragmented and damaged DNA had either small halos or none at all. Thus, halosperm can be used as a potential prognostic marker of embryo quality and successful pregnancy with IVF13. This example emphasizes the potential clinical applications of this technique. In our work we have used HALO-FISH to assess changes in genome behavior and the effect of specific drug treatments in the premature ageing disease Hutchinson-Gilford Progeria Syndrome (HGPS)15.
Together these, and other studies, highlight the breadth of processes/applications that the DNA halo technique can be used to study and utility of the technique.
Protocol
1. Slide preparation, sterilization and cell culture
- Prepare 500 mL of 10% HCl (v/v) and pour into a large beaker.
- Drop microscope slides individually into the acid and incubate for 1 h at room temperature on a shaker set at 2 x g.
CAUTION: HCl is corrosive and an irritant. It may cause severe skin burns and eye damage and irritation of the skin, eyes, and respiratory system. Ensure appropriate personal protection is worn including nitrile gloves, eye protection and a laboratory coat. - Decant acid from the beaker and wash slides ten times in tap water and then a further ten times in deionized water.
- Rinse slides in methanol twice and keep in methanol until sterilization by flaming.
CAUTION: Methanol is a highly flammable liquid and toxic if swallowed, in contact with skin or if inhaled. In addition, methanol may cause damage to organs, is corrosive and an irritant. Adhere to the workplace exposure limits and ensure appropriate personal protection is worn including butyl-rubber gloves, eye protection and a laboratory coat. Where possible handle in a local exhaust ventilation (LEV) fume cupboard. - Using metal tongs or long forceps, remove a microscope slide from the beaker containing methanol. Flame over a Bunsen burner to sterilize, and transfer into a rectangular cell culture vessel containing four compartments for slides, located close to the Bunsen burner.
CAUTION: Flaming allows immediate sterilization of microscope slides prior to use; however, this method does have associated hazards. As methanol is highly flammable it is important that the beaker containing the slides is positioned away from the Bunsen burner. Long tongs or forceps should be used that tightly grip the slides. The methanol level in the beaker should just cover the slides, to both minimize the amount of methanol used and so that only the ends of the forceps/tongs are in contact with the methanol. Always ensure the methanol has evaporated from the forceps or tongs after use and that these have cooled before placing back into the beaker containing the slides and methanol. The beaker should be covered by a piece of aluminum foil to starve the oxygen should the methanol catch fire. Never flame slides within a Class II laminar flow hood where the air is being circulated. - Alternatively, perform steps 1.1 to 1.4, but rather than flaming the slides after incubating with methanol, place slides on lint-free tissue to air dry. Once dry, wrap in aluminum foil and place in a sterilizer oven or autoclave.
- Grow cells in the appropriate medium with serum for at least 48 h at 37 °C in 5% CO2 until 60-70% confluency is reached. This protocol was performed on an early passage of human dermal fibroblasts (HDFs) and on classical Hutchinson-Gilford progeria syndrome (HGPS) fibroblasts (AG06297) and atypical type 2 HGPS fibroblasts (AG08466). Harvest each cell type and count using a hemocytometer to determine cell density. Seed 1 x 105 cells in 10 mL medium per slide.
NOTE: The cell density is important as DNA loops from different nuclei may converge if cells become too confluent. Seeding densities may need to be optimized depending on the cell type used as transformed cells may proliferate more quickly, whilst later passage cell cultures may take a longer amount of time to reach the desired confluency. - If cells need to be arrested in G0 to become quiescent, then seed 1 x 105 cells (in 10 mL medium) per slide and leave to grow for 24 h. Wash cells twice with serum-free medium and incubate in standard medium that contains a lower concentration of serum at 0.5% (newborn calf serum, NCS; or fetal bovine serum, FBS) for 7 days.
- If the proliferative status of the cells is required for the DNA Halo assay, then determine cells that have passed through S-Phase by the incorporation of 5-bromo-2'-deoxy-uridine (BrdU) into the DNA during replication.
- Seed cells as normal and grow for 24 h. Remove the culture medium and replace with medium containing BrdU and 5-fluoro-2'-deoxyuridine (3 µg/µL). After a further 24 h, remove the medium, wash the cells once with medium (10% NCS) and then re-feed with fresh medium (10% NCS). Incubate for an additional 24 h and then prepare the slides for the DNA halo assay.
2. Probe preparation
- Chromosome whole and arm painting probes
- Make chromosome probes from the amplification of flow sorted or microdissected chromosomes by degenerate oligonucleotide primed polymerase chain reaction (DOP-PCR) using the method by Telenius et al.16. Use DOP-PCR to label chromosome probes with either Biotin-16-dUTP or Digoxigenin-11-dUTP as shown in Table 1. Please check manufacturer's instructions for amplification profile, however, the conditions used for this experiment are shown in Table 2.
- Prepare arm or whole chromosome probe by adding together 8 µL of labeled PCR product, 7 µL of Cot-1 DNA, 3 µL of herring sperm, 1/20th volume of 3 M sodium acetate (pH 5.4) and 2 volumes of 100% ethanol. Incubate the probe solution for a minimum of 30 min at -80 °C.
NOTE: This method can be used to create single chromosome probes, or multiple chromosome probes if different labels (i.e., Biotin-16-UTP and Digoxigenin-11-dUTP) are used for each chromosome of interest.
CAUTION: Ethanol is a highly flammable liquid and vapor and may cause serious eye irritation. Keep away from heat, hot surfaces, and ignition sources. Adhere to the workplace exposure limits and ensure appropriate personal protection is worn including butyl-rubber gloves, eye protection and a laboratory coat. Where possible handle in a LEV fume cupboard. - Centrifuge probe solution at 13,700 x g for 15 min at 4 °C and then wash with 70% ethanol. Repeat the centrifugation procedure and discard the supernatant, taking care not to disturb or lose the DNA pellet. Allow the DNA pellet to dry.
- Add 12 µL of hybridization buffer (50% formamide, 10% dextran sulphate, 10% 20x saline sodium citrate (SSC; 3 M NaCl, 0.3 M tri-sodium citrate; pH 7.0), 1% (v/v) polyoxyethylene sorbinal monolaurate (Tween-20)) to the DNA pellet. Leave at 37 °C for at least 2 h for the DNA pellet to dissolve into the hybridization buffer.
CAUTION: Formamide is carcinogenic and teratogenic so may cause serious harm to an unborn child. If a woman is pregnant or suspects she is pregnant they should avoid working with formamide. Formamide should be used in a LEV fume hood. Adhere to the workplace exposure limits and ensure appropriate personal protection is worn including butyl-rubber gloves, eye protection and a laboratory coat.
- DNA isolation from Bacterial Artificial Chromosomes (BACs)
- Streak a small portion of the glycerol stock from the BAC clone onto a Luria-Bertani (LB) agar plate (1% (W/V) NaCl; 1% (w/v) tryptone, 0.5% (w/v) yeast extract, 1.5% (w/v) Agar Technical, 12.5 µg/mL (w/v) chloramphenicol). Incubate overnight at 37 °C.
- Select a single colony from the plate and inoculate 10 mL of LB broth (1% (w/v) NaCl, 1% (w/v) bactotryptone, 0.5% (w/v) yeast extract, 12.5 µg/mL (w/v) chloramphenicol). Leave the solution in a shaking incubator overnight at 37 °C.
CAUTION: Chloramphenicol is suspected of causing cancer. Handle with care and reduce exposure. - Centrifuge culture at 1,700 x g for 10 min at room temperature.
- Discard supernatant and add 300 µL of P1 solution (15 mM Tris (pH 8), 10 mM EDTA, 100 µg/mL RNase A) to the pellet. Vortex vigorously and transfer cells to a 2 mL microcentrifuge tube.
- Add 300 µL of P2 solution (0.2 M NaOH, 1% (w/v) sodium dodecyl sulphate (SDS) dropwise to the cells. Invert the closed microcentrifuge tube 5 times and leave at room temperature for a maximum of 5 min.
CAUTION: Sodium hydroxide is corrosive and may cause severe skin burns and eye damage. It may be corrosive to metals. Handle with care and reduce exposure. Adhere to the workplace exposure limits and ensure appropriate personal protection is worn including nitrile-rubber gloves, eye protection and a laboratory coat. Where possible handle in a LEV fume cupboard.
CAUTION: Sodium dodecyl sulfate is a flammable solid, harmful if swallowed and may cause skin and respiratory irritation. It may also cause serious eye damage. Ensure appropriate personal protection is worn including nitrile-rubber gloves, eye protection and a laboratory coat. Where possible handle in a LEV fume cupboard. - Add 300 µL of P3 (3 M potassium acetate) slowly to the cells and gently mix. Place the microcentrifuge tube on ice for 10 min.
- Centrifuge at 8,100 x g for 10 min at 4 °C and transfer the supernatant to a tube containing 800 µL of ice-cold isopropanol. Invert the tube several times and incubate at -20 °C overnight.
CAUTION: Isopropanol is a highly flammable liquid and vapor and may cause serious eye irritation, drowsiness, or dizziness. Keep away from heat, hot surfaces, and ignition sources. Adhere to the workplace exposure limits and ensure appropriate personal protection is worn including nitrile-rubber gloves, eye protection, and a laboratory coat. Where possible handle in a LEV)fume cupboard. - Centrifuge at 8,100 x g for 15 min at 4 °C. Remove the supernatant and transfer to another tube. Add 500 µL of ice-cold 70% ethanol. Invert tube several times and centrifuge at 8,100 x g for 5 min at 4 °C.
- Remove the supernatant and air-dry the pellet at room temperature. Once the pellet is dry re-suspend in 40 µL of diethyl pyrocarbonate-treated water (DEPC-treated) and leave at 4 °C overnight. Once fully resuspended remove 5 µL of solution and load on a 1% agarose gel to check for the presence of DNA.
- Single gene probe preparation of BACs via nick translation
- Use commercially available nick translation labeling kits. Alternatively, use the following protocol. See Table 3 for constituents and volumes.
- Add the constituents from Table 3 together in a microcentrifuge tube adding the DNA Polymerase I last, gently mix and centrifuge briefly for a few seconds. Incubate the solution at 15 °C for 2 h.
- To verify fragment sizes, load 5 µL of the solution onto a 2% agarose gel. The DNA fragment size range should be between 200-600 bp. If the DNA fragment sizes are larger, then continue to incubate the solution for a further 15 min at 15 ˚C and run products on 2% agarose gel.
- Stop the nick translation reaction by adding 10 mM EDTA, 0.1% SDS (2.5 µL of 0.5 M EDTA, pH 8.0 in 100 µL and 1 µL of 10% SDS in 100 µL). Heat the solution at 65 °C for 5 min.
- To remove unincorporated nucleotides apply BAC probe to a spin column. Commercial spin columns can be purchased, or they can be created using a syringe as follows:
- Add 30 g of Sephadex G-50 to 500 mL of column buffer (10 mM Tris-HCl (pH8), 1 mM EDTA, 0.1% SDS). Autoclave the mixture. Also, make 500 mL of column buffer (without Sephadex G-50) and autoclave.
- Make spin columns by adding autoclaved glass wool to the bottom of a 1 mL syringe. Fill the 1 mL syringe with Sephadex G-50 in column buffer. Place 1 mL syringe into a 15 mL centrifuge tube that has a microcentrifuge tube with no lid at the bottom. Centrifuge at 1,600 x g for 5 min.
- Remove syringe and discard the microcentrifuge tube at the bottom. Add a fresh microcentrifuge tube back into the 15 mL centrifuge tube. Add column buffer (without Sephadex G-50) to the 1 mL syringe and reinsert back into 15 mL centrifuge tube. Centrifuge at 1,600 x g for 5 min. Repeat this step again twice.
- Remove syringe and insert into a 15 mL centrifuge tube that contains a new clean microcentrifuge tube. Apply probe to the syringe and collect probe in the microcentrifuge tube.
- To precipitate the DNA probe add 5 µL of herring sperm DNA (10 mg/mL), 10 µL of sodium acetate and 2.25 volume of 100% ethanol to the DNA solution. Gently mix the solution and incubate at -80 ˚C for a minimum 1 h. Centrifuge at 13,700 x g for 15 min at 4 °C.
- Discard the supernatant and wash the pellet with 200 µL of ice-cold 70 % ethanol for 15 minutes at 4 °C. Remove the supernatant and air dry. Once dry re-suspend pellet in 20 µL of DEPC-treated water at room temperature for several hours or overnight at 4 °C. The probe is now ready to be used or may be stored at -20 °C.
- For each slide mix 5 µL probe DNA with 5 µL Cot-1 DNA and dry using a Speed Vac vacuum concentrator. Once the pellet has dried re-suspend in 12 µL of hybridization mix.
3. DNA Halo preparation
- Remove the square culture dish containing the slides and attached cells from the incubator. Discard medium, label slides using a pencil and place in a Coplin jar containing 50 mL of ice-cold cytoskeleton (CSK) buffer: 100 mM NaCl, 3 mM MgCl2, 0.3 M sucrose, 10 mM 1, 4-piperazinediethanesulfonic acid (PIPES; pH 7.8), 0.5% (v/v) Triton X-100 made up in deionized water. Incubate for 15 min on ice or at 4 °C.
CAUTION: Triton X-100 can cause skin irritation and serious eye damage. Handle using appropriate personal protective equipment including nitrile gloves, goggles, and laboratory coat. - Discard the CSK buffer and quickly rinse slides in 50 mL of 1x DNA halo buffer (DHB; 140 mM NaCl, 27 mM KCl, 110 mM NaHPO4, 15 mM KH2PO4; pH7.4) three times, i.e., dip slide into Coplin jar containing DHB and remove.
- Transfer slides to a Coplin jar containing 50 mL of extraction buffer: 2 M NaCl, 10 mM PIPES (pH 6.8), 10 mM ethylenediaminetetraacetic acid (EDTA), 0.1% (w/v) digitonin, 0.05 mM (v/v) spermine, 0.125 mM (v/v) spermidine. Incubate for 4 min at room temperature.
CAUTION: Digitonin is toxic if swallowed or in contact with skin and is fatal if inhaled. Ensure digitonin is handled in a LEV fume cupboard and wear a laboratory coat, nitrile gloves (double gloved), safety glasses and mask. Both spermine and spermidine may cause severe skin burns and eye damage, whilst EDTA causes serious eye irritation, so handle each chemical with care.
NOTE: Prepare digitonin separately by dissolving the powder in water at a temperature of 60-70 °C. Add dissolved digitonin to the extraction buffer once cooled. Add spermine, spermidine and digitonin last to the extraction buffer to preserve the biological activity. - Incubate slides consecutively in 50 mL of 10x DHB (1.4 M NaCl, 270 mM KCl, 1.1 M NaHPO4, 150 mM KH2PO4; PH7.4), 5x, 2x and 1x DHB for 1 min each.
- Dip slides (straight-in and straight-out) through a 50 mL sequential ethanol series of 10%, 30%, 70% and 95% (v/v) ethanol.
- Air dry slides and store at -80 °C until two-dimensional fluorescence in situ hybridization (2D FISH) is performed.
4. Two-dimensional fluorescence in situ hybridization
- Make 20x SSC: 3 M NaCl, 0.3 M tri-sodium citrate, pH 7.0. This buffer can be autoclaved, stored at room temperature, and diluted as needed.
- Make 70% (v/v) formamide, 2x SSC pH 7.0 and heat to 70 °C in a water bath.
- Incubate slides, for 5 min each, through a sequential 50 mL ethanol series of 70, 90 and 100% ethanol.
- Air dry slides on a warming plate and bake in a 70 °C oven for 5 min.
- Denature slides by placing in the 70% formamide, 2x SSC solution for 2 min at 70 °C.
NOTE: The temperature and timing are critical for step 4.5. If the temperature is the DNA will not denature and the probes will not hybridize, and no signal will be obtained from DNA halo FISH. - Place the denatured slide in 50 mL of ice-cold 70% ethanol for 5 min and take through an ethanol series of 90%, 95% and 100% at room temperature for 5 min each.
- Air dry on a warming plate
- Handle directly labeled total human chromosome probes according to manufacturer's instructions. For these experiments we used human whole chromosome paints 1, 13, 15, 17 and 18. In addition, in this experiment, CCND1 and CTNNA1 gene probes were used.
NOTE: Both the whole chromosome probes and BAC gene-specific probes were labeled with biotin-11-dUTP and detected by streptavidin conjugated to Cyanine 3 (Cy3). For the chromosome painting probes made by (DOP-PCR) and BAC DNA labeled by nick translation, these will be referred to as DNA probes from this point forward in the protocol and treated as follows. - Denature DNA probe (whole chromosome paint or gene-specific probe) at 75 °C for 10 min in a hot-block or water bath.
- Warm DNA probes at 37 °C for 30 min in a hot-block or water bath before pipetting 10 µL onto the appropriate slide.
NOTE: This step is important to block repetitive chromosomal sequences. If not performed non-specific signals may be produced in the DNA Halo FISH. - Overlay probe with a 21 mm x 21 mm coverslip and seal using rubber cement.
- Incubate slides for a minimum of 18 h at 37 ˚C in a humidified hybridization chamber.
NOTE: Humidified hybridization chambers can be made from sandwich boxes that contain several layers of moistened tissue and a raised platform constructed from cut 10 mL plastic pipettes to rest the slides upon. This is covered in aluminum foil to minimize exposure to light. - Remove the rubber cement carefully using forceps.
- Incubate slides in 50 mL 50% (v/v) formamide, 2x SSC, pH 7.0 solution that has been pre-warmed to 45 ˚C for three 5 min incubations.
NOTE: Allow the coverslip to fall away from the slide in the first incubation in 50% (v/v) formamide, 2x SSC, pH 7.0 solution. This prevents damage to the DNA Halo preparation that could be caused by 'dragging' the coverslip away. The slides can be agitated in the buffer via gripping by forceps to help detach the coverslip. - Next, place slides in 50 mL of 0.1x SSC, pH 7.0 solution that has been preheated to 60 ˚C but placed in a 45 ˚C water bath. Incubate for 5 min and replace the buffer two more times with 5 min incubations.
- Place slides into a Coplin jar containing 50 mL of 4x SSC, pH 7.0 solution at room temperature and incubate for 15 min with three changes of buffer.
- Apply 100 µL of 4% BSA, 4x SSC solution to each slide and overlay with a piece of paraffin film. Incubate at room temperature for 10 min. This prevents non-specific binding of the antibody.
- To detect the labeled probe (biotin-16-dUTP), incubate with 100 µL of a 1:200 (made in 1% BSA, 4x SSC) streptavidin-Cy3 for 1 h at room temperature.
NOTE: Follow manufacturer's instructions with dilutions of antibodies and test dilution prior to experiment to ensure a good signal is produced - Place slides into a Coplin jar containing 50 mL of 4x SSC (0.5% Tween-20) pH 7.0 solution at room temperature and incubate for 15 min with three changes of buffer. Slides can be mounted at this stage as shown in step 4.21 if immunofluorescence is not required.
- If the proliferative status of cells made into DNA halos is required, stain with anti-pKi67 antibodies after the FISH steps, prior to mounting or stain for incorporated BrdU.
- Wash slides 3 times for 5 min each in 50 mL of 1x phosphate buffered saline (PBS), followed by blocking with 4% NCS in PBS for 1 h at room temperature.
- Apply 200 µL of the necessary primary antibody (rabbit anti-human pKi67; mouse anti-BrdU) to the slide, overlay with a strip of paraffin film and incubate at room temperature for 1 h.
- Wash slides 3 times for 5 min in 1x PBS and incubate at room temperature for 1 h in 200 µL of fluorochrome-conjugated secondary antibody (pKi67: swine anti-rabbit TRITC; BrdU: donkey anti-mouse Cy3). Perform 3 more 5 min washes with PBS. All dilutions to be made using 1% (v/v) NCS in PBS at the manufacturers' range of suggested dilutions.
- Mount slides in 20 µL of mountant containing DAPI and overlay with a 22 mm x 50 mm coverslip.
5. Telomere PNA FISH
- To detect Telomeres, use telomere PNA FISH kit - FITC; perform the procedure with the manufacturer's instructions. The procedure should be executed at room temperature, unless stated otherwise.
- Immerse slides in tris-buffered saline (TBS, pH 7.5) for 2 min and then place in 3.7% formaldehyde (in TBS; v/v) for exactly 2 min.
CAUTION: TBS solution contains 10-30% trometamol and 10-30% 2-amino-2-(hydroxymethyl) propane-1,3-diol hydrochloride. This may cause serious eye and skin irritation, so wear protective gloves and goggles/face protection. Use in a well-ventilated area. - Wash slides in a Coplin jar twice with TBS for 5 min each.
- Immerse slides in pre-treatment solution for 10 min and then wash twice with TBS for 5 min per wash.
- Next, take the slides through an ice-cold ethanol series comprising of 50 mL of 70%, 85% and 95% (v/v) ethanol for 2 min per concentration. Afterwards allow slides to air dry.
- Apply 10 µL of Telomere PNA Probe/FITC (or Cy3) depending on the choice of fluorescent tag coloration, to each slide and cover overlay with a coverslip. Incubate in a pre-heated oven set at 80 °C for 5 min and then place in the dark for approximately 1 h.
CAUTION: Telomere PNA Probe/FITC contains 6-100% formamide, which causes serious eye irritation and is teratogenic so may cause serious harm to an unborn child. If a woman is pregnant or suspects she is pregnant they should avoid working with formamide. Formamide should be used in a LEV fume hood and appropriate eye or face protection should be worn. - To remove the coverslips, immerse the slides in 'Rinse Solution' for 1 min and then place in the 'Wash Solution' for 5 min at 65 °C.
CAUTION: Wash solution contains 1-5% polyoxyethylene octyl phenyl ether and 1-5% sodium chloride. This is corrosive and can cause serious eye damage. Ensure that goggles or face protection is worn when handling the wash solution. - Incubate slides through a 50 mL of ice-cold ethanol series (70%, 85% and 95% (v/v)) for 2 min per concentration and then air dry. Once dry mount slide with mountant containing DAPI and overlay with a coverslip.
6. Image capture and analysis
- To visualize DNA halos and chromosome territories use an epifluorescence microscope (e.g., Leica DM4000 microscope) capturing images using a HC PL FLUOTAR 100X/1.30 oil objective and DFC365FX camera.
- Capture grey-scale images and define color for each channel captured to enable pseudocoloring of images. A commercial software was used in this experiment (e.g., LAS AF version 4.5.0 software). The individual color channels were exported as TIFFs.
- Analyze images using the Java image processing program Fiji ImageJ. Upload image by pressing File and Open.
- Load separate image channels or split a composite image into separate greyscale channels by clicking Image | Color | Split Channels. Select an image channel and click Image | Adjust and then select Brightness & Contrast. Alter accordingly and repeat with other channels.
- Create a mask of the residual nucleus by selecting the DAPI-stained channel depicting the nucleus. Click Image | Adjust and then select Threshold. A dialogue box will appear where threshold can be altered, check the Dark background box. Alter until residual nucleus is clear and press Apply and close the dialogue box.
NOTE: This creates a binary mask based on the pixel intensity, with white pixels showing regions of interest and black pixels showing background. Repeat the same procedure on the probe channel. - Use the freehand selection to outline the periphery of the residual nucleus then click Edit and Clear Outside. Overlay the probe channel onto the residual nucleus. This can be done by pressing Image| Color | Merge Channels.
- To set the measurement scale in ImageJ, draw a line on the scale bar or between the points of two known distances. Go to Analyze and press Set Scale. In the dialogue box add the distance length and click OK. To measure distances, draw a line between the points being measured and click Analyze | Measure. This will transfer the distance values to a data window.
- Measure the brightest DAPI intensity as this coincides with the center of the nucleus. From this measure the distance from the nuclear center to the furthest chromosome territory edge (CTE). Measure the distance of the nuclear center to the nuclear edge (NE).
- Ensure that results are portrayed as a CTE/NE ratio. Here the distance from the nuclear center to each furthest chromosome territory edge (CTE) is divided by the distance from the nuclear center to each respective nuclear edge (NE). This should be performed on a minimum of 50 nuclei. This may be depicted as a bar or box chart.
- For analysis of the telomeres, analyze a minimum of 30 nuclei per dataset. Images can be analyzed using Fiji ImageJ or manually to count the number of telomeres within the residual nucleus and within the DNA halo. BrdU or pKi67 enabled differentiation of proliferating (BrdU/piK67+) and senescent/quiescent (BrdU/pKi67-) nuclei. Data may be depicted in bar charts with error bars corresponding to standard error of the mean (SEM).
- Use student's t-test (unpaired) to statistically compare the results with p> 0.05 considered significant.
Representative Results
This method of DNA halo preparation has helped us in our endeavors to determine differences in genome behavior within young and old cells, but also in cells derived from premature ageing diseases with aberrant nucleoskeletal proteins15. Figure 1 displays examples of DNA halos where it is possible to see the edge of a residual nucleus, the DNA remaining within the residual nucleus and the unattached DNA that has spooled out into the surrounding area creating a DNA halo. It also depicts the analysis showing how the residual nucleus is obtained and the NE and CTE measurements. It is possible to differentiate between proliferating and non-proliferating cells by either incorporating a labeled nucleotide such as BrdU when cells are in S-phase or employing the diagnostic proliferation marker anti-pKi67, which reveals nucleoli, and regions of heterochromatin in G1 cells17,18. Primary cells grown in high serum without achieving confluency, that are negative for the proliferation markers, are assumed to be senescent. Primary cells grown in low serum or have become confluent i.e., contact inhibited that are negative for the proliferation markers are deemed quiescent and would be able to reenter the proliferative cell cycle given the correct nutrients and situation. Being able to differentiate between Ki67 positive and negative cells has enabled us to determine differences between proliferating, quiescent and senescent human dermal fibroblasts. Figure 2 displays DNA halos of proliferating human dermal fibroblasts created from cells where BrdU was incorporated into them during DNA replication, a mechanism that does not occur in non-proliferating cells, and subsequently stained with anti-BrdU antibody. Staining with the proliferative marker anti-pKi67 antibody is also visible in Figure 2. This is a robust antigen and survives the FISH protocol and so can be stained for post-FISH and pre-mounting. Thus, proliferating cells are positive (red) for BrdU and anti-pKi67 (red) in the left-hand column and non-proliferating cells, indeed senescent cells in Figure 2 are displayed in the right-hand column. The green signals are individual telomeres revealed with a telomere PNA FISH/FITC kit. Combining immunofluorescence with DNA halos enables analysis during different cell states, as shown in Figure 2 when investigating proliferating, quiescent and senescent cells. Depending on the antibody chosen other conditions can be examined, such as differentiation, DNA damage via irradiation etc.
Chromosome territories can also be visualized within DNA halos using FISH. Due to the preparation permitting spooling of DNA out the nuclei, the chromosome territory shape can be disturbed, with smaller or larger amounts of the chromosome found in the DNA halo, depending on the anchorage of the genome inside the residual nucleus and its structures. Figure 3 reveals a panel of DNA halos whereby individual chromosomes have been revealed with specific whole arm chromosome painting probes (red) for chromosomes 1, 13, 17 and 18. Anti-pKi67 (green) has been used to mark proliferating cells and its absence within the same culture, upon the same slide, denoting senescent cells. It is very obvious from the images and the data presented as CTE/NE that the small gene-poor chromosome 18 is a chromosome that has few attachments and spools further out into the DNA halo away from the residual nuclei and is significantly further from the center of the residual nuclei than the other chromosomes. However, this is also true for chromosome 1 as well. Using the proliferative marker anti-pKi67 it has also been possible to compare proliferating with senescent cells, within the same culture, and on the same slide, and this analysis has revealed that chromosomes within these two very different cell statuses are not significantly different from one another, with respect to attachment with the residual nuclear structures.
Interestingly, genes also are showing statistically significant differences between proliferating and senescent cells with respect to remaining within a residual nucleus or being located in the DNA Halo. Figure 4 demonstrates this with gene loci delineated by labeled BAC probes in red and anti-Ki67 in green. There are no significant differences between gene locations in the proliferating versus the senescent cells, after a DNA Halo preparation. However, there are significantly more catenin alpha 1 CTNNA1 loci within the DNA halo than cyclin D1 CNDD1 loci, where there are very few. Figure 5 displays DNA halo preparations with telomeres in green. The background is left deliberately high to enable telomere signals to be visualized within the DNA halo. In this set of data quiescent cells i.e., cells that have been serum starved for 7 days have been included and interestingly there are significantly more telomeres unattached and located within the DNA halos in quiescent cells than for proliferating and senescent cells. In Figure 5a the proportion of telomeres in the DNA halo can be observed, particularly for the image 'Experiment 2'. This corresponds with Figure 5b where the mean percentage of telomeres in DNA halo is approximately 17% in quiescent cells. There is some evidence that not all telomeres in senescent cells can be seen as some of them maybe very short.
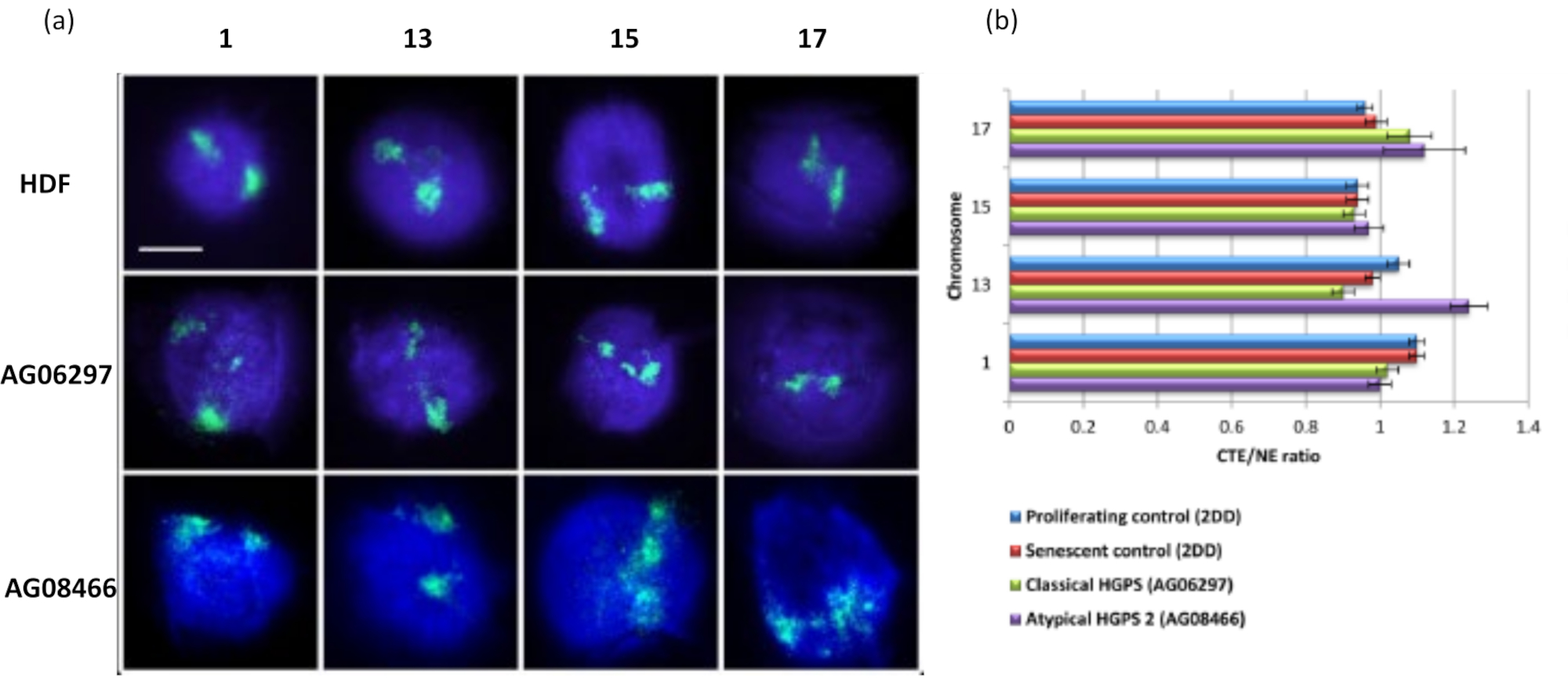
This method of DNA halo has been successful for us to investigate genome interaction alterations within nuclei in diseased cells15. Figure 6 demonstrates differences in chromosome attachment in primary control fibroblasts and in diseased cells with typical (lamin A mutation) and atypical Hutchinson-Gilford Progeria Syndrome, expressing a different SUN1 isoform and no lamin A mutation19. Chromosomes 1 and 13 show statistically significant differences in their attachment within the residual nuclei when compared to control DNA halos. Figure 6b correlates the position of the whole chromosome territory to the residual nucleus and DNA Halo. Values of 1 or less indicates the chromosome is located within the residual nucleus and values over 1 demonstrate chromosomes or portions of chromosomes within the DNA Halo.
Overall, this highlights the utility of HALO-FISH in investigating genomic interactions of whole chromosomes, specific genes and telomeres under a variety of conditions that affect the cell cycle (proliferation, quiescence and senescence) or within disease cells e.g., progeria and cancer cell lines. Indeed, the differences in interactions between these states implies the nucleoskeleton has an important role in regulating key processes within the nucleus.

Figure 1: HDF extracted nucleus displaying the residual nucleus and DNA halo and overview of analysis method. (a) An HDF nucleus prepared via DNA halo assay and counterstained with DAPI. The brightly stained residual nucleus shows DNA anchored to the nucleoskeleton and this is surrounded by the non-attached DNA which forms a halo of DNA. Magnification = x 100; scale bar 10 µm. (b) The blue channel captures the DAPI-stained nucleus and surrounding DNA. The residual nucleus is selected and removed using ImageJ. The arrow depicts the distance from the nuclear center to the residual nuclear edge (NE). (c) The red channel shows the probe signal. (d) The image denoted 'Result' is the outcome of superimposing the red channel on the blue channel image; this allows the distance from the nuclear center to the furthest chromosome territory edge (CTE). Please click here to view a larger version of this figure.
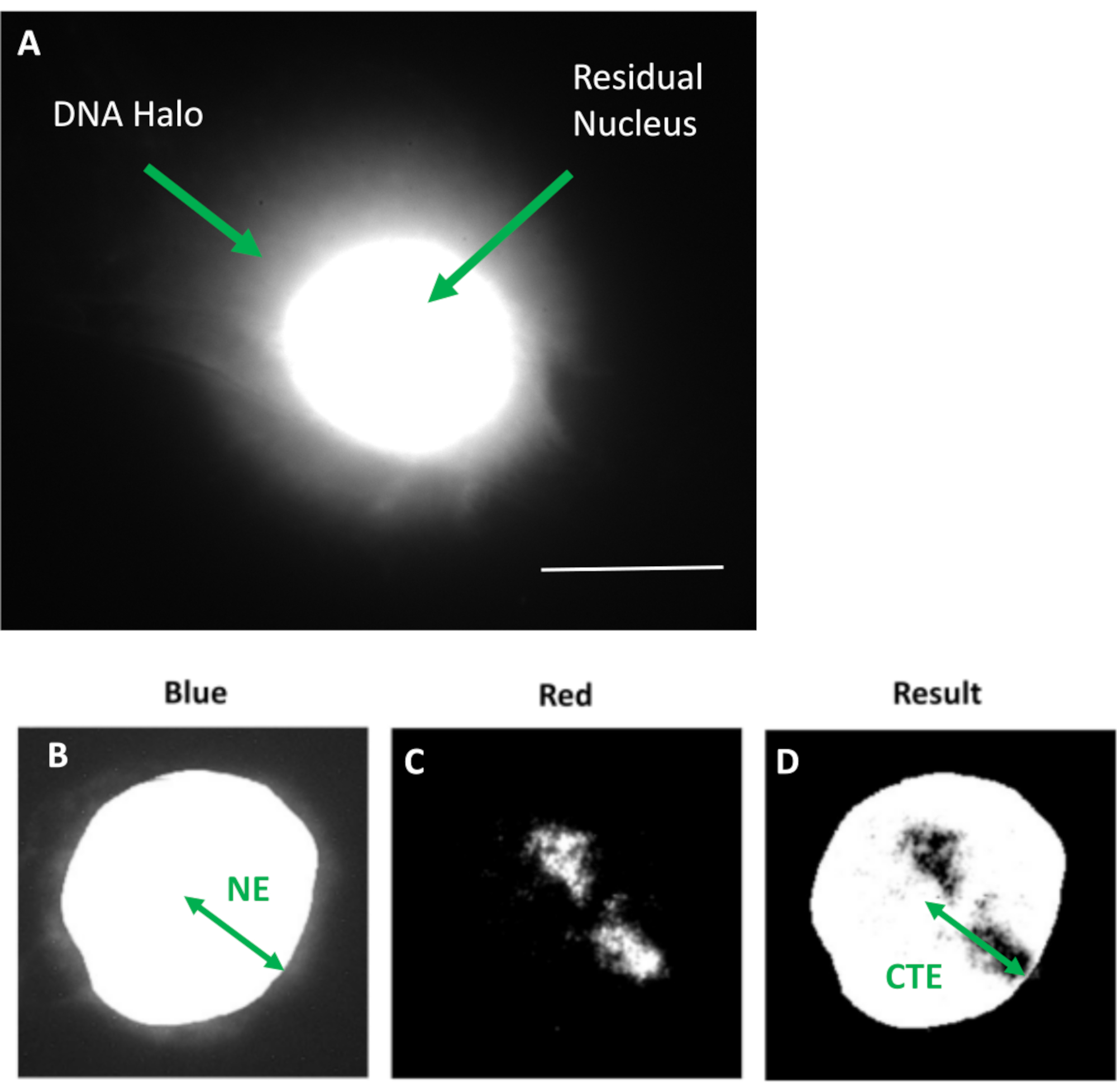{kind=link}

Figure 2: DNA halo preparation with telomere PNA FISH on proliferating and senescent HDFs. Telomere PNA FISH on HDFs subjected to DNA halo assay. Telomere signals are visualized in green (FITC), residual and halo DNA was counterstained using DAPI (blue) and proliferating nuclei were detected using either anti-BrdU or anti-pKi67 antibodies via indirect immunofluorescence in red (TRITC). Magnification = x 100; scale bar 10 µm. Please click here to view a larger version of this figure.
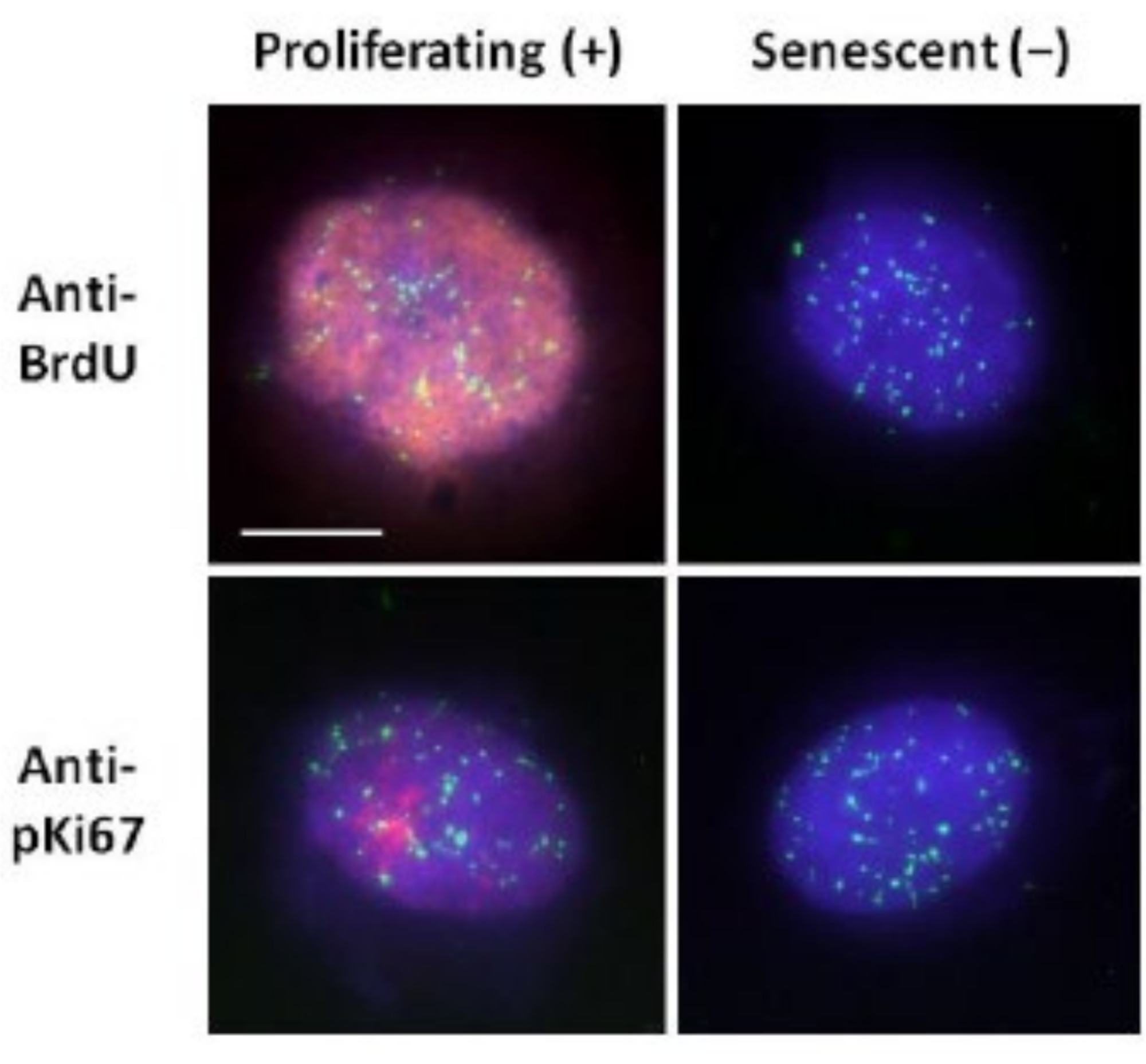{kind=link}

Figure 3: Nucleoskeleton-chromosome interactions and analysis using DNA halo assay. (a) 2D-FISH with probes specific for chromosomes 1, 13, 15, 17 and 18 was performed on HDFs subjected to DNA halo preparation. Whole chromosomes were painted in red (Cy3) and nuclei were probed with pKi67 to determine if they were proliferating or senescent. Proliferating cells (pKi67+) were delineated in green (FITC), whereas senescent cells remained unstained (pKi67-) i.e. no green signal detected. Magnification = x 100; scale bar 10 µm. (b) Chromosome anchorage by the nucleoskeleton in proliferating and senescent HDFs that had undergone HALO-FISH. Measurements show the ratio of the furthest chromosome territory edge (CTE) to respective nuclear edge (NE) for chromosomes 1, 13, 15, 17 and 18 in proliferating (pKi67+) and senescent (pKi67-) cells. Error bars represent ± SEM. (c) Modified box plot representation of chromosome territory edge (CTE) to respective nuclear edge (NE) of specific chromosomes in pKi67+ and pKi67- nuclei. Q1 = lower quartile; Min = lowest value recorded; Med = median; Max = maximum value recorded; Q3 = upper quartile. Please click here to view a larger version of this figure.
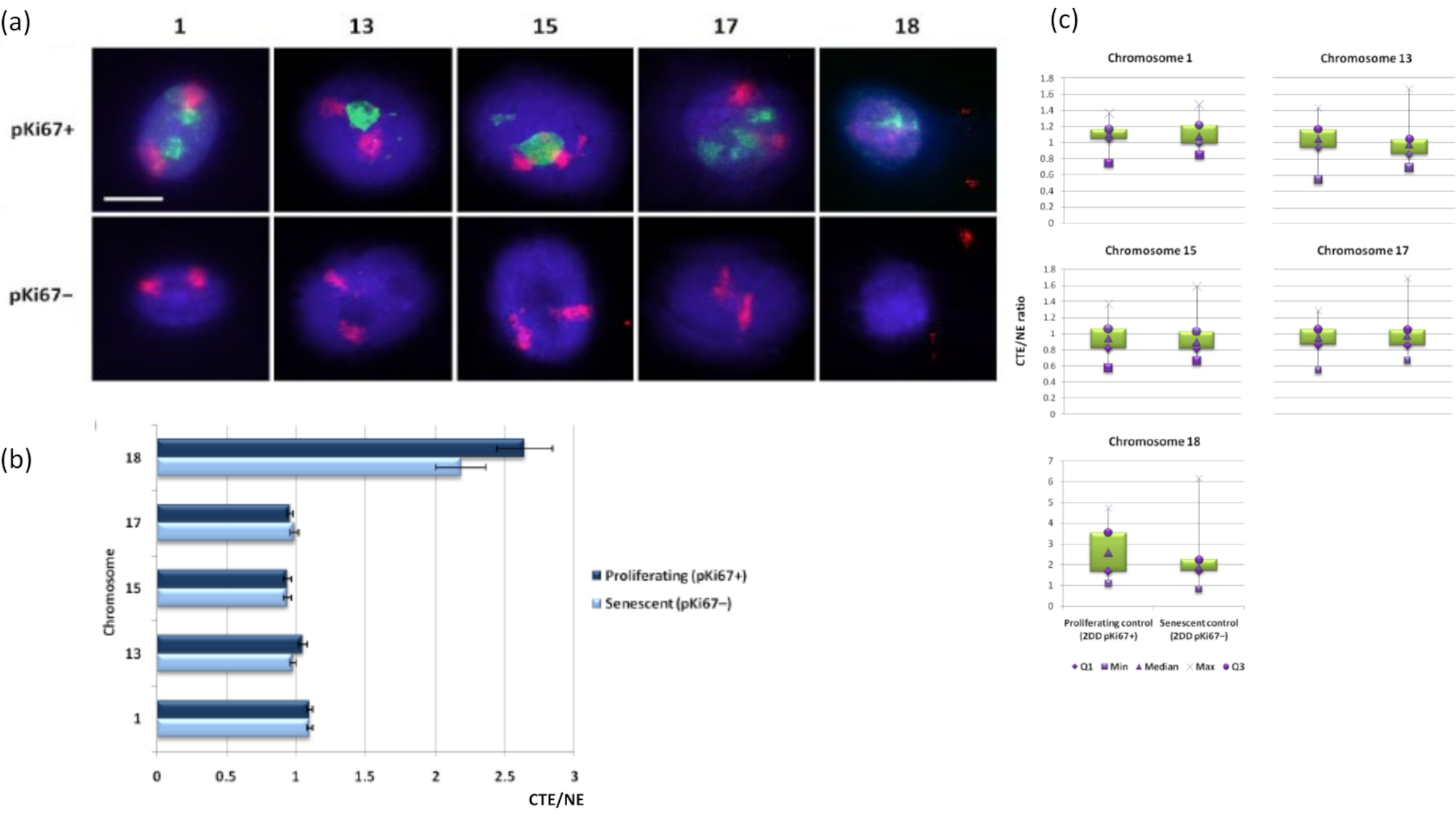{kind=link}

Figure 4: Gene-specific interactions in HDFs using HALO-FISH. (a) DNA halo extracted nuclei were probed with gene specific probes (CCND1 and CTNNA1) to investigate their anchorage to the NM on proliferating and senescent cells. The gene signals are shown in red (Cy3) and anti-pKi67 depicts proliferating cells and signal is visualized in green (FITC). For the proliferating CCND1 image, the residual nucleus is enclosed within the white circle, and the space between the white and green circle depicts the DNA Halo. Magnification = x 100; scale bar 10 µm. (b) Gene-specific signals for CCND1 and CTNNA1 are compared between the residual nucleus and DNA halo, and also, between proliferating and senescent cells. Error bars represent ± SEM. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: DNA halo assay on quiescent HDFs probed with telomere PNA-FISH. (a) Quiescence of HDFs was induced by culture in low serum medium for 7 days. The DNA halo assay was performed, and PNA-FISH enabled visualization of telomeres by FITC signal (green) and the residual nucleus and surrounding DNA halo was counterstained with DAPI (blue). Cells were also stained with anti-pKi67 antibody to ensure nuclei were non-proliferating. This was repeated on two separate occasions. Magnification = x 100; scale bar 10 µm. (b) Comparison of the mean percentage of telomeres localized within the DNA halo in proliferating, senescent and quiescent HDF cells. Error bars represent ± SEM. Please click here to view a larger version of this figure.
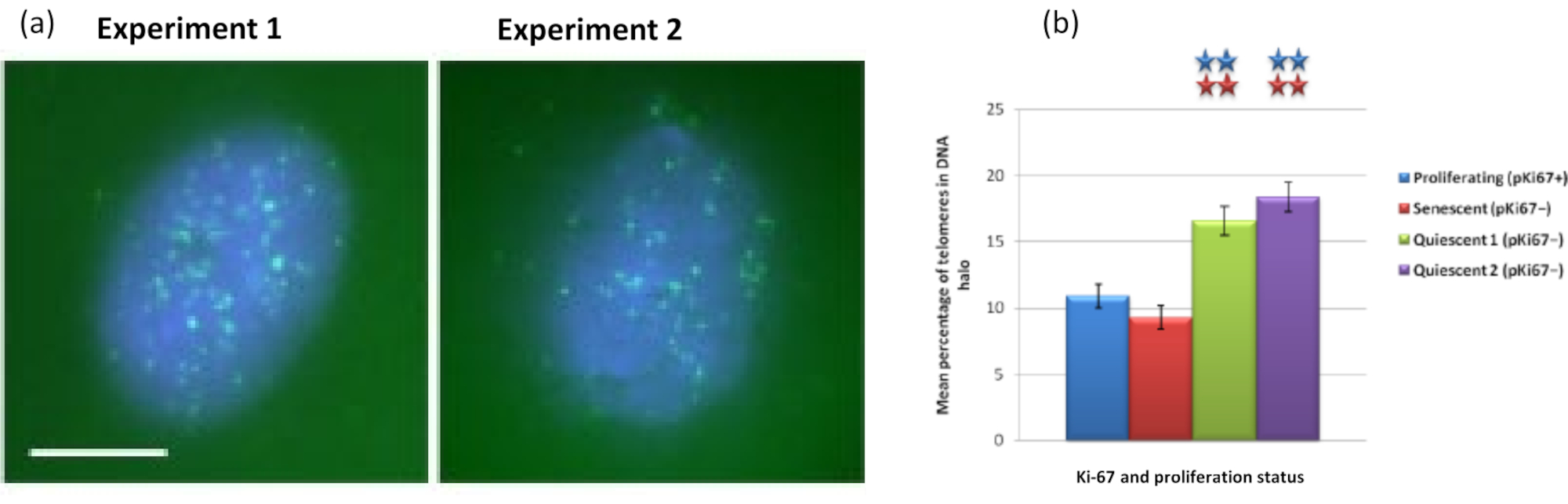{kind=link}

Figure 6: Examining whole chromosome anchorage to the nucleoskeleton in HGPS cells using HALO-FISH26. (a) Control HDF (2DD), classical HGPS (AG06297) and atypical type 2 HGPS (AG08466) nuclei underwent DNA halo preparation and then 2D-FISH using whole chromosome paints for chromosome 1, 13, 15 and 17. Whole chromosomes are depicted in green (FITC) and DNA was counterstained with DAPI (blue). Magnification = x 100; scale bar 10 µm. (b) Positioning of chromosomes within extracted nuclei was determined by measuring the ratio of the mean chromosome territory edge (CTE) to the nuclear edge (NE). A ratio above 1 demonstrates that the furthest CTE lies outside the corresponding NE within the DNA halo, while a ratio below 1 signifies that the furthest CTE lies within the NE within the residual nucleus. Please click here to view a larger version of this figure.
{kind=link}
| Constituents | Volume(μL) |
| 5XDOP-PCRbuffer | 10 |
| dNTPmix(withoutdTTP)(2mM) | 5 |
| dTTP(2mM) | 2 |
| Biotin-16-dUTPorDigoxigenin-11-dUTP | 10 |
| DOPprimer(20μM) | 5 |
| TaqDNAPolymerase(1U/μL) | 1 |
| PCRgradewater | 12 |
| Template | 5 |
Table 1: Table showing the DOP-PCR components and volumes for a 1x reaction
| Step | Cycles | Temp (degree Centigrade) | Time |
| Initial Denaturation | 1 | 95 | 3 min |
| Denaturation | 34 | 98 | 20 s |
| Primer Annealing | 62 | 1 min | |
| Extension | 72 | 30 s | |
| Final Extension | 1 | 72 | 5 min |
| Cooling | 4 | Hold |
Table 2: Table showing the DOP-PCR cycle, temperature, and time profile.
| Constituent | Volume(μL) |
| 10x NT buffer (0.5M Tris-HCl pH 8,50 mM MgCl2, 0.5 mg/ml BSA) | 5 |
| 0.1 M beta-mercaptoethanol | 5 |
| 10X Nucleotide stock (0.5 mM dATP, 0.5 mM dCTP, 0.5 mM dGTP, 0.5 mM dTTP, 0.5 mg/ml biotin-16-dUTP) | 5 |
| Dnase I (1 ng/ml) | 2 |
| DNApolymerase I | 5U per μg of DNA |
| DNAtemplate (1 μg) | 1 |
| DEPC-treated water | To 50 μL |
Table 3: Table showing the nick translation components and volumes for a one probe.
Discussion
The DNA halo method is an excellent method of choice when analyzing interactions between the nucleoskeleton and genome, however, there are some critical steps that must be adhered too. One of the most important parameters is the optimization of the cell seeding density. If cells become over confluent, then the DNA halos will overlap with neighboring cells making it impossible to perform the analysis. The CSK and extraction buffers must always be made fresh on the day of use with spermine, spermidine and digitonin being added to the extraction buffer at the end of the preparation process to maintain their biological activity. If performing Halo-FISH it is extremely important to use the correct denaturation temperature of the DNA halos to enable the probe or paint to subsequently hybridize.
Electron microscopy has been used to visualize the nuclear matrix, with filamentous structures being identified20. However, electron microscopy is limited as matrix associations with chromatin cannot easily be deduced. Indeed, the DNA Halo method is more versatile compared with electron microscopy as specific genes, chromosomes and cell states can all be examined. Furthermore, proteomic analysis of nuclear matrix proteins is being studied21,22. This method is good for comparing nuclear matrix components, particularly when comparing diseased cells, however, it doesn't provide the spatial distribution and attachments highlighted by the standard DNA Halo technique.
DNA Halo assays do have limitations. Firstly, as the matrix is extracted, this can only be performed on fixed cells so live imaging is not possible. Although the DNA Halo method is relatively quick and easy to perform, the overall process may be time consuming when cell culture, probe generation, Halo-FISH and analysis is all taken into account.
Image capture of DNA Halos and HALO-FISH using super-resolution microscopy would greatly improve the resolution of DNA specific probes and antibodies. In addition, as fluorochromes can be more easily spectrally resolved, it may be possible to use a number of DNA probes in a single experiment, providing even more information. Improvements in molecular biology techniques such as chromosome conformation capture (3C) have been used to determine interactions of gene loci and analyze the spatial organization on chromatin in the cell. DNA Halo assays and 3C can be combined, a term known as M3C23, again demonstrating the adaptability of the DNA Halo technique.
The original data presented here are to demonstrate the possibilities for genome behavior interrogation and how to present those data. With these data we have demonstrated that it is possible to determine significant differences in genome attachment using (1) chromosome painting probes, in this study revealing chromosome 18 being the least attached chromosome out of those analysed (Figure 3); (2) Gene loci with significant differences between two gene loci and (Figure 4) (3) Telomeres, which are less strongly attached in quiescent cells compared to proliferating and senescent cells (Figure 5). We are able to differentiate between proliferating and non-proliferating cells via the presence of the proliferation marker Ki67 antigen which is an insoluble protein so remains with the residual nuclei or using the incorporation of nucleotides to highlight cells that have been through S-phase within a specific time period (Figure 2). This technique has also enabled us to analyze genome behavior in cells that are compromised in their nucleoskeletons i.e. laminopathy cells and here and in Bikkul et al., 2018 we reveal that the genome can be less tightly attached when compared to control cells and can be restored when treating with specific drugs that ameliorate the effect of the lamin A mutation in classical HGPS cells15. However, we show new data here for the atypical HGPS AGO8466 cells, lacking a lamin A mutation but containing an unusual form of the LINC complex protein SUN119 that chromosome 13 is less tightly attached in (Figure 6).
HALO-FISH is a unique method by enabling the study of genomic interactions with the nucleoskeleton in combination with indirect immunofluorescence to resolve proteins not removed from the extraction procedure. It has been demonstrated that the nucleoskeleton is modified in various diseases such as certain cancer types19 and the importance of some nucleoskeleton-associated proteins as diagnostic biomarkers24,25. Thus, this technique has an important role in examining the effect of the nucleoskeleton on chromatin organization/disorganization in disease15,24,25,27 and is not restricted to human cells, with chromosomal painting probes from other animals, the same DNA-halo protocol could be employed28.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to thank Prof Michael Bittner for the kind gift of chromosome arm painting probes. LG was supported by EU funded EURO-Laminopathies project and the Brunel Progeria Research Fund.
Materials
| Name | Company | Catalog Number | Comments |
| 10X PBS | Thermo Fisher Scientific | 10388739 | Used to create DNA halos |
| 5-bromo-2′-deoxy-uridine | Sigma-Aldrich | B5002-100MG | Labelled nucleotide |
| 5-Fluoro-2′-deoxyuridine | Sigma-Aldrich | F0503-100MG | Labelled nucleotide |
| Agar Technical | Thermo Fisher Scientific | 15562141 | DNA isolation of BAC clones |
| Agarose | Sigma-Aldrich | A939-50G | Check product size of DOP-PCR and nick translation |
| Atypical type 2 HGPS fibroblasts (AG08466) | Coriell Institute | AG08466 | Cell line |
| Bacto tryptone | Thermo Fisher Scientific | 16269751 | DNA isolation of BAC clones |
| Biotin-16-dUTP | Roche Diagnostics | 11093711103 | Labelled nucleotides |
| Chloramphenicol | Sigma-Aldrich | C0378-25G | DNA isolation of BAC clones |
| Classical Hutchinson-Gilford progeria syndrome (HGPS) fibroblasts (AG06297) | Coriell Institute | AG0297 | Cell line |
| Coplin jar | Thermo Fisher Scientific | 12608596 | Holds 5 slides or 8 slides back to back |
| Cot-1 DNA | Thermo Fisher Scientific | 15279011 | Block nonspecific hybridization in HALO FISH |
| DEPC-treated water | Sigma-Aldrich | 693520-1L | DNA isolation of BAC clones |
| Dextran sulphate | Sigma-Aldrich | S4030 | Hybridisation mixture |
| Digitonin | Sigma-Aldrich | D141 | Component of extraction buffer |
| Digoxigenin-11-dUTP | Sigma-Aldrich | 11093088910 | Labelled nucleotides |
| Donkey anti-mouse Cy3 | Jackson Laboratory | 715-165-150 | Secondary antibody |
| EDTA | Sigma-Aldrich | E6758 | Component of extraction buffer |
| Ethanol | Component of extraction buffer | ||
| Ethanol | Sigma-Aldrich | 443611 | Probe precipitation and HALO FISH |
| Fetal bovine system | Thermo Fisher Scientific | 26140079 | Cell culture serum |
| Formamide | Thermo Fisher Scientific | 10523525 | 2D FISH of DNA halos |
| Glass wool | Sigma-Aldrich | 18421 | Spin column |
| Herring sperm | Sigma-Aldrich | D7290 | Probe precipitation |
| HXP™ Lamp (metal halide microscope lamp) | OSRAM | HXP-R120W45C VIS | Image capture of DNA halos |
| Hydrochloric acid | Thermo Fisher Scientific | 10313680 | Cleaning microscope slides |
| Isopropanol | Sigma-Aldrich | I9516-25ML | DNA isolation of BAC clones |
| KAPA HiFi PCR Kit | KAPA Biosystems | KK2103 | PCR Kit |
| Leica DM4000 fluorescent microscope with DFC365 FX camera and LAS AF (Version: 4.5.0) image acquisition software. | Leica Microsystems | Image capture of DNA halos | |
| Luria-Bertani agar | Thermo Fisher Scientific | 13274843 | DNA isolation of BAC clones |
| Magnesium chloride | Sigma-Aldrich | M8266 | Component of CSK buffer |
| Methanol | Thermo Fisher Scientific | 10284580 | Cleaning and sterilizing microscope slides |
| Mouse anti-BrdU antibody | BD Pharmingen | B2531-100UL | BrdU visualisation |
| Newborn calf serum | Thermo Fisher Scientific | 16010159 | Cell culture serum and blocking reagent |
| Nick translation kit | Invitrogen | ||
| PCR grade water | Sigma-Aldrich | 693520-1L | PCR and DNA isolation of BAC clones |
| PCR Primers | Sigma-Aldrich | ||
| PIPES | Sigma-Aldrich | P1851 | Component of CSK and extraction buffers |
| Potassium acetate | Sigma-Aldrich | P1190-100G | DNA isolation of BAC clones |
| QuadriPERM® 4 X 12 | SARSTEDT | 94.6077.307 | Square cell culture dish, polysterene with four compartments. This has hydrophobic surface, is sterile, non-pyrogenic/endotoxin-fee and non-cytotoxic. |
| Rabbit Anti-Ki67 antibody | Sigma-Aldrich | ZRB1007-25UL | Proliferation marker |
| Rnase A | Sigma-Aldrich | R6513 | DNA isolation of BAC clones |
| Rubber cement | Halford's | 101836 | 2D FISH of DNA halos |
| Sephadex G-50 | Sigma-Aldrich | S6022-25G | Spin column |
| Sodium acetate | Sigma-Aldrich | S2889 | Probe precipitation |
| Sodium chloride | Sigma-Aldrich | S5886 | Component of CSK, extraction and SSC buffers |
| Sodium citrate | Sigma-Aldrich | C8532 | Component of SSC buffer |
| Sodium dodecyl sulphate | L3771-100G | DNA isolation of BAC clones | |
| Sodium hydroxide | Sigma-Aldrich | S8045-500G | DNA isolation of BAC clones |
| Spermidine | Sigma-Aldrich | S2626 | Component of extraction buffer |
| Spermine | Sigma-Aldrich | S4264 | Component of extraction buffer |
| Streptavidin-Cy3 | Amersham Life Sciences Ltd, Scientific Laboratory Supplies | pa43001 | Probe antibody |
| Sucrose | Sigma-Aldrich | S0389 | Component of CSK buffer |
| Sucrose | Sigma-Aldrich | S0389 | CSK buffer+A66:D68 |
| SuperFrost™ microscope slides | Thermo Fisher Scientific | 12372098 | Microscope slides: 1 mm thickness, 76 mm length, 26 mm width. Uncoated. |
| Swine anti-rabbit TRITC | Dako | ||
| TELO-PNA FISH KIT | Agilent Dako | K532511-8 | Delineation of telomeres |
| Tris-HCl | Sigma-Aldrich | T3253-100G | Column buffer |
| Triton™ X-100 | Sigma-Aldrich | T9284 | Component of CSK buffer |
| Tryptone | Thermo Fisher Scientific | 10158962 | DNA isolation of BAC clones |
| Tween-20 | Sigma-Aldrich | P9416- 100ML | Detergent |
| Vectashield mountant containing DAPI | Vector Laboratories | H-1200 | 2D FISH of DNA halos |
| Whole human chromosome probes | Calbiochem | 2D FISH of DNA halos | |
| Yeast extract | Thermo Fisher Scientific | 10108202 | DNA isolation of BAC clones |
References
- Berezney, R., Coffey, D. S. Identification of a nuclear protein matrix. Biochemical Biophysical Research Communications. 60 (4), 1410-1417 (1974).
- Haaf, T., Ward, D. C. High resolution ordering of YAC contigs using extended chromatin and chromosomes. Human Molecular Genetics. 3 (4), 629-633 (1994).
- Parra, I., Windle, B. High resolution visual mapping of stretched DNA by fluorescent hybridization. Nature Genetics. 5 (1), 17-21 (1993).
- Senger, G., et al. Released chromatin: linearized DNA for high resolution fluorescence in situ hybridization. Human Molecular Genetics. 3 (8), 1275-1280 (1994).
- Florijn, R. J., et al. High-resolution DNA Fiber-FISH for genomic DNA mapping and colour bar-coding of large genes. Human Molecular Genetics. 4 (5), 831-836 (1995).
- Elcock, L. S., Bridger, J. M. Fluorescence in situ hybridization on DNA halo preparations and extended chromatin fibres. Methods Molecular Biology. 659, 21-31 (2010).
- Heiskanen, M., et al. Visual mapping by fiber-FISH. Genomics. 30 (1), 31-36 (1995).
- Bensimon, A., et al. Alignment and sensitive detection of DNA by a moving interface. Science. 265 (5181), 2096-2098 (1994).
- Michalet, X., et al. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science. 277 (5331), 1518-1523 (1997).
- Wilson, R. H., Coverley, D. Relationship between DNA replication and the nuclear matrix. Genes Cells. 18 (1), 17-31 (2013).
- Wilson, R. H. C., Coverley, D. Transformation-induced changes in the DNA-nuclear matrix interface, revealed by high-throughput analysis of DNA halos. Science Reports. 7 (1), 6475 (2017).
- Iarovaia, O. V., Akopov, S. B., Nikolaev, L. G., Sverdlov, E. D., Razin, S. V. Induction of transcription within chromosomal DNA loops flanked by MAR elements causes an association of loop DNA with the nuclear matrix. Nucleic Acids Research. 33 (13), 4157-4163 (2005).
- Tandara, M., et al. Sperm DNA integrity testing: big halo is a good predictor of embryo quality and pregnancy after conventional IVF. Andrology. 2 (5), 678-686 (2014).
- Repping, S., et al. The use of spermHALO-FISH to determine DAZ gene copy number. Mol Human Reproduction. 9 (4), 183-188 (2003).
- Bikkul, M. U., et al. Farnesyltransferase inhibitor and rapamycin correct aberrant genome organisation and decrease DNA damage respectively, in Hutchinson-Gilford progeria syndrome fibroblasts. Biogerontology. 19 (6), 579-602 (2018).
- Telenius, H., et al. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 13 (3), 718-725 (1992).
- Bridger, J. M., et al. Association of pKi-67 with satellite DNA of the human genome in early G1 cells. Chromosome Research. 6, 13-24 (1998).
- Sales Gil, R., Vagnarelli, P. Ki-67: More Hidden behind a 'Classic Proliferation Marker'. Trends in Biochemical Sciences. 43 (10), 747-748 (2018).
- Bikkul, M. U., et al. Telomere elongation through hTERT immortalization leads to chromosome repositioning in control cells and genomic instability in Hutchinson-Gilford progeria syndrome fibroblasts, expressing a novel SUN1 isoform. Genes Chromosomes Cancer. 58 (6), 341-356 (2019).
- Jackson, D. A., Cook, P. R. Visualization of a filamentous nucleoskeleton with a 23 nm axial repeat. EMBO Journal. 7, 3667-3677 (1988).
- Albrethsen, J., et al. Unravelling the nuclear matrix proteome. Journal of Proteomics. 72, 71-81 (2009).
- Mika, S., Rost, B. NMPdb: Database of nuclear matrix proteins. Nucleic Acids Research. 33, 160-163 (2005).
- Gavrilov, A. A., et al. of the nuclear matrix-bound chromatin hubs by a new M3C experimental procedure. Nucleic Acids Research. 38, 8051-8060 (2010).
- Sjakste, N., et al. Role of the nuclear matrix proteins in malignant transformation and cancer diagnosis. Experimental Oncology. 26 (3), 170-178 (2004).
- Leman, E. S., Getzenberg, R. H. Nuclear structure as a source of cancer specific biomarkers. Journal of Cellular Biochemistry. 104 (6), 1988-1993 (2008).
- Volpi, E. V., Bridger, J. M. FISH glossary: an overview of the fluorescence in situ hybridization technique. Biotechniques. 45 (4), 385-386 (2008).
- Bridger, J. M., Foster, H. A. Senescence and the Genome. Human Interphase Chromosomes. , (2021).
- Foster, H. A., Griffin, D. K., Bridger, J. M. Interphase chromosome positioning in in vitro porcine cells and ex vivo porcine tissues. BMC Cell Biology. 13 (1), 30 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved