
Single Myofiber Culture Assay for the Assessment of Adult Muscle Stem Cell Functionality Ex Vivo
In This Article
Summary
In this protocol an in vitro culturing and functional analysis method for muscle stem cells is described, which preserves most of their interactions with their endogenous niche.
Abstract
Adult skeletal muscle tissue harbors a stem cell population that is indispensable for its ability to regenerate. Upon muscle damage, muscle stem cells leave their quiescent state and activate the myogenic program ultimately leading to the repair of damaged tissue concomitant with the replenishment of the muscle stem cell pool. Various factors influence muscle stem cell activity, among them intrinsic stimuli but also signals from the direct muscle stem cell environment, the stem cell niche. The isolation and culture of single myofibers with their associated muscle stem cells preserves most of the interaction of the stem cell with its niche and is, therefore, the closest possibility to study muscle stem cell functionality ex vivo. Here, a protocol for the isolation, culture, siRNA transfection and immunostaining of muscle stem cells on their respective myofibers from mouse EDL (extensor digitorum longus) muscles is provided. The experimental conditions outlined here allow the study and manipulation of muscle stem cells ex vivo including investigation of myogenic activity without the inherent need for in vivo animal experiments.
Introduction
Skeletal muscle in the adult is a postmitotic tissue mainly composed of multinucleated myofibers, which are the effector cells for voluntary movements. It has a remarkable ability to regenerate, a process that resembles embryonic myogenesis and undergoes impairments in age and disease1. This striking regenerative capacity of skeletal muscle depends on muscle stem cells (MuSCs), which are also termed satellite cells due to their location between the sarcolemma and the basal lamina of myofibers2,3. Under resting conditions MuSCs are quiescent and characterized by the expression of the transcription factor Pax7 and quiescence markers such as Sprouty14,5,6,7,8. Upon activation, e.g., after injury, MuSCs leave the quiescent state and upregulate the myogenic regulatory factor MyoD9. The Pax7/MyoD double positive MuSCs proliferate and differentiate thereby generating myogenic precursor cells, which are also often referred to as myoblasts. Those myoblasts then further differentiate into elongated myocytes, a process coinciding with molecular and morphological changes, e.g., loss of Pax7 and upregulation of Myogenin expression10. Myocytes then eventually fuse to each other or to existing myofibers thereby repairing the damaged tissue. Importantly, a small fraction of muscle stem cells reverts MyoD upregulation and is able to self-renew11. The status of MuSC differentiation and myogenic progression can be readily observed by investigation of myogenic markers such as Pax7, MyoD and Myogenin10.
The culture of single myofibers with their adjacent MuSCs is an excellent method to investigate MuSC functionality in an ex vivo setting since MuSCs are remaining in their endogenous niche12,13. The behavior of MuSCs is regulated by intrinsic signals as well as extrinsic signals provided by the niche, a specialized anatomical location comprising components of the extracellular matrix (ECM) surrounding the MuSCs and the myofiber itself. For instance, one of the extrinsic regulators of MuSC quiescence is Notch signaling. Here, signaling cues are received by MuSCs from both the myofiber and the ECM14,15,16. Moreover, the MuSC niche is important to control the division axis of MuSCs thus regulating the cell fate of MuSC daughter cells17,18. Reasonably, parameters like asymmetric MuSC divisions, myogenic progression and self-renewal can be uniquely assessed in this experimental setup. For instance, a multicellular cluster can form arising from one MuSC after a 72 h culture period, which can be investigated for the occurrence and percentage of distinct myogenic populations such as self-renewing, proliferating and further differentiated MuSCs8,19,20,21. The differentiation state of MuSCs can be determined by investigation of the expression/co-expression of Pax7, MyoD and Myogenin. After 72 h of culture the cells in a cluster can be discriminated by the following parameters: Pax7 only cells are self-renewing MuSCs, while Pax7/MyoD double positive cells are proliferating/activated MuSCs and further differentiated myogenic cells are Myogenin positive22. Furthermore, MuSC numbers or re-entry into the cell cycle/activation can be investigated in addition to myogenic progression, e.g., through immunofluorescence-based analyses based on the parameters described above.
Here, the unique features of the myofiber isolation and culture protocol, e.g., the preservation of the interaction of the MuSC with its niche, are described. Mouse whole EDL (extensor digitorum longus) muscles are carefully dissected, digested by collagenase, and physically triturated to obtain single myofibers with their associated MuSCs for further culture. Moreover, the protocol delineates the steps to transfect MuSCs with siRNA for functional analyses of candidate genes and consecutive immunofluorescence-based analyses without the necessity of transgenic animals.
Protocol
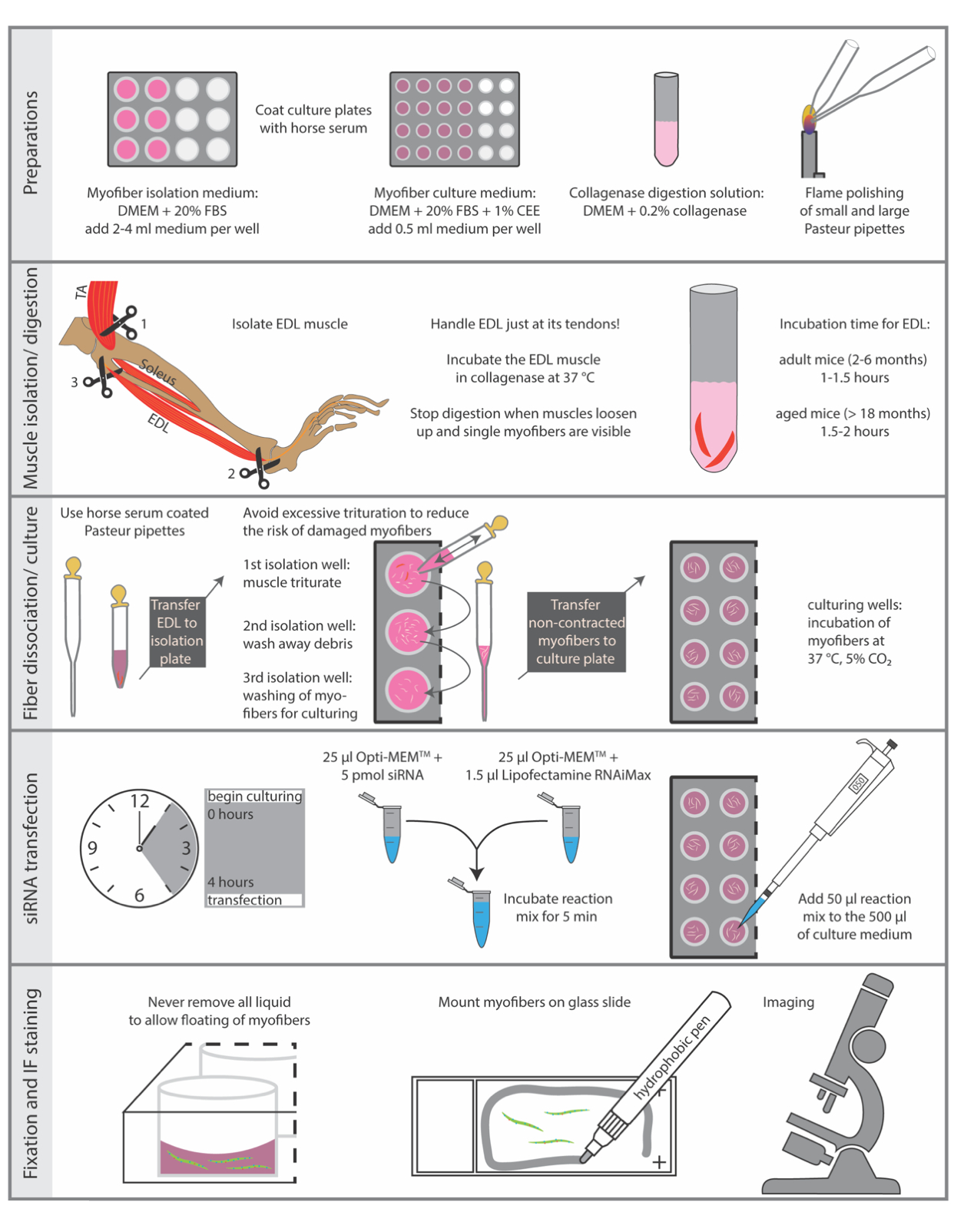
Sacrificing of animals must be performed in accordance with the national regulations for animal experimentation. The protocol described here was performed in accordance with the guidelines of the Leibniz Institute on Aging - Fritz Lipmann Institute and the European Union (EU) directive 2010/63/EU (license number for organ harvesting: O_JvM_18-20). Essential steps of the protocol are summarized in Figure 1.
1. Preparation of culture plates, media, and Pasteur pipettes
NOTE: All materials and equipment necessary to isolate and culture single myofibers need to be as sterile as possible. Therefore, it is recommended to isolate single myofibers under a semi-sterile dissection hood.
- Use sterile HS (horse serum) to coat tissue culture plates. Coating prevents the attachment of single myofibers to the plastic surface. For each mouse, 4 wells of a 12-well plate for the isolation and a dedicated number of wells of a 24-well plate for culturing are required. Incubate the wells of a 12-well plate with 1 mL and the wells of a 24-well plate with 0.5 mL of HS for 5 min at RT (room temperature), then remove the HS and let the plates dry for another 5 min.
NOTE: HS can be collected and re-used for coating purposes multiple times, if kept sterile. - Prepare myofiber isolation medium by supplementing DMEM (Dulbecco's modified Eagle's medium with 4.5 g/L glucose and sodium pyruvate) with 20% FBS (fetal bovine serum), filter through 0.22 µm filter. Add medium to the pre-coated isolation plates (12-well plate, 2-4 mL isolation medium per well) approximately 30 min before the isolation and equilibrate in a humidified 37 °C incubator with 5% CO2.
- Prepare myofiber culture medium by supplementing DMEM (Dulbecco's modified Eagle's medium with 4.5 g/L glucose and sodium pyruvate) with 20% FBS (fetal bovine serum) and 1% chicken embryo extract, filter through 0.22 µm filter. Add 0.5 mL medium to each well of the pre-coated culture plates (24-well plate) approximately 30 min before the isolation and equilibrate in a humidified 37 °C incubator with 5% CO2.
- Prepare collagenase digestion solution by dissolving 0.2% (w/v) collagenase type 1 (from Clostridium histolyticum) in DMEM (Dulbecco's modified Eagle's medium with 4.5 g/L glucose and sodium pyruvate), filter through 0.22 µm filter. For two EDL (extensor digitorum longus) muscles use 2.5 mL collagenase solution in a sterile 15 mL reaction tube. Additionally, pre-heat the solution ~10 min in a circulating water bath at 37 °C before starting the isolation.
- Prepare sterile Pasteur pipettes for trituration of the collagenase-digested muscles. Use a diamond pen to cut the Pasteur pipettes.
- For each mouse, use one large bore pipette with an opening of about 0.3 cm and a length of about 10-12 cm and a second glass pipette with a small opening of about 0.1 cm and a length of approximately 22 cm (Figure 2F).
- Smoothen the edges of both pipettes by heat polishing with the flame of a Bunsen burner. Hold the pipette tip for 5-10 s into the flame with gentle movement to allow equal heat distribution until the sharp glass edges smoothen.
- Immediately before use, coat both types of glass pipettes with sterile HS by filling the whole pipette with ~ 2 mL of HS for 5 min, thereafter, eject the HS and let the pipettes dry for 5 min at RT.
2. EDL muscle isolation and collagenase digestion
- Spray all equipment with 70% ethanol to avoid contamination.
- Sacrifice the mouse in accordance with the national regulations for animal experimentation.
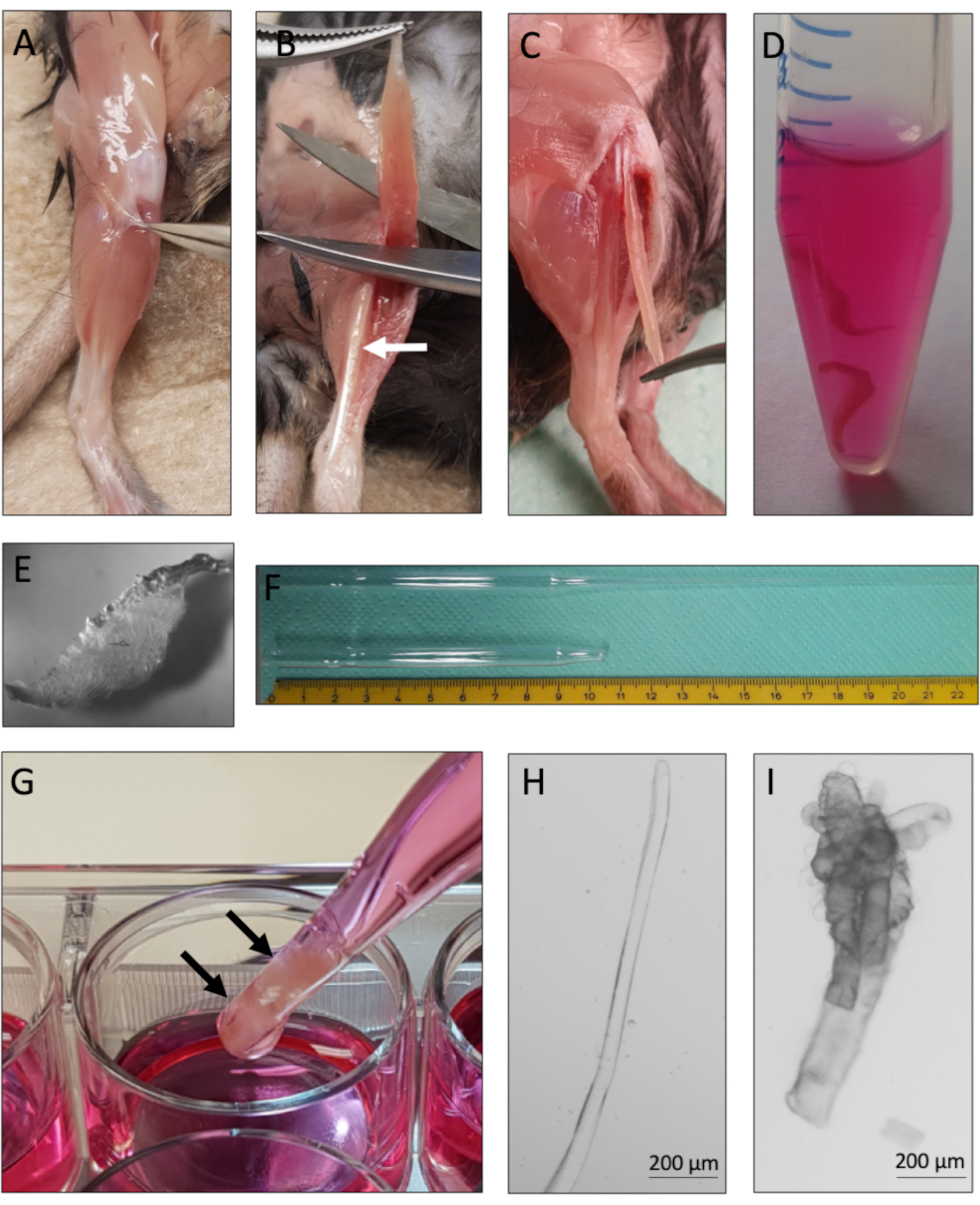
- Spray the hindlimbs of the mouse with 70% ethanol. Use hardened fine curved scissors (24 mm cutting edge) and fine forceps (Dumont 7, curved or straight) to remove the skin and to expose the underlying muscles. Avoid any contact of the underlying muscles with fur (increases the risk of contamination).
- Remove the surrounding fascia with fine curved forceps without damaging the underlying muscles (Figure 2A). Close the forceps to avoid bending.
- Use curved forceps to expose the distal tendons of the TA (tibialis anterior) and EDL muscle. To remove the TA, grab the distal TA tendon with forceps and cut with fine Vannas spring scissors (5 mm cutting edge, 0.35 mm tip diameter). While holding the TA at the tendon, pull it towards the knee and cut the muscle close to the knee (Figure 2B), the EDL muscle is now exposed.
NOTE: Make sure that only the TA tendon is grabbed in this step, otherwise the underlying EDL might get damaged. When cutting off the TA muscle make sure that the tendons at the knee can be seen easily afterwards. - Lift the distal EDL tendon with fine curved forceps and cut with fine Vannas spring scissors (Figure 2C). Expose the proximal EDL tendon by carefully pulling the EDL towards the knee. Cut the proximal tendon with fine Vannas spring scissors. Transfer the EDL muscle to the 37 °C preheated 2.5 mL of collagenase digestion solution in the 15 mL reaction tube from step 1.4. (Figure 2D).
NOTE: Make a small incision at the outer knee connective tissue to fully expose the proximal EDL tendon. Make sure to only grab the tendons and not to stretch the EDL too much. - Repeat steps 2.3. to 2.6. with the second EDL. Add both EDL muscles to the same 15 mL reaction tube filled with 2.5 mL collagenase digestion solution.
- Incubate the EDL muscles in the reaction tube at 37 °C in a circulating water bath.
NOTE: Incubation time depends on several factors, such as collagenase activity, age of the mouse and amount of fibrotic tissue. The typical incubation time for EDL muscles of adult mice (2-6 months of age) is 1-1.5 h and for aged mice (18 months of age) 1.5-2 h. - To avoid excessive digestion of the muscles, check the muscles during the digestion time. Stop the digestion when muscles loosen up and single myofibers are visible (Figure 2E). Transfer muscles carefully with the large bore pipette to the first well of the prewarmed 12-well plate containing myofiber isolation medium (Figure 2G).
3. Myofiber dissociation and culture
- For the following steps use a stereo binocular microscope, preferentially equipped with a heating plate (37 °C). Use the large bore pipette to flush the muscles with warm isolation medium until single myofibers are released. Dissociate the muscles with the large bore pipette until the desired number of myofibers are floating freely in the solution.
NOTE: Avoid excessive trituration to reduce the risk of damaged myofibers. Using a heating plate for myofiber dissociation is strongly advised since temperature drops during the isolation process, which will result in myofiber death. - Transfer non-contracted myofibers (Figure 2H) with the HS coated small bore glass pipette to the second well filled with isolation medium to wash away debris and contracted (damaged) myofibers (Figure 2I).
NOTE: To avoid excessive movement of isolated myofibers, they can be transferred to the second well and then the trituration process can be continued. - Transfer non-contracted myofibers to the next (3rd) well filled with isolation medium to wash again.
- Use the small-bore glass pipette, coated with HS, to transfer approximately 50-100 non-contracted myofibers to the 24-well plate containing myofiber culture medium.
- Incubate myofibers at 37 °C, 5% CO2 for dedicated time (96 h maximum are recommended).
NOTE: MuSCs divide once either planar or apical-basal after 42 h of culture. Additionally, after 72 h of culture MuSCs form multicellular clusters consisting of self-renewing, proliferating or committed (differentiated) MuSCs.
4. siRNA transfection
- 4 h after myofiber isolation, transfect myofiber associated MuSCs (50-100 non-contracted myofibers in one well of the 24-well plate filled with 500 µL culture medium) with lipid based transfection reagent, e.g. RNAiMAX according to the manufacturer's protocol with a final concentration of 5 pmol siRNA. Therefore, add 25 µL of Opti-MEM with the respective volume of siRNA to 25 µL Opti-MEM containing 1.5 µL of the transfection reagent. Incubate the reaction mix for 5 min and add it then to the 500 µL of culture medium.
NOTE: A second transfection after 24 h or 48 h is recommended for longer culture periods, e.g., more than 48 h. A change of medium after transfection is not necessary.
5. Fixation and IF staining
- For immunostaining use a stereo binocular microscope. All volumes in the following steps are adjusted to one well of a 24-well plate. Perform all following steps using a HS-coated small-bore pipette.
- Carefully discard the myofiber culture medium while leaving some solution in the well (approximately 100 µL per 24-well). Do this for all further steps unless stated otherwise to allow floating of the myofibers. Add 500 µL of 2% PFA to fix the myofibers with their adjacent MuSCs, incubate for 5 min at room temperature (RT).
- Remove the supernatant carefully and wash the myofibers three times with PBS (pH 7.4, 500 µL for 5 min at RT each).
- Add 500 µL permeabilization solution (0.1% Triton X-100, 0.1 M Glycine in PBS, pH 7.4), incubate for 10 min at RT.
- Remove the permeabilization solution and add 500 µL blocking solution (5% HS in PBS, pH 7.4) for 1 h at RT.
NOTE: Check the recommended blocking solution for primary antibodies to avoid unspecific binding. - Remove the blocking solution and add 300 µL of primary antibody dilution (e.g., anti-Pax7 (PAX7, DSHB, undiluted), anti-MyoD (clone G-1, Santa Cruz, 1:200)) per well. Incubate at 4 °C overnight.
- Wash three times with 500 µL of PBS per well (5 min at RT).
- Remove PBS and add 300 µL of secondary antibody dilution (e.g., anti-mouse-IgG1-546 and anti-mouse-IgG2b-488 1:1000) per well. Incubate 1 h at RT protected from light. For the following steps, light reduced conditions are recommended.
- Wash two times with 500 µL of PBS per well (5 min at RT).
- Perform DAPI staining by using 500 µL of the solution per well (final DAPI concentration 10 µg/mL) for 5 min at RT.
- Wash two times with 500 µL of PBS per well (5 min at RT).
- Use a hydrophobic pen to draw a circle on a microscopic glass slide to create a water repellant barrier that will prevent spilling of liquid containing myofibers. Transfer the myofibers in the smallest volume possible to the microscopic glass slide and disperse them on the glass slide.
NOTE: Avoid physical pulling of the myofibers over the microscopic glass slide since this might cause friction resulting in detachment of clusters from myofibers. - Remove the residual liquid with the small-bore Pasteur pipette or a 200 µL pipette.
- Use two drops of aqueous mounting medium and cover the myofibers with a coverslip. Let the slides dry for the time recommended by the manufacturer. Store the slides at 4 °C in the dark.
Representative Results
This protocol provides instructions for successful derivation and culture of single myofibers with their associated MuSCs from murine EDL muscles. Essential steps of the protocol are summarized in Figure 1. Careful tendon-to-tendon dissection of the EDL muscles (Figure 2A-C) is critical for a high yield of viable myofibers. Muscle dissociation is achieved first by collagenase digestion (Figure 2D) followed by physical trituration (Figure 2G). Intact myofibers (Figure 2H) are cultured, whereas hypercontracted and dead myofibers (Figure 2I) should be excluded from culture and analysis.
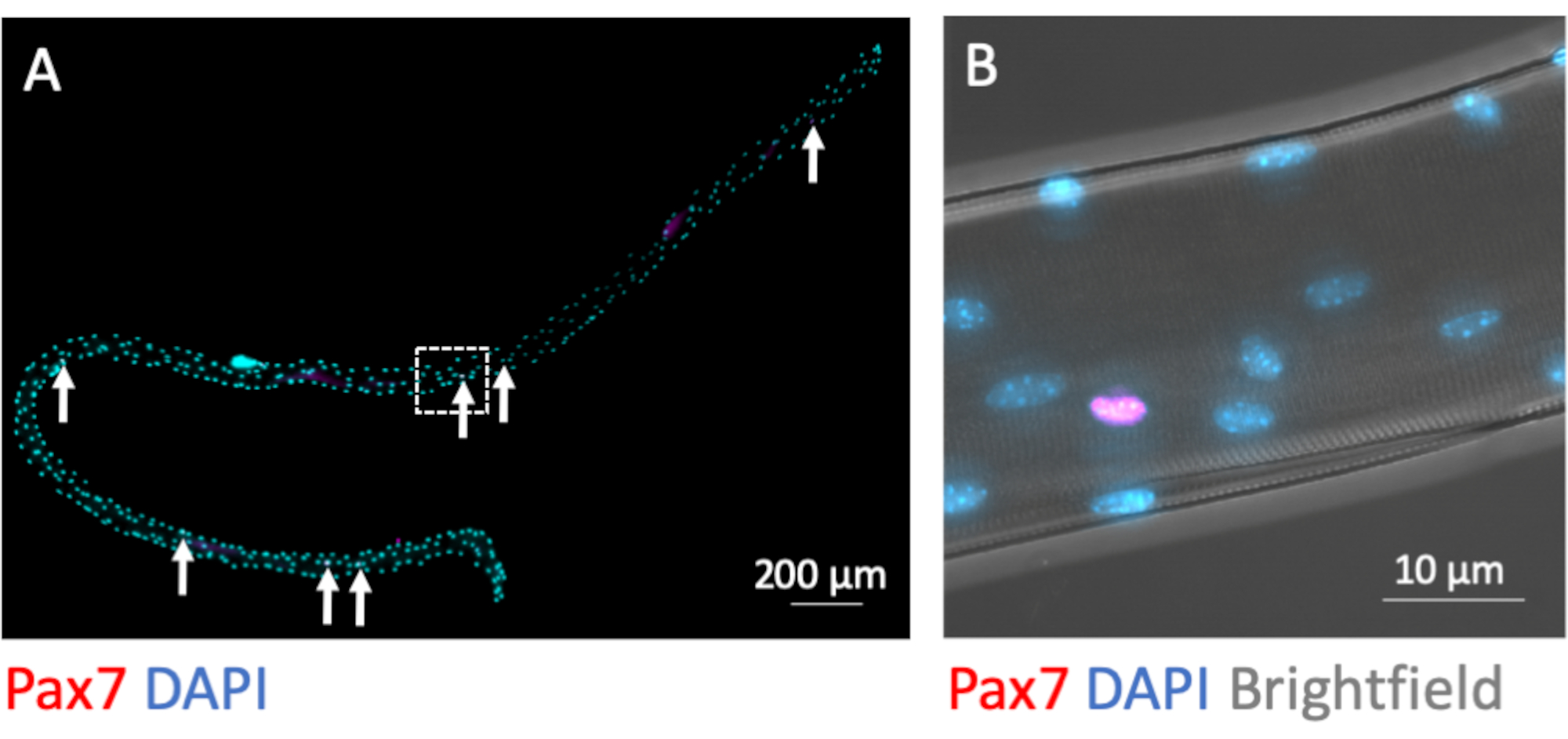
MuSCs remain myofiber-associated during the isolation process. Immunofluorescence staining for the transcription factor Pax7 identifies and distinguishes 7-9 MuSC nuclei from the plethora of myonuclei per myofiber when fixed directly after completion of the isolation process (Figure 3A). Figure 3B shows a magnified area from Figure 3A with the additional brightfield channel, which exposes the subcellular myofibrillary structure of the myotube and demonstrates Pax7 immunofluorescence signal in the nucleus of a MuSC.
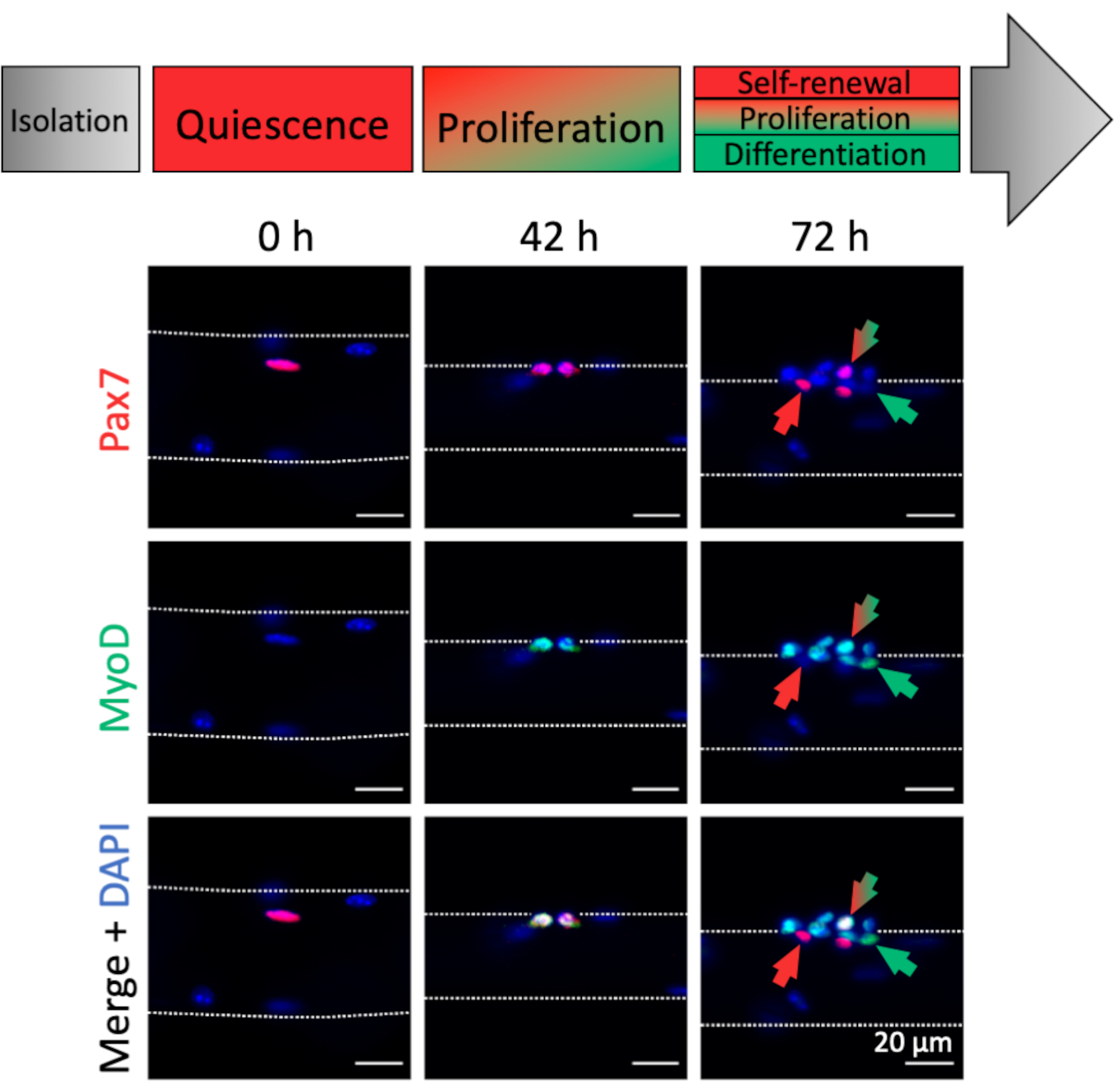
Activation and myogenic progression of myofiber associated MuSCs can be analyzed by myogenic marker expression. Associated with freshly isolated myofibers (0 h), mostly quiescent MuSCs can be found, which are characterized by Pax7 expression and absence of MyoD expression, thereby resembling an in vivo homeostatic condition (Figure 4, 0 h). Due to the dissection/dissociation procedure and the myofiber culture media composition, the MuSCs rapidly activate and upregulate the transcription factor MyoD to facilitate proliferation as can be observed at 42 hours, when MuSCs have undergone their first division (Figure 4, 42 h). After 72 hours of culture, MuSCs form clusters of progenies with different myogenic fates that is paralleled by the expression patterns of different myogenic markers (Figure 4, 72 h). Pax7+ only cells resist differentiation and become self-renewing stem cells. Pax7+ and MyoD+ double positive cells are proliferative, whereas MyoD+ only cells have further progressed along the myogenic lineage and will differentiate.
The myofiber culture system allows for efficient interference of MuSC activity by various interventions, which one of them is siRNA transfection as described in detail in this protocol. To monitor the transfection efficiency of myofiber associated MuSCs a fluorescently labeled non-targeting siRNA was transfected. Pax7 positive MuSCs accumulated cytoplasmic siRNA in a granule-like fashion, indicating efficient uptake (Figure 5A). No fluorescent granules were observed in the cytoplasm of myofibers suggesting a natural uptake barrier in myofibers at 4 h after isolation and that siRNA transfection specifically targets MuSCs. Quantification of positively transfected Pax7+ cells per myofiber revealed that more than half of all MuSCs had taken up visible amounts of fluorescently labelled siRNA just before completion of the first round of division at 24 hours. The number of transfected Pax7+ cells increased further up to 74% after 30 hours (Figure 5B). Furthermore, there was no difference in the number of Pax7+ cells per myofiber of transfected or non-transfected conditions at both timepoints, demonstrating no adverse effects on the stem cell numbers due to the transfection procedure (Figure 5C).
In summary, the protocol provides a detailed description of the isolation and culture of EDL single myofibers with their adjacent muscle stem cells. It enables the study of myofiber-associated muscle stem cell activity ex vivo, e.g., by immunofluorescence-based analyses. Manipulation of muscle stem cells by siRNA transfection is efficient and provides a methodological basis for functional analyses.

Figure 1: Schematic summary of essential steps. The essential steps of the isolation and immunostaining procedure are summarized in this figure. Abbreviations: DMEM, Dulbecco's Modified Eagle's Medium; FBS, Fetal Bovine Serum; CEE, Chicken Embryo Extract; TA, tibialis anterior; EDL, extensor digitorum longus Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Mouse EDL dissection and single myofiber isolation. (A) The skin of the mouse hindlimb is removed and the muscle surrounding fascia is pulled away to expose the TA (tibialis anterior) muscle. (B) The TA muscle is removed to expose the EDL (extensor digitorum longus) muscle, marked by the white arrow. (C) The EDL muscle is dissected by cutting its tendons. (D) Two EDL muscles are collagenase digested. (E) Appearance of collagenase digested muscle with visible single myofibers loosening up from the tissue core. (F) Large and small-bore Pasteur pipette with scale. (G) The EDL muscles (marked by black arrows) are physically triturated using a large bore Pasteur pipette. (H) Single intact myofibers are thin and shiny and can be individually collected for culture and analysis. (I) Hypercontracted and dead myofibers are unsuitable for culture and analysis. The microscopic images of 2H and 2I were captured with a microscope using a N-Achroplan 5x objective. Scale bars are 200 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Microscopic image of a whole single myofiber with its associated MuSCs. Single myofibers from EDL of young C57BL/6 mice were prepared and PFA-fixed after isolation (0 h). (A) Pax7 and DAPI immunofluorescence staining identifies myofiber associated muscle stem cells (MuSCs, marked by arrows). The microscopic image was captured using the tiles and z-stack function of a microscope equipped with a Plan-Apochromat 40x oil objective. Scale bar is 200 µm. (B) Magnification from (A) showing the myonuclei, one Pax7 positive nucleus and the bright-dark pattern from myofibrillary structures in brightfield. Scale bar is 10 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Immunofluorescence images of MuSC activation and myogenic progression during single myofiber culture. Muscle stem cells (MuSCs) of freshly isolated myofibers (0 h) represent a homeostatic state close to in vivo quiescence, characterized by expression of Pax7 and lack of MyoD expression. During single myofiber culture most MuSCs upregulate MyoD and re-enter the cell cycle to divide and proliferate (42 h). After 72 h of culture the MuSC fate can be discriminated based on myogenic marker expression. Cells with Pax7 only expression will self-renew (red arrow) whereas Pax7 and MyoD double positive cells (red/green arrow) continue to proliferate. MyoD only positive cells (green arrow) have committed to myogenic differentiation. Microscopic images were captured using a microscope with a Plan-Apochromat 20x objective (0 h, 42 h) or a LD Plan-Neofluar 40x objective (72 h). Scale bars are 20 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Fluorescent siRNA transfection of MuSCs and uptake analysis. EDL single myofibers were prepared and transfected with fluorescently labeled siRNA (siGLO) following the steps provided by this protocol. (A) Cytoplasmatic accumulation of siGLO granules specifically in a Pax7 positive muscle stem cell (MuSC) on a single myofiber at 30 h of culture. The microscopic image was captured using the z-stack and apotome function of a microscope with a Plan-Apochromat 100x oil objective. Scale bars are 5 µm. (B) Quantification of siRNA uptake by Pax7 positive muscle stem cells at 24 or 30 h of culture. (C) Number of Pax7 positive cells per single myofiber comparing non-transfected and transfected conditions at 24 or 30 hours of culture. Data are shown as mean with standard deviation of n = 4 mice. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Here, a method to functionally investigate the role of a specific gene in MuSCs using an in vitro approach is presented. Importantly, in the system described here the MuSCs are cultured in conditions, which resemble the in vivo situation as much as possible preserving most of the interactions of the MuSCs with their niche. This is accomplished by culturing isolated myofibers with their adjacent MuSCs under floating conditions and consecutive siRNA transfection. The procedures of myofiber isolation, siRNA transfection and investigation of MuSC populations during 72 h of culture through immunofluorescence analyses are described. Furthermore, it was demonstrated that around 74% of all MuSCs were transfected with a fluorescently labelled control siRNA after 30 h of culture.
Particular attention should be focused on the careful dissection of the EDL muscle since extensive stretching, pinching, or squeezing will lead to contraction and consecutive death of myofibers. Furthermore, it is important to investigate clusters of MuSCs from at least 20 different myofibers per replicate per condition. This is necessary since MuSC numbers and properties are varying due to the existence of MuSC subpopulations. When investigating the effect of a specific siRNA on MuSCs using the floating myofiber culture method a comparison of the condition with the targeting siRNA to the non-targeting control should be performed within the same mouse and muscle. This is recommended to avoid mouse-specific differences which might cover or amplify effects of the siRNA. The knockdown efficiency can be determined by immunofluorescence analyses with antibodies directed against the target gene using single myofibers with their adjacent MuSCs. If this is not an option, one can test the siRNA knockdown efficiency in primary myoblasts followed by quantitative RT-PCR or immunoblot analyses. The efficiency of the siRNA should be determined before analyzing the effect of the siRNA on MuSCs on single myofibers. The use of a smart pool consisting of 4 different siRNAs versus a single one increases the knockdown efficiency but also increases the risk of unspecific targeting. A non-targeting siRNA should be used as a control. To directly monitor the transfection efficiency, one can use a fluorescently labelled non-targeting siRNA as performed here. The time point for transfection with the siRNA is around 4 hours after isolation, a time point when the basal lamina surrounding the MuSCs is already permeable for the siRNA. If the effect of a specific siRNA on MuSCs after 72 or 96 h should be investigated, it is recommended to perform a second siRNA transfection after 24 h or 48 h to maintain a high knockdown efficiency.
The myofiber culture assay displays diverse advantages compared to the investigation of MuSCs with conventional cell culture methods. MuSCs stay attached to the myofibers during the whole isolation process, thereby preserving the crucial interaction of the MuSC with its niche19,23,24,25. The preserved interaction of MuSCs with the myofiber is a prerequisite for studying niche dependent effects on MuSC functionality, which cannot be recapitulated in conventional 2D myoblast cultures. For example, during aging MuSCs display impaired myogenic capacity resulting in a reduced efficiency to regenerate muscle tissue after damage20,26. This impairment is at least partially attributable to changes in the MuSC niche, in particular changes in the ECM composition27,28. The myofiber culture protocol allows the study and interference with these aberrant niche changes.
In contrast to the method described here, purification of MuSCs by immunolabeling and -sorting techniques like FACS (fluorescence activated cell sorting) or MACS (magnetic cell sorting) involves the removal of the MuSCs from their niche. Interestingly, 2D cultures of isolated MuSCs from aged muscles loose their extrinsic cues and behave similar to MuSCs isolated from young muscles thereby not recapitulating the in vivo situation appropriately29. Furthermore, the complete dissociation of the muscle tissue and labeling of MuSCs with surface markers results in transcriptomic changes and activation of the cells30,31,32. Another advantage of the myofiber culture system is the possibility to interfere with MuSC functionality at various levels. Manipulation of MuSCs on cultured myofibers can be effectively achieved by siRNA-mediated gene knockdown as described here in detail. Likewise, the application of chemical compounds or the delivery of recombinant proteins is very effective to interfere with stem cell pathways20,28. Furthermore, retro- or lentiviral expression vectors permit the introduction of exogenous genes, i.e., constitutive active mutants33. Additionally, the influence of extrinsic factors on MuSC functionality can be explored in the system described here, e.g., the culture conditions can be supplemented with supernatant from different physiological or pathological sources to model different states like cancer cachexia34,35.
One limitation of the method described here is the fact, that the single myofiber culture system cannot completely recapitulate the impact of all systemic factors or influence of other cell types on MuSCs. Also, the time in which the myofibers can be kept viable in culture is limited and therefore the study of MuSC related processes focuses on early events like activation and myogenic commitment. Furthermore, the investigation of the MuSC interaction with other niche cells such as immune cells or fibro-adipogenic progenitor cells is not possible. To investigate systemic effects on MuSC functionality one can either perform muscle injury experiments followed by the analysis of muscle regeneration in vivo or perform transplantation experiments36,37.
Taken together, the myofiber isolation and culture protocol provides great opportunity for genetic or mechanistic studies on adult MuSCs without the requirement of transgenic mouse models and can potentially reduce animal experimentation.
Disclosures
The authors declare no competing financial interests.
Acknowledgements
We thank Christine Poser and Christina Picker for excellent technical assistance and critical reading of the manuscript. This work was supported by a grant from the Deutsche Forschungsgemeinschaft to JvM (MA-3975/2-1), the Carl Zeiss foundation and the Deutsche Krebshilfe (DKH-JvM-861005).
Materials
| Name | Company | Catalog Number | Comments |
| Alexa Fluor 488 goat anti-rabbit IgG 2b | ThermoScientific | A-21141 | use 1:1000 for IF |
| Alexa Fluor 546 goat anti-mouse IgG1 | ThermoScientific | A-21123 | use 1:1000 for IF |
| chicken embryo extract | Seralab | CE-650-J | chicken embryo extract containing growth factors etc. |
| collagenase type 1 | Sigma | C0130 | |
| DMEM (Dulbecco’s modified Eagle’s medium with 4.5 g/l glucose and sodium pyruvate) | GibCo | 41966029 | cell culture medium |
| fetal bovine serum | Gibco | 10270-106 | fetal bovine serum |
| horse serum | Gibco | 26050-088 | |
| Lipofectamine RNAiMax | ThermoScientific | 13778150 | transfection reagent |
| MyoD antibody clone G-1 | Santa Cruz | sc-377460 | dilute 1:200 for IF |
| Pax7 antibody | DSHB | PAX7 | use undiluted |
| siGLO Red Transfection Indicator | horizon discovery | D-001630-02-05 | non targeting siRNA |
References
- Henze, H., Jung, M. J., Ahrens, H. E., Steiner, S., von Maltzahn, J. Skeletal muscle aging - Stem cells in the spotlight. Mechanisms of Ageing and Development. 189, 111283 (2020).
- Chang, N. C., Rudnicki, M. A. Satellite cells: the architects of skeletal muscle. Current Topics in Developmental Biology. 107, 161-181 (2014).
- Mauro, A. Satellite cell of skeletal muscle fibers. Journal of Biophysics and Biochemical Cytolology. 9, 493-495 (1961).
- Lepper, C., Partridge, T. A., Fan, C. M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 138 (17), 3639-3646 (2011).
- Murphy, M. M., Lawson, J. A., Mathew, S. J., Hutcheson, D. A., Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 138 (17), 3625-3637 (2011).
- Sambasivan, R., et al. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 138 (17), 3647-3656 (2011).
- Seale, P., et al. Pax7 is required for the specification of myogenic satellite cells. Cell. 102 (6), 777-786 (2000).
- Shea, K. L., et al. Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell. 6 (2), 117-129 (2010).
- Chal, J., Pourquie, O. Making muscle: skeletal myogenesis in vivo and in vitro. Development. 144 (12), 2104-2122 (2017).
- Schmidt, M., Schuler, S. C., Huttner, S. S., von Eyss, B., von Maltzahn, J. Adult stem cells at work: regenerating skeletal muscle. Cellular and Molecular Life Sciences. 76 (13), 2559-2570 (2019).
- Motohashi, N., Asakura, A. Muscle satellite cell heterogeneity and self-renewal. Frontiers in Cell and Developmental Biology. 2, 1 (2014).
- Huttner, S. S., et al. Isolation and culture of individual myofibers and their adjacent muscle stem cells from aged and adult skeletal muscle. Methods in Molecular Biology. 2045, 25-36 (2019).
- Pasut, A., Jones, A. E., Rudnicki, M. A. Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. Journal of Visualized Experiments. (73), e50074 (2013).
- Baghdadi, M. B., et al. Reciprocal signalling by Notch-Collagen V-CALCR retains muscle stem cells in their niche. Nature. 557 (7707), 714-718 (2018).
- Mourikis, P., et al. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells. 30 (2), 243-252 (2012).
- Pisconti, A., Cornelison, D. D., Olguin, H. C., Antwine, T. L., Olwin, B. B. Syndecan-3 and Notch cooperate in regulating adult myogenesis. Journal of Cell Biology. 190 (3), 427-441 (2010).
- Kuang, S., Kuroda, K., Le Grand, F., Rudnicki, M. A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 129 (5), 999-1010 (2007).
- Troy, A., et al. Coordination of satellite cell activation and self-renewal by Par-complex-dependent asymmetric activation of p38alpha/beta MAPK. Cell Stem Cell. 11 (4), 541-553 (2012).
- Chakkalakal, J. V., Jones, K. M., Basson, M. A., Brack, A. S. The aged niche disrupts muscle stem cell quiescence. Nature. 490 (7420), 355-360 (2012).
- Price, F. D., et al. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nature Medicine. 20 (10), 1174-1181 (2014).
- Bernet, J. D., et al. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nature Medicine. 20 (3), 265-271 (2014).
- Wang, Y. X., Dumont, N. A., Rudnicki, M. A. Muscle stem cells at a glance. Journal of Cell Science. 127, 4543-4548 (2014).
- Bentzinger, C. F., Wang, Y. X., Dumont, N. A., Rudnicki, M. A. Cellular dynamics in the muscle satellite cell niche. EMBO Reports. 14 (12), 1062-1072 (2013).
- Eliazer, S., et al. Wnt4 from the Niche Controls the Mechano-Properties and Quiescent State of Muscle Stem Cells. Cell Stem Cell. 25 (5), 654-665 (2019).
- Goel, A. J., Rieder, M. K., Arnold, H. H., Radice, G. L., Krauss, R. S. Niche cadherins control the quiescence-to-activation transition in muscle stem cells. Cell Reports. 21 (8), 2236-2250 (2017).
- Sousa-Victor, P., et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 506 (7488), 316-321 (2014).
- Lukjanenko, L., et al. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nature Medicine. 22 (8), 897-905 (2016).
- Lukjanenko, L., et al. Aging disrupts muscle stem cell function by impairing matricellular wisp1 secretion from fibro-adipogenic progenitors. Cell Stem Cell. 24 (3), 433-446 (2019).
- Alsharidah, M., et al. Primary human muscle precursor cells obtained from young and old donors produce similar proliferative, differentiation and senescent profiles in culture. Aging Cell. 12 (3), 333-344 (2013).
- Machado, L., et al. In situ fixation redefines quiescence and early activation of skeletal muscle stem cells. Cell Reports. 21 (7), 1982-1993 (2017).
- van Velthoven, C. T. J., de Morree, A., Egner, I. M., Brett, J. O., Rando, T. A. Transcriptional profiling of quiescent muscle stem cells in vivo. Cell Reports. 21 (7), 1994-2004 (2017).
- van Velthoven, C. T. J., Rando, T. A. Stem cell quiescence: Dynamism, restraint, and cellular idling. Cell Stem Cell. 24 (2), 213-225 (2019).
- Judson, R. N., et al. The Hippo pathway member Yap plays a key role in influencing fate decisions in muscle satellite cells. Journal of Cell Science. 125, 6009-6019 (2012).
- He, W. A., et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. Journal of Clinical Investigation. 123 (11), 4821-4835 (2013).
- Schmidt, M., Poser, C., von Maltzahn, J. Wnt7a counteracts cancer cachexia. Molecular Therapy Oncolytics. 16, 134-146 (2020).
- Feige, P., Rudnicki, M. A. Isolation of satellite cells and transplantation into mice for lineage tracing in muscle. Nature Protocols. 15 (3), 1082-1097 (2020).
- Garry, G. A., Antony, M. L., Garry, D. J. Cardiotoxin induced injury and skeletal muscle regeneration. Methods in Molecular Biology. 1460, 61-71 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved