A Microfluidics Approach for the Functional Investigation of Signaling Oscillations Governing Somitogenesis
In This Article
Summary
A protocol for the culture and manipulation of mouse embryonic tissue on a microfluidic chip is provided. By applying pulses of pathway modulators, this system can be used to externally control signaling oscillations for the functional investigation of mouse somitogenesis.
Abstract
Periodic segmentation of the presomitic mesoderm of a developing mouse embryo is controlled by a network of signaling pathways. Signaling oscillations and gradients are thought to control the timing and spacing of segment formation, respectively. While the involved signaling pathways have been studied extensively over the last decades, direct evidence for the function of signaling oscillations in controlling somitogenesis has been lacking. To enable the functional investigation of signaling dynamics, microfluidics is a previously established tool for the subtle modulation of these dynamics. With this microfluidics-based entrainment approach endogenous signaling oscillations are synchronized by pulses of pathway modulators. This enables modulation of, for instance, the oscillation period or the phase-relationship between two oscillating pathways. Furthermore, spatial gradients of pathway modulators can be established along the tissue to study how specific changes in the signaling gradients affect somitogenesis.
The present protocol is meant to help establish microfluidic approaches for the first-time users of microfluidics. The basic principles and equipment needed to set up a microfluidic system are described, and a chip design is provided, with which a mold for chip generation can conveniently be prepared using a 3D printer. Finally, how to culture primary mouse tissue on a microfluidic chip and how to entrain signaling oscillations to external pulses of pathway modulators are discussed.
This microfluidic system can also be adapted to harbor other in vivo and in vitro model systems such as gastruloids and organoids for functional investigation of signaling dynamics and morphogen gradients in other contexts.
Introduction
Development is controlled by intercellular communication via signaling pathways. There is only a limited number of signaling pathways that orchestrate the complex formation of tissues and proper cell differentiation in space and time. To regulate this multitude of processes, information can be encoded in the dynamics of a signaling pathway, the change of a pathway over time, such as the frequency or duration of a signal1,2.
During somitogenesis, somitic tissue is periodically segmented off from the presomitic mesoderm (PSM)3. The PSM is spatially organized by gradients of Wnt, Fibroblast Growth Factor (FGF), and Retinoic acid signaling. In anterior PSM at the determination front, where Wnt and FGF signals are low, cells are primed for differentiation into somites. Differentiation occurs when a wave of transcriptional activation reaches this determination front. Within the PSM, Wnt, FGF, and Notch signaling oscillate. Neighboring cells oscillate slightly out of phase, which results in waves of oscillatory transcriptional activation downstream of the Wnt, FGF, and Notch pathways traveling from posterior to anterior PSM. In mouse embryos, a transcriptional wave reaches the determination front approximately every 2 h and initiates somite formation. Studying somitogenesis by perturbing or activating signaling pathways can illustrate the importance of these pathways4,5,6,7,8,9. However, to be able to investigate the function of signaling dynamics in the control of cellular behavior, it is essential to subtly modulate signaling pathways instead of permanently activating or inhibiting them.
To temporally modulate signaling pathway activity within the segmenting mouse embryo, Sonnen et al. have developed a microfluidic system10. This system allows the tight control of fluid flows within microchannels of a chip that contains the biological sample11. To study the importance of signaling dynamics for proper segmentation of PSM, this microfluidics setup is utilized to modulate signaling dynamics of the mouse segmentation clock ex vivo. By sequentially pulsing pathway activators or inhibitors into the culture chamber, external control of the dynamics of Wnt, FGF, and Notch signaling is achieved10. For instance, it is possible to modify the period of individual pathways and the phase relationship between multiple oscillatory signaling pathways. Using concomitant real-time imaging of dynamic signaling reporters, the effect of entrainment on the pathways themselves, on differentiation and somite formation can be analyzed. Using this level of control over signaling dynamics, the importance of the phase relationship between Wnt- and Notch-signaling pathways during somitogenesis was highlighted10.
Personalized chip designs allow for a plethora of options for spatiotemporal perturbations within the local environment, e.g., stable gradient formation12,13,14,15, pulsatile activation/inhibition10,16,17,18 or localized perturbations19,20. Microfluidics can also enable a more reproducible read-out and higher throughput due to automation of experimental handling21,22,23. The present protocol is meant to bring microfluidics and entrainment of endogenous signaling oscillations within tissues to every standard life sciences lab. Even in the absence of sophisticated equipment for chip generation, such as clean room and equipment for soft-lithography, microfluidic chips can be manufactured and used to address biological questions. Molds can be designed using freely available computer-aided design (CAD) software. A mold for the generation of microfluidic chips, usually consisting of polydimethylsiloxane (PDMS), can be printed with a 3D printer, or be ordered from printing companies. This way, microfluidic chips can be produced within one day without the requirement of expensive equipment24. Here, a chip design is provided, with which a mold for the entrainment of the mouse segmentation clock in two-dimensional (2D) ex vivo cultures25 can be printed with a 3D printer.
On-chip cultures and precise perturbations, enabled by microfluidics, hold outstanding potential in unravelling the molecular mechanisms of how signaling pathways control multicellular behavior. Signaling dynamics and morphogen gradients are required for many processes in development. Previously, labs had cultured cells, tissues and whole organisms in microfluidic chips and protocols for spatiotemporal perturbation of primarily 2D cell culture are provided elsewhere12,26,27,28,29. Applying microfluidics to modulate local environments in multicellular systems opens new perspectives for high-throughput and precise spatiotemporal perturbations. The field of microfluidics has now reached a point that it has become a non-specialist, inexpensive, and easily applicable tool for developmental biologists.
Here, a protocol for the entrainment of the mouse segmentation clock to pulses of a Notch signaling inhibitor is provided. Such an experiment consists of the following steps: (1) generation of microfluidic chip, (2) preparation of tubing and coating of the chip, and (3) the microfluidic experiment itself (Figure 1A). Research involving vertebrate model systems requires prior ethical approval from the responsible committee.
Protocol
The research presented here has been approved by the EMBL ethics committee10 and the Dutch committee for animal research (dierenexperimentencommissie, DEC), and follows the Hubrecht guidelines for animal care.
Wear gloves while working with liquid PDMS. Cover surfaces and equipment. Remove any spills immediately, as cleaning becomes difficult once it's hardened.
1. Generation of the chip
NOTE: Microfluidic chips are generated by casting PDMS (Polydimethylsiloxane) in a mold. Molds can be designed using CAD software (e.g., uFlow or 3DµF30). A repository of free designs is provided by the MIT (Metafluidics.org). Molds are either printed with a 3D printer or can be generated by soft lithography using a photomask that contains the desired design (for further information see, e.g., Qin et al., 201031). Here, a design for a mold is provided that can be printed with a 3D printer for the on-chip culture of mouse embryo tissue (Figure 1B,C, Supplementary Files 1 and 2). To allow printing with lower resolution 3D printers, the design has been modified compared to the previously published one10. A mold without air bubble traps and one with air bubble traps is provided (Supplementary Files 1 and 2, respectively). As the procedure for printing is dependent on the printer available, the details for printing will not be described. However, one must make sure that all unpolymerized resin is entirely removed. It is often required to perform extra washing with solvents to remove unpolymerized resin from the small <200 µm holes within the microfluidic chamber. These holes will form PDMS pillars to trap the embryonic tissue within the chip during the experiment. PDMS is generally used for microfluidics, as it is cheap, biocompatible, and transparent, and has low autofluorescence. After curing, the PDMS chip is cut out of the mold and bonded onto a glass slide. Plasma treatment of both the glass and the PDMS chip activates the surfaces and allows the formation of covalent bonds, when brought into contact.
- Prepare the required amount of PDMS by mixing the monomer with the catalyst in a 9:1 ratio (w:w) to induce polymerization. Use disposable tools to mix, such as plastic cups and forks. Ensure that the mixing is properly achieved.
- Place the PDMS mixture in a desiccator and apply vacuum for approximately 30 min to remove air from the PDMS mixture. Due to the vacuum within the desiccator, air will be removed from the PDMS mixture.
NOTE: Cover the inside of the desiccator with tissues in case PDMS flows over. - Pour the PDMS mixture into the chip mold (a PDMS layer of approximately 3-5 mm is enough).
- Place the mold filled with PDMS back into the desiccator and apply vacuum to remove any remaining bubbles. Make sure that air is removed from all smaller structures of the mold.
- Cure the mold by placing it in a 65 °C (or lower depending on the mold material) oven overnight. Cut the chip out of the mold using a scalpel. Cut the hardened PDMS out of the mold with sufficient space (1-2 cm) around the design to allow for later bonding. Make sure to cut the PDMS completely to prevent breaking of the chip when taking it out. Lift the PDMS chip off carefully, first with the blunt end of the scalpel, and when possible, with hands.
- Punch inlet and outlet holes, starting from the inside of the microfluidic chamber using a 1 mm biopsy punch.
- Clean the PDMS chip briefly with compressed air and stick adhesive tape on both sides to remove any stuck bits of PDMS and dust and keep it clean until further use.
NOTE: The chip can be kept like this until further use. - Clean the glass slide with compressed air. Be careful that it does not break. Remove adhesive tape from the PDMS chip.
- Bond the chip to the glass slide using a plasma oven. The following instructions are optimized for air plasma.
NOTE: Consult the manual of the lab's plasma oven and optimize the procedure for the specific experiment.- Place the chip and glass slide into the plasma oven with the sides to be bonded facing up.
- Place the lid onto the plasma oven and hold it in place while applying vacuum and wait until vacuum has established.
- Turn the gas on by pressing the Gas button and wait for approximately 1 min (pressure should be at around 0.37 mbar).
- Generate plasma by pressing Generator (Power: 9.0, Time: 1 min).
- When finished, turn the pump and gas off; ventilate and open the door.
- Bond the chip to the glass by placing the activated surfaces onto each other and applying pressure evenly. Be careful that the glass does not break.
NOTE: A water test can be performed to confirm that plasma treatment has worked. Place a drop of water on the glass surface. If plasma treatment has worked, the water should spread out immediately instead of forming a droplet.
- Place the bonded chip in an oven at 65 °C for 5 min.
- Seal the exposed side of the chip with adhesive tape to prevent dust from entering the inlets.
NOTE: The chip can be kept until use. Openings should be closed with tape until then.
2. Preparing microfluidics experiment
NOTE: The following preparatory steps are necessary to perform the microfluidics experiment: First, when performing the microfluidics experiment, a fluid flow is created from the inlets to the outlets. Any holes in the chip will function as outlets due to the pressure build-up from the inlets. Therefore, all holes that are not used must be closed; for instance, holes that are used to load tissue onto the chip. If these are not closed, the tissue might flow out with the fluid. To close these holes, PDMS-filled tubing is used. The PDMS-filled tubing can be kept indefinitely and, therefore, will not need to be prepared for each microfluidics experiment separately but can be made in larger batches. Second, before the start of the microfluidic experiment, tubing, PDMS-filled tubing, and the chip, required for the experiment, are prepared and UV-sterilized. Finally, the chip has to be coated with fibronectin, so that the embryonic tissue can attach to the glass25.
- Making PDMS-filled tubing.
- Take ±1 m of tubing. More tubing can be filled with PDMS by taking multiple pieces of tubing.
- Prepare the required amount of PDMS by mixing the monomer with the catalyst in a 9:1 ratio (w:w) to induce polymerization.
NOTE: Use disposable tools to mix, such as plastic cups and forks. Mix properly. - Place the PDMS mixture in a desiccator and apply vacuum for approximately 30 min to remove air from the PDMS mixture.
NOTE: Cover the inside of the desiccator with tissues in case PDMS flows over. - Fill a 3 mL syringe with PDMS. Fill carefully to prevent the formation of air bubbles in the mixture.
- Use forceps to insert the tip of a 22 G needle into the tubing.
NOTE: Be careful not to puncture the tubing or your fingers with the needle. - Attach the tubing to the PDMS-filled syringe via the needle. Use the syringe pump to fill the tubing, with a flow rate of approximately 500 µL/h. Be careful not to apply too high pressure to prevent breaking of the pump.
- Once the tubing is entirely filled with PDMS, place it in a 15 cm dish and cure at room temperature (at least overnight).
- Prepare the tubing and chip for the experiment.
NOTE: The following tubing is required for the experiment: first, approximately 50 cm of tubing per outlet and second, approximately 2-3 m per inlet (the exact size depends on the spatial organization of the pumps, incubator or microscope used later on). The inlet tubing needs to be long to allow the culture medium to equilibrate with CO2 for embryo culture during the experiment.- Cut the tubing at a 45° angle so that pointed ends are created; this will make it easier to insert the tubing into the holes of the chip. Attach one needle to each of the inlet tubing. See step 2.1.5.
- Cut PDMS-filled tubing into ±1 cm plugs. Cut at a 45° angle so that pointed ends are created; this will make it easier to insert the tubing into the holes of the chip. Enough plugs are needed to fill each hole that is not attached to any tubing.
- Remove the adhesive tape from the chip.
- Place all the tubing with needles attached, the chip, and the plugs in a dish and sterilize by exposing to UV light for ±15 min. Any cell culture hood with the possibility for UV sterilization can be used.
- Coating of chip with fibronectin.
NOTE: For culture of 2D ex vivo cultures of posterior PSM, coat the chip with fibronectin.- Place the chip in a beaker containing PBS + 1% Penicillin/Streptomycin at room temperature. Make sure the chip is completely covered with PBS. Flush the chip with PBS to remove air with a P200 pipette.
NOTE: All further handling of the chip will be done in this beaker until the start of the experiment to prevent entrance of air into the chip. Be careful not to break the glass slide of the chip. - Prepare 2 mL of 1:20 Fibronectin in PBS and fill into a beaker.
- Load a 3 mL syringe for each chamber of the chip.With forceps, insert a 22 G needle into the outlet tubing. Attach the needle to the syringe containing fibronectin.
- Connect the syringe to the syringe pump. Flush the tubing, until there is no air left in the tubing.
- Attach the outlet tubing to the outlet of the chip. Make sure to push the tubing all the way to the bottom. Set a low flow rate to coat the chip. All other openings can remain open. Let the syringe pump run for at least 2 h, can also run overnight.
- Stop the pump. Cut off the tubing right after the needle. The remaining tubing will serve as the outlet during the experiment later.
- Place the chip in a beaker containing PBS + 1% Penicillin/Streptomycin at room temperature. Make sure the chip is completely covered with PBS. Flush the chip with PBS to remove air with a P200 pipette.
3. Microfluidics experiment
NOTE: Here, a protocol for the entrainment of the mouse segmentation clock in 2D ex vivo cultures by applying pulses of the Notch signaling inhibitor DAPT is presented. Applying pulses with a period of 130 min, close to the natural period of the mouse segmentation clock (137 min25), allows efficient entrainment. Pulses of 100 min of medium and 30 min of 2 µM DAPT are applied (Figure 2A). The presence of drug within the chip is monitored using a fluorescent dye. The excitation and emission spectra of this dye and the fluorescent signaling reporter must be different enough to prevent bleed-through during fluorescence real-time imaging. When yellow fluorescent proteins are used as signaling reporter, such as the Notch signaling reporter LuVeLu5 (Lunatic fringe-Venus-Lunatic fringe), the dye, Cascade Blue, can be applied. To identify other possible combinations of fluorescent dye and reporter, freely available spectra viewer can be used. The effect of entrainment can be detected by real-time imaging of dynamic signaling reporters5,10,32. Each chip has two incubation chambers. This allows the direct comparison of drug pulses (DAPT) to control pulses (DMSO).
- Make medium and degas.
- On the day of the experiment, prepare 50 mL of culture medium (DMEM-F12 supplemented with 0.5 mM glucose, 2 mM glutamine, and 1% BSA, for further information of mouse tailbud culture see25).
- Prepare syringes filled with the medium for the experiment. Prepare culture medium in beakers: To entrain Notch signaling oscillations, prepare 6 mL of medium with 1.2 µL of DMSO + 10 µM Cascade Blue; 6 mL of culture medium with 2 µM DAPT + 10 µM Cascade Blue; two tubes with 6 mL of medium each. Use at least 6 mL of medium per syringe. Load syringes with the medium. Make sure to label the syringes containing drug and DMSO.
- Degas the chip within PBS and the syringes containing medium in a desiccator. Place the syringes into a beaker with the tip facing up, so that the air can escape at the top. It might occur that the chip floats and is still filled with bubbles. These will be removed subsequently.
- Try to flush out/suck up most of the air bubbles from the chip using a P200 pipette. If some air remains, this will be removed during the following experiment.
- Install syringes in the pumps and attach inlet tubing to the syringes. To entrain Notch signaling, use two syringe pumps, one carrying the medium syringes, one carrying the drug/DMSO syringes. Let this run at a higher flow rate (0.5 mL/h) until the start of the experiment to remove air bubbles from the tubing. Be careful to set the syringe details (diameter) properly in the pump.
- Loading of tissue onto the chip.
- Dissect mouse embryo tissue. Dissect the most posterior tip of the tail (tailbud). Place the dissected tissue into culture medium + 25 µM HEPES.
- Flush the chip with culture medium + 25 µM HEPES using a P200 to remove PBS.
- Load the tissue into the chip using a P200 pipette (Figure 1C). Make sure not to introduce air bubbles into the chip.
NOTE: Efficient loading requires some practice. If the tissue is not oriented properly, it might be possible to turn it by carefully sucking out and flushing in again. However, this often results in quick cell death. - After each tissue-loading step, close the corresponding tissue-loading inlet using a piece of PDMS-filled tubing. Make sure the plugs are sufficiently pushed down to the bottom of the chip using blunt tweezers.
- After all samples have been loaded, close any unused inlets/outlets with pieces of PDMS-filled tubing using blunt forceps.
- Assembly of microfluidic setup.
- Lower the flow rate of the microfluidic pumps. 60-100 µL/h works well for mouse embryo tailbuds. At higher flow rates, cell death was observed – presumably due to too high cell shearing. The cumulative flow rate of multiple pumps should not exceed these 60-100 µL/h per microfluidic chamber at a given time.
- Attach tubing to the microfluidic chip. Do this without getting air bubbles inside the chip (this can be achieved by ensuring that there is a drop of medium present at the end of the tubing). Outlets are still present from the fibronectin coating (see 2.3).
- Once all tubing is attached to the chip, take it out of the beaker, dry it properly, place it in a dish, and put this together with approximately 1.5 m of inlet tubing in an incubator (37 °C, 20% O2, 5% CO2) for overnight culture. The tubing is gas permeable; this will allow equilibration of O2 and CO2 in the medium. Alternatively, for simultaneous fluorescence real-time imaging, the chip and approximately 1.5 m inlet tubing are placed into the microscope directly. A holder for the chip, which fits into standard 96-well-plate holders, is provided in Supplementary File 3.
NOTE: Make sure the humidity is high enough during incubation. If necessary, add a moist tissue to the imaging chamber or incubator to prevent evaporation in the microfluidics set-up. If humidity in the microscope incubator is too low, this results in air bubble formation in the tubing and the microfluidic chip. A closed imaging box, in which chip and tubing are placed, can then be used. The simplest way of making such an imaging box is by placing a big lid over the chip and adding a wet tissue, it does not have to be closed off entirely. Make sure that the height difference between pump and chip is not too big and, if necessary, change heights slowly to prevent the formation of air bubbles due to gravity. - Let all pumps flow at a flow rate of 20 µL/h for 20 min, after the setup has been placed to its final location. Because the chamber and pump have most likely moved in height, this is necessary to re-establish the correct pressure in the tubing.
- Turn the drug pump off, let only the medium pump run at 60 µL/h until the start of experiment/real-time imaging.
- Start of the experiment.
- Start the planned pumping program for the experiment. To entrain Notch signaling, use a pumping program of 100 min medium and 30 min drug pulses, which is repeated until the end of the experiment, typically for 24 h.
NOTE: Any programmable microfluidic pump can be used. In the simplest format, the pumps can be programmed and started manually. Alternatively, the pumps can be controlled by a computer software. - In case real-time imaging is performed, start imaging after a period of at least 30 min. This will allow the temperature of the chip to adjust to the temperature within the incubator and prevent a too strong drift during imaging. Autofocus is still beneficial.
- Perform standard confocal imaging via the glass slide of the chip using an inverted microscope. Use an imaging interval of 10 min to sufficiently detect signaling oscillations with a period of 130 min.
- For shorter periods, shorten the interval for sufficient sampling. Include a low-resolution imaging track for detection of Cascade Blue to visualize the presence of drug on the chip.
- For excitation of a Venus reporter, use a 515 nm laser or a 2-photon laser at a wavelength of 960 nm.
- Acquire a z stack of 6-8 planes with a distance of 8 µm through a 20x plan objective (resolution 512 x 512 pixels, 1.38 µm/pixel). Use a motorized stage to image multiple samples within one experiment.
- Excite Cascade Blue with a 405 nm laser and acquire a single z plane every 10 min (resolution 32 x 32 pixel, 22.14 µm/pixel).
NOTE: Further information on imaging and image analysis are provided in the Representative Results and can be found in reference10.
- Start the planned pumping program for the experiment. To entrain Notch signaling, use a pumping program of 100 min medium and 30 min drug pulses, which is repeated until the end of the experiment, typically for 24 h.
Results
With this protocol, a method for the external entrainment of signaling oscillations of the mouse segmentation clock using microfluidics is presented. By applying pulses of Notch signaling inhibitor, signaling oscillations in independent embryo cultures get synchronized to each other10. A prerequisite for the application of this system to study the functionality of the segmentation clock is that signaling dynamics and segmentation are still present on-chip. It was shown previously that both Wnt and Notch signaling dynamics are maintained despite constant medium flow and physical boundary formation persists in peripheral 2D cultures, representing anterior PSM10.
To confirm entrainment of signaling oscillations to external drug pulses, real-time imaging of, for instance, the Notch signaling reporter LuVeLu5, expressing the yellow fluorescent protein Venus, during the microfluidic experiment is performed. Since orientation of the 2D cultures on chip is difficult to control, signaling oscillations in anterior tissue are analyzed. The periphery of the 2D culture can reproducibly be detected. Orientation of the tissue sections on-chip cannot be controlled at will and the whole inner surface of the chip gets coated with Fibronectin. Therefore, it occasionally happens that cultures attach to the sides or ceiling of the chip. In case the samples move out of the field of view (in x, y, and z direction) or they attach with the posterior end of the tail facing downwards, generally these samples are excluded.
For further analysis, multiple of such entrainment experiments are combined. Independent experiments can be aligned to each other using the timing of the drug pulses visualized by the dye, Cascade Blue, at approximately 400 nm. To analyze and visualize synchronization, quantified oscillations can be detrended (Figure 2B,D) and then either displayed as mean and standard deviation or phases of the oscillations can be calculated. This allows the analysis of the phase-relationship between oscillations of independent posterior embryo cultures to each other and to the external drug pulses (Figure 2C,E). The python-based program pyBOAT33 is a straightforward and user-friendly tool to determine period, phase, and amplitude of such signaling oscillations. To confirm entrainment, one can, for instance, determine the period of the endogenous signaling oscillations. When applying pulses with a period of 130 min, Notch signaling oscillations also show a period of 130 min (Figure 2F)10. In addition, using Cascade Blue pulses, independent experiments with different signaling reporters can be aligned to each other. This way the phase-relationship between oscillations of multiple signaling reporters can indirectly be determined10.
Thus, the presented microfluidic system allows the control of signaling oscillations in ex vivo cultures of the developing mouse embryo. In combination with imaging of markers for segment formation and differentiation, this system can now be applied to dissect how signaling pathways of the segmentation clock interact and how they control somite formation.

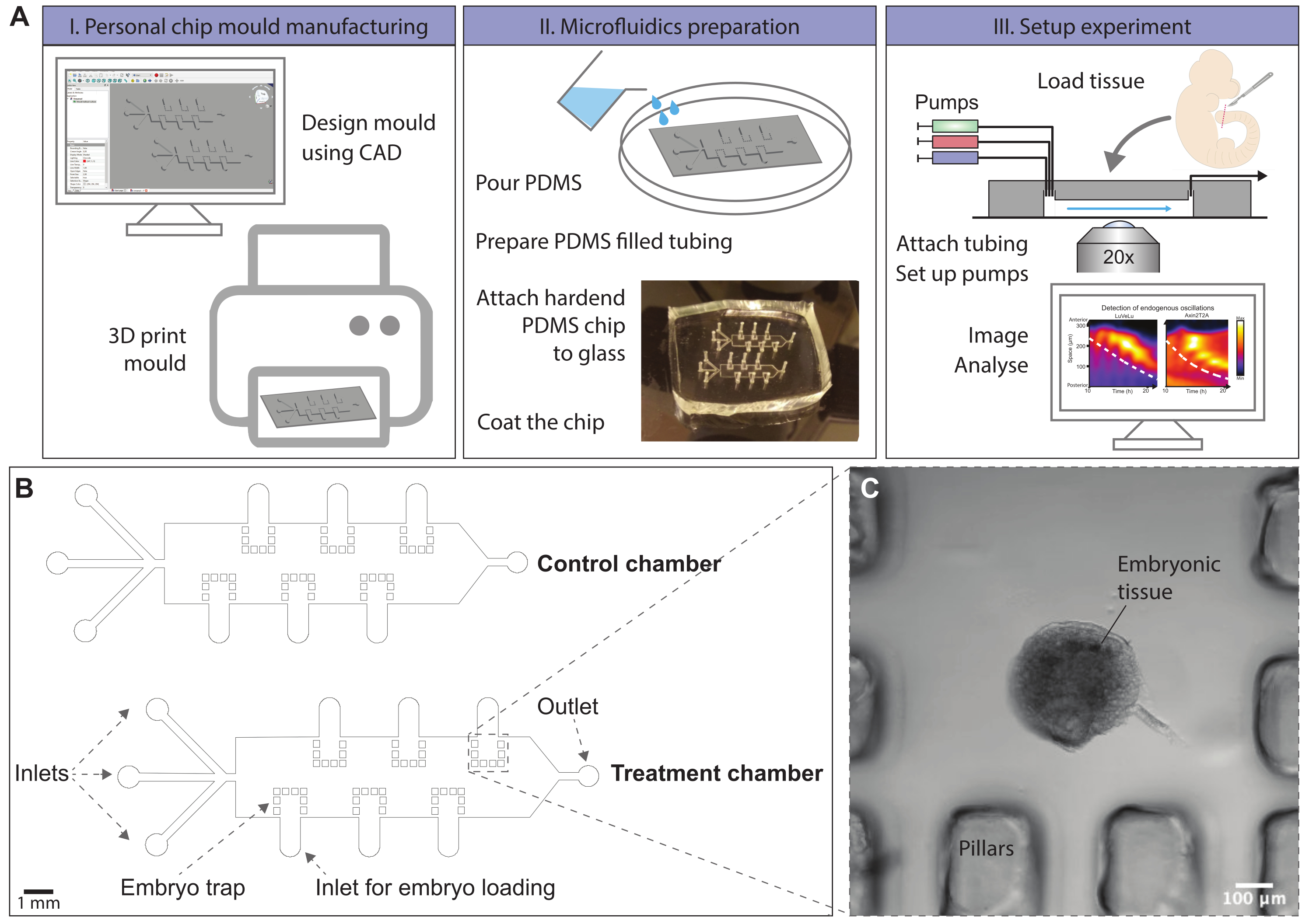
Figure 1: Schematic overview of the microfluidics protocol. (A) Chip molds can be designed using computer-aided-design (CAD) software. A chip design to entrain signaling oscillations in ex vivo models of mouse somitogenesis is provided. Molds can, for instance, be generated by 3D printing or ordered. Microfluidic chips are produced by molding of PDMS. A hardened PDMS chip is cut out and covalently bonded to a glass slide using a plasma oven. The glass surface within the chip is coated with fibronectin to allow dissected tissue to attach. Embryonic tissue is loaded onto the chip via loading inlets, which are subsequently closed. Pumps with different media conditions are attached to the inlets of the chip and a flow program is initialized. Tissue can be imaged using a confocal microscope during the experiment. (Kymographs adapted from Sonnen et al., 201810. Reprinted and modified from Cell according to Creative Commons Attribution CC BY-NC-ND 4.0.) (B) Schematic drawing of a mold design for on-chip culture of posterior mouse embryo tissue. The height of the microfluidic channels is 500 µm, sufficient for the culture of posterior embryonic mouse tails. The file to print this mold is provided in Supplementary File 1, and an alternative is given in Supplementary File 2. (C) Brightfield image of an E10.5 sectioned mouse tail enclosed by PDMS pillars on-chip. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative results from a microfluidics experiment. (A) Schematic representation of entrainment experiment with medium and drug pump. Pulses of signaling pathway modulator are applied to ex vivo cultures of mouse embryos and signaling oscillations can be visualized by real-time imaging of dynamic signaling reporters (for instance, the Notch signaling reporter LuVeLu5 and Wnt signaling reporter Axin2T2A10). Dashed lines indicate the region that is used for analyses over time. (B-F) LuVeLu reporter oscillations are entrained by periodic pulses of the Notch signaling inhibitor DAPT. (B,D) Quantifications of Notch signaling oscillations in anterior PSM upon entrainment using DAPT or a DMSO control. (C,E) Phase-phase relation plots between the phase of Notch signaling oscillations and the timing of external DAPT/DMSO pulses. In case of entrainment, a stable phase-relationship is established. (F) Quantification of the mean period and SEM of reporter oscillations comparing samples entrained with DMSO and DAPT pulses. (Figure adapted from Sonnenet al., 201810. Reprinted and modified from Cell according to Creative Commons Attribution CC BY-NC-ND 4.0.). Please click here to view a larger version of this figure.
{kind=link}
| Problem | Suggested solution(s) |
| The PDMS does not bond to the glass. | - Make sure both the PDMS and glass are clean. |
| - Make sure there is enough PDMS around the chamber for bonding. | |
| - Optimize the settings of the plasma oven. | |
| There are air bubbles on my chip. | - Check if the pump was turned on. |
| - Degas the chip before loading tissue onto the chip. | |
| - Degas the medium before start of the experiment. | |
| - Make sure the humidity in the incubation chamber is high enough. | |
| The tissue dies at the beginning of the experiment. | - Add HEPES to the medium when loading and assembling the chip. |
| - Do not flush the tissue in and out too often during loading. | |
| - Check that the flow rate is not too high. | |
| The tissue dies during imaging. | - Make sure there is no phototoxicity from the imaging |
| - Make sure there is no contamination. | |
| - The flow rate should not be too high. | |
| The focus changes during imaging. | - Use autofocus during imaging to adjust for drift of the imaging slide. |
| - Let the chip equilibrate to the temperature within the microscope for at least 30 minutes. | |
| Outlets/inlets detach. | - Push all tubing completely down to the bottom. |
| The glass of the chip breaks. | - Be more careful, the glass is very fragile. |
| - The thin glass is only required for imaging. If imaging is not performed, a normal thicker glass slide can be used. | |
| - If the break is outside of the chip, it might not be a problem. If the glass seal to the chip is broken, liquid will leak out and air will come in. | |
| There is a contamination on my chip. | - Add antibiotics to the culture medium. |
| - Sterilize all equipment and tubing. | |
| - Work fast and clean. |
Table 1: Troubleshooting
Supplementary File 1: STL file for printing a mold with a 3D printer. This mold is used to generate a microfluidic chip for the on-chip culture of posterior embryonic tissue (design shown in Figure 1). Please click here to download this File.
Supplementary File 2: STL file for printing a mold with a 3D printer to generate a microfluidic chip for the on-chip culture of posterior embryonic tissue. In contrast to Supplementary File 1 and the design shown in Figure 1, this design contains bubble traps at all inlets to prevent small amounts of air from entering the main microfluidic chamber. Please click here to download this File.
Supplementary File 3: Design file for the generation of a holder to place the chip into the microscope. This holder has the dimensions of a 96-well plate and should fit into standard holders of most microscopes. Please click here to download this File.
Discussion
How signaling dynamics control multicellular systems has been a longstanding question in the field. Functional investigation poses a key challenge, because these dynamics have to be subtly modulated to allow this11. Such temporal control over pathway perturbations can in principle be achieved with optogenetics, which also enables a high spatial control34. However, optogenetics require the establishment of sophisticated genetic tools for the analysis of each signaling pathway in question. The current protocol described here provides a highly versatile tool for the perturbation of any signaling pathway. The microfluidics experiment allows the application of temporally controlled drug pulses to functionally investigate the dynamics of signaling pathways in tissue cultures. Here, the focus is on the study of somitogenesis in ex vivo cultures, but the protocol can be adapted to fit any other model systems.
Certain steps in the protocol are critical to perform a successful microfluidics experiment (summarized in Table 1). Key points are discussed as follows. Due to medium flow during the course of the experiment, the tissue culture experiences shear stress. Exposure of the model system to continuous flow can cause cell stress and in extreme cases cell death due to shearing effect. For ex vivo tissue cultures of mouse tailbuds and the current chip design, a flowrate of 60 µL/h (maximum of 100 µL/h) was found to work well with minimal shearing effect. When multiple pumps are turned on simultaneously for the same chip, flow rate of individual pumps needs to be adjusted accordingly to not cause an increase in total flow rate. When the current protocol is adapted to other model systems and chip designs, the flow rate should be optimized. Alternatively, it is possible to not flush medium continuously, but periodically change medium on the chip35. Such a setup can also be applied to control signaling oscillations in mouse somitogenesis (unpublished, data not shown). Moreover, a setup can also be envisioned, in which tissue cultures are not exposed to direct fluid flow, but drugs reach the tissue by diffusion from a neighboring channel. Such systems have been used successfully to apply gradients of growth factors to in vitro models of ESC differentiation14,36.
One major issue of microfluidic experiments is the occurrence of air bubbles on-chip during the experiment. Air bubbles within the chip or tubing can interfere with uniform liquid flow on the chip and can lead to removal of the embryo culture from its location. To prevent the presence of air bubbles, various steps of the protocol are indispensable. First, culture medium within syringes and the microfluidic chip itself must be degassed using a desiccator (step 3.1.3). Second, when loading samples into the loading inlets, one must be careful not to pipet air into the chip together with the sample (step 3.2.3). Third, when filling the tubing with medium, all air bubbles must be pumped out before attaching the chip (step 3.3.2). Otherwise, these bubbles will be pushed into the main chip during the experiment. Lastly, during the experiment, the medium is equilibrated within the imaging chamber or incubator due to the semi-permeable tubing. The permeability of the tubing also allows for evaporation of the medium, so make sure that the humidity is regulated during the experiment to prevent the formation of new bubbles in the tubing.
In general, microfluidics is a highly versatile tool that can be adapted to the researcher's specific question. The current design approach allows for imaging up to 12 ex vivo cultures in parallel per chip. This number is mainly limited by the number of embryos per experiment and the time it takes to image each embryo culture with sufficient resolution. If multiple conditions need to be compared in a single experiment, multiple chips can be mounted on a single glass slide. A chip design is provided, which is ideal for imaging of multiple tissue explants in parallel, but personalized designs can overcome limitations in sample number to allow high throughput analysis and increase the combination of more conditions within a single experiment16,17. Microfluidics is limited to models that can be cultured on a chip and on-chip culture will have to be optimized for each model system individually27,37,38.
Over the last 5-10 years, microfluidics has been applied to address various biological questions due to its potential for high throughput approaches and subtle modulations of signaling dynamics and gradients. Nowadays, microfluidics has become an easily applicable, cheap, and versatile tool that laboratories can establish with ease. Here, the current protocol is optimized for a specific application, namely, studying signaling dynamics governing somitogenesis. It is straightforward to adapt this protocol and design to suit individual research questions in tissue biology.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We are grateful to Yang Li and Jos Malda from the UMC Utrecht for help with 3D printing of molds, Karen van den Anker from the Sonnen group for very useful feedback on the protocol, and Tjeerd Faase from the mechanical workshop at the Hubrecht Institute for the holder for microfluidic chips within the microscope. We would like to thank the entire Sonnen group for critical reading of the manuscript and the reviewers for their constructive feedback. This work received funding from the European Research Council under an ERC starting grant agreement no. 850554 to K.F.S.
Materials
| Name | Company | Catalog Number | Comments |
| 10 mL syringe | Becton, Dickinson and company | 300912 | |
| 184 Silicon Elastomer curing agent | SYLGARD | 1673921 | |
| 184 Silicon Elastomer PDMS | SYLGARD | 1673921 | |
| 3 ml syringes | Becton, Dickinson and company | 309657 | |
| Adhesive tape | Scotch Magic tape or similar | ||
| Blunt-tip forceps Bochem 18/10 Stainless steel | Fisher Scientific | 10663341 | |
| Bovine Serum albumin Lyophilised pH~7 | Biowest | P6154 | |
| Compressed air system | |||
| Crystallizing dish | VWR | 10754-7 | Sterilized |
| D-(+)-Glucose 45% | Sigma | G8769 | |
| Desiccator | VWR | 467-0104 | |
| Disposable cups | Duni | 173 941 | |
| Disposable forks | Staples | 511514 | |
| Dissection equipment | |||
| DMEM/F12 | Cell culture technologies | custom | DMEM/F-12, no phenol red, no L-glutamine, no Glucose |
| Fibronectin | Sigma | F1141-5MG | |
| Flow hood BioVanguard Greenline | CleanAir by Baker | 511514 | For UV light source |
| Glass slide No 1.5H high precision, 70x70 mm | Marienfeld | 107999098 | |
| HEPES Buffer | Gibco | 15630-056 | |
| L-glutamine | Gibco | 25030024 | |
| Microscope | Leica | Leica SP8 MP | |
| Needles 22G | Becton, dickinson and company | z412139-100EA | |
| Oven | VWR | 390-09 | |
| PBS0 | |||
| Penicillin-Streptomycin | Gibco | 15070063 | |
| Plasma oven Femto | Diener electronic | ||
| Punch Ø 1mm | Uni-Core | WB100073 | |
| Scale | Sartorius | Entris | |
| Syringe pump | WPI | AL-4000 | |
| Tubing Ø 1mm | APT | AWG24T | |
| Vacuum pump | VWR | 181-0308 |
References
- Manning, C. S., et al. Quantitative single-cell live imaging links HES5 dynamics with cell-state and fate in murine neurogenesis. Nature Communications. 10 (1), (2019).
- Seymour, P. A., et al. Jag1 modulates an oscillatory Dll1-Notch-Hes1 Signaling module to coordinate growth and fate of pancreatic progenitors. Developmental Cell. , 1-17 (2020).
- Hubaud, A., Pourquié, O. Signaling dynamics in vertebrate segmentation. Nature Reviews Molecular Cell Biology. 15 (11), 709-721 (2014).
- Aulehla, A., et al. Wnt3a plays a major role in the segmentation clock controlling somitogenesis. Developmental Cell. 4 (3), 395-406 (2003).
- Aulehla, A., et al. A β-catenin gradient links the clock and wavefront systems in mouse embryo segmentation. Nature Cell Biology. 10 (2), 186-193 (2008).
- Niwa, Y., et al. The initiation and propagation of Hes7 oscillation are cooperatively regulated by Fgf and Notch signaling in the somite segmentation clock. Developmental Cell. 13 (2), 298-304 (2007).
- Niwa, Y., et al. Different types of oscillations in notch and Fgf signaling regulate the spatiotemporal periodicity of somitogenesis. Genes and Development. 25 (11), 1115-1120 (2011).
- Sonnen, K. F., Aulehla, A. Dynamic signal encoding - From cells to organisms. Seminars in Cell and Developmental Biology. 34, 91-98 (2014).
- Wahl, M. B., Deng, C., Lewandowski, M., Pourquié, O. FGF signaling acts upstream of the NOTCH and WNT signaling pathways to control segmentation clock oscillations in mouse somitogenesis. Development. 134 (22), 4033-4041 (2007).
- Sonnen, K. F., et al. Modulation of phase shift between Wnt and Notch signaling oscillations controls mesoderm segmentation. Cell. 172 (5), 1079-1090 (2018).
- Sonnen, K. F., Merten, C. A. Microfluidics as an emerging precision tool in developmental biology. Developmental Cell. 48 (3), 293-311 (2019).
- Baker, B. M., Trappmann, B., Stapleton, S. C., Toro, E., Chen, C. S. Microfluidics embedded within extracellular matrix to define vascular architectures and pattern diffusive gradients. Lab on a Chip. 13 (16), 3246-3252 (2013).
- Cimetta, E., et al. Microfluidic device generating stable concentration gradients for long term cell culture: Application to Wnt3a regulation of β-catenin signaling. Lab on a Chip. 10 (23), 3277-3283 (2010).
- Demers, C. J., et al. Development-on-chip: In vitro neural tube patterning with a microfluidic device. Development (Cambridge). 143 (11), 1884-1892 (2016).
- Frank, T., Tay, S. Flow-switching allows independently programmable, extremely stable, high-throughput diffusion-based gradients. Lab on a Chip. 13 (7), 1273-1281 (2013).
- Tay, S., et al. Single-cell NF-B dynamics reveal digital activation and analogue information processing. Nature. 466 (7303), 267-271 (2010).
- Kellogg, R. A., Gómez-Sjöberg, R., Leyrat, A. A., Tay, S. High-throughput microfluidic single-cell analysis pipeline for studies of signaling dynamics. Nature Protocols. 9 (7), 1713-1726 (2014).
- Kellogg, R. A., Tay, S. Noise facilitates transcriptional control under dynamic inputs. Cell. 160 (3), 381-392 (2015).
- Takayama, S., et al. Subcellular positioning of small molecules. Nature. 411 (6841), 1016 (2001).
- Takayama, S., et al. Selective chemical treatment of cellular microdomains using multiple laminar streams. Chemistry & Biology. 10 (2), 123-130 (2003).
- Agarwal, A., Goss, J. A., Cho, A., McCain, M. L., Parker, K. K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab on a Chip. 13 (18), 3599-3608 (2013).
- Li, C. Y., Wood, D. K., Huang, J. H., Bhatia, S. N. Flow-based pipeline for systematic modulation and analysis of 3D tumor microenvironments. Lab on a Chip. 13 (10), 1969-1978 (2013).
- Tumarkin, E., et al. High-throughput combinatorial cell co-culture using microfluidics. Integrative Biology. 3 (6), 653-662 (2011).
- Berthier, E., Young, E. W. K., Beebe, D. Engineers are from PDMS-land, biologists are from polystyrenia. Lab on a Chip. 12 (7), 1224-1237 (2012).
- Lauschke, V. M., Tsiairis, C. D., François, P., Aulehla, A. Scaling of embryonic patterning based on phase-gradient encoding. Nature. 493 (7430), 101-105 (2013).
- Lo, J. F. J., et al. Quantitative and temporal control of oxygen microenvironment at the single islet level. Journal of Visualized Experiments: JoVE. (81), (2013).
- Keil, W., Kutscher, L. M., Shaham, S., Siggia, E. D. Long-term high-resolution imaging of developing C. elegans larvae with microfluidics. Developmental Cell. 40 (2), 202-214 (2017).
- Lucchetta, E. M., Lee, J. H., Fu, L. A., Patel, N. H., Ismagilov, R. F. Dynamics of Drosophila embryonic patterning network perturbed in space and time using microfluidics. Nature. 434 (7037), 1134-1138 (2005).
- Nikolaev, M., et al. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature. , (2020).
- Sanka, R., Lippai, J., Samarasekera, D., Nemsick, S., Densmore, D. 3DµF - Interactive design environment for continuous flow microfluidic devices. Scientific Reports. 9 (1), 1-10 (2019).
- Qin, D., Xia, Y., Whitesides, G. M. Soft lithography for micro- and nanoscale patterning. Nature Protocols. 5 (3), 491-502 (2010).
- Yoshioka-kobayashi, K., et al. Coupling delay controls synchronized oscillation in the segmentation clock. Nature. 16, (2019).
- Mönke, G., Sorgenfrei, F. A., Schmal, C., Granada, A. E. Optimal time frequency analysis for biological data - pyBOAT. bioRxiv. , (2020).
- Toettcher, J. E., Voigt, C. A., Weiner, O. D., Lim, W. A. The promise of optogenetics in cell biology: Interrogating molecular circuits in space and time. Nature Methods. 8 (1), 35-38 (2011).
- Dettinger, P., et al. Automated microfluidic system for dynamic stimulation and tracking of single cells. Analytical Chemistry. 90 (18), 10695-10700 (2018).
- Manfrin, A., et al. Engineered signaling centers for the spatially controlled patterning of human pluripotent stem cells. Nature Methods. 16 (7), 640-648 (2019).
- Park, S. E., Georgescu, A., Huh, D. Organoids-on-a-chip. Science. 965, 960-965 (2019).
- Krenger, R., Cornaglia, M., Lehnert, T., Gijs, M. A. M. Microfluidic system for: Caenorhabditis elegans culture and oxygen consumption rate measurements. Lab on a Chip. 20 (1), 126-135 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved