
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Extraction and Purification of FAHD1 Protein from Swine Kidney and Mouse Liver
In This Article
Summary
This protocol describes how to extract fumarylacetoacetate hydrolase domain-containing protein 1 (FAHD1) from swine kidney and mouse liver. The listed methods may be adapted to other proteins of interest and modified for other tissues.
Abstract
Fumarylacetoacetate hydrolase domain-containing protein 1 (FAHD1) is the first identified member of the FAH superfamily in eukaryotes, acting as oxaloacetate decarboxylase in mitochondria. This article presents a series of methods for the extraction and purification of FAHD1 from swine kidney and mouse liver. Covered methods are ionic exchange chromatography with fast protein liquid chromatography (FPLC), preparative and analytical gel filtration with FPLC, and proteomic approaches. After total protein extraction, ammonium sulfate precipitation and ionic exchange chromatography were explored, and FAHD1 was extracted via a sequential strategy using ionic exchange and size-exclusion chromatography. This representative approach may be adapted to other proteins of interest (expressed at significant levels) and modified for other tissues. Purified protein from tissue may support the development of high-quality antibodies, and/or potent and specific pharmacological inhibitors.
Introduction
The eukaryotic FAH domain-containing protein 1 (FAHD1) acts as bi-functional oxaloacetate (OAA) decarboxylase (ODx)1 and acylpyruvate hydrolase (ApH)2. It is localized in mitochondria2 and belongs to the broad FAH superfamily of enzymes1,2,3,4,5,6. While its ApH activity is only of minor relevance, the ODx activity of FAHD1 is involved in the regulation of the TCA cycle flux1,7,8,9. OAA is not only required for the central citrate synthase reaction in the tricarboxylic acid cycle but also acts as a competitive inhibitor of succinate dehydrogenase as part of the electron transport system and as a cataplerotic metabolite. Downregulation of FAHD1 gene expression in human umbilical vein endothelial cells (HUVEC) resulted in a significant reduction in the rate of cell proliferation10, and significant inhibition of mitochondrial membrane potential, associated with a concomitant switch to glycolysis. The working model refers to mitochondrial dysfunction associated senescence (MiDAS)11-like phenotype8, where mitochondrial OAA levels are tightly regulated by FAHD1 activity1,8,9.
Recombinant protein is easier to obtain via expression and purification from bacteria12 rather than from tissue. However, a protein expressed in bacteria may be biased by possible lack of post-translational modifications, or may simply be problematic (i.e., due to plasmid loss, bacterial stress responses, distorted/unformed disulfide bonds, none or poor secretion, protein aggregation, proteolytic cleavage, etc.). For certain applications, protein needs to be obtained from cell lysate or tissue, in order to include such modifications and/or to exclude possible artifacts. Purified protein from tissue supports the development of high-quality antibodies, and/or potent and specific pharmacological inhibitors for selected enzymes, such as for FAHD113.
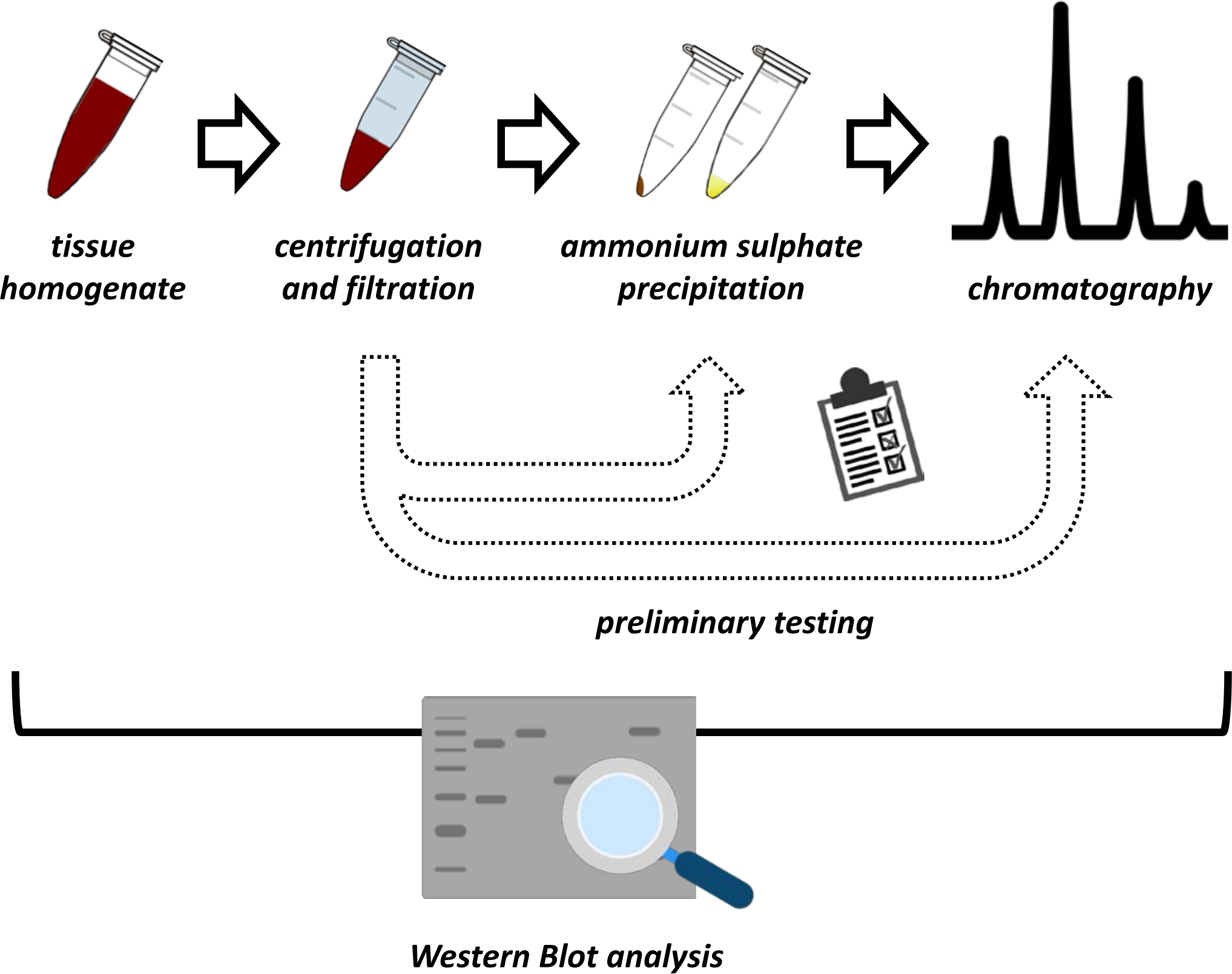
This manuscript presents a series of methods for the extraction and purification of FAHD1 from swine kidney and mouse liver. The described methods require fast protein liquid chromatography (FPLC) but otherwise use common laboratory equipment. Alternative methods may be found elsewhere14,15,16,17. After total protein extraction, the proposed protocol involves a testing phase, in which sub-protocols for ammonium sulfate precipitation and ionic exchange chromatography are discussed (Figure 1). After defining these sub-protocols, the protein of interest is extracted via a sequential strategy using ionic exchange and size-exclusion chromatography with FPLC. Based on these guidelines, the final protocol may be adapted individually for other proteins of interest.

Figure 1: The overall strategy of this protocol. From top to bottom: Protein is extracted from tissues. Tissue homogenate is prepared, centrifuged, and filtrated. For each pair of supernatant and pellet-derived samples, tests for ammonium sulfate precipitation and ionic exchange chromatography (FPLC) need to be performed to probe for optimal conditions. After establishing these sub-protocols, the protein may be extracted via a sequential procedure of ammonium sulfate precipitation, ionic exchange chromatography, and repetitive size exclusion chromatography (FPLC) at varying pH and salt concentrations. All steps need to be controlled by western blot. Please click here to view a larger version of this figure.
{kind=link}
Protocol
All experiments were performed in compliance with institutional guidelines. Swine kidney was obtained fresh from the local supermarket. Liver tissues were harvested from C57BL6 wild-type mice maintained at the Institute for Biomedical Aging Research at Innsbruck University, Rennweg 10, 6020 Innsbruck, Austria under the supervision of Univ.-Doz. Dr. Pidder Jansen-Dürr, covered by ethical permission as project leader issued in 2013 (BMWF-66.008/0007-II/3b/2013). Maintenance and use of the mice for the project are covered under ethical permission No. 2020-0.242.978 from May 5th, 2020, issued by the Austrian Ministry for Education, Science, and Research (BMBWF).
1. Preparations
NOTE: Before the protocol starts, several things need to be prepared, i.e., the protein lysis buffer, the crude tissue sample, and a specific antibody, besides general chemicals, and materials.
- Prepare 250 mL of protein lysis buffer per 100 g of net weight of tissue: 250 mL of 1x PBS with 50 mM NaF, 1 mM PMSF, 2 µg/mL aprotinin, and 1 mM activated orthovanadate (see Table 1). Filter the solution using a 0.22 µm syringe filter unit.
NOTE: Activation of orthovanadate is required before use to convert it into a more potent inhibitor of protein tyrosine phosphatases18. Activated orthovanadate may be obtained from commercial suppliers, but also prepared as follows.- Prepare a 200 mM stock solution of (sodium) orthovanadate in ddH2O. For preparing 10 mL of solution, add 368 mg of Na3VO4 to 9 mL of water and dissolve by stirring. Once dissolved, make up the volume to 10 mL with ddH2O.
NOTE: The starting pH of the sodium orthovanadate solution may vary with the source of material, and the pH needs to be adjusted to 10 in a repetitive approach as follows. - Depending on the initial pH of the solution, adjust the pH to 10 with NaOH or HCl. At pH > 10, the solution will have a yellow color. Boil the solution until it turns colorless, cool it down to room temperature, and check the pH. If pH is >10, add a small volume of HCl to adjust the pH to 10. At this point, the solution may turn yellow again.
- Repeat the boiling and cooling until the solution remains colorless and the pH stabilizes at 10 (approximately 5-7 times). At this point, adding HCl results in a faint appearance of yellow color in the solution. Store activated orthovanadate in 1 mL aliquots at -20 °C.
- Prepare a 200 mM stock solution of (sodium) orthovanadate in ddH2O. For preparing 10 mL of solution, add 368 mg of Na3VO4 to 9 mL of water and dissolve by stirring. Once dissolved, make up the volume to 10 mL with ddH2O.
- Prepare tubes with 2 mL of lysis buffer per gram of tissue and place them on ice.
NOTE: This protocol used eight 50 mL tubes, each filled with 30 mL of lysis buffer in total for one swine kidney (about 100-150 g), and two tubes each filled with 40 mL of lysis buffer for 20 mouse livers (each 1-2 g) in total. - Prepare the tissue: dissect the tissue on a pre-cleaned glass plate placed on ice in a polystyrene foam box. Cut tissue pieces of about 100 mg each to be easily transferred into respective tubes for subsequent lysis. Transfer the tissue pieces into the prepared tubes (step 1.2).
- Prepare a saturated ammonium sulfate solution: heat 500 mL of ddH2O to 70 °C and while stirring, gradually add ammonium sulfate powder (see Table of Materials) until no more ammonium sulfate is dissolved. Cool this (over)saturated solution to room temperature and store it at 4 °C overnight.
2. Total protein extraction
NOTE After preparing the sample in cold protein lysis buffer (see step 1.3), homogenize the tissue as best as possible via sonication by an ultrasonic probe, or using an electric homogenizer as follows.
- Homogenization of tissues
- In the case of a swine kidney, sonicate the suspension preferably by an ultrasonic probe while keeping the sample on ice (10 cycles of 15 s pulse, with intervals of 30 s between the pulses to cool the sample on ice, at medium amplitude with 50% duty cycle).
- In the case of mouse organs, homogenize the suspension using an electric homogenizer (starting with a low force, and slowly accelerating to medium force) while keeping the sample on ice. Regularly wash the electric homogenizer in PBS to remove any organic material clogging the device.
- Take 20 µL out of the samples and check under the microscope whether the cells of the homogenized tissue are properly destroyed; otherwise, repeat the homogenization.
- Centrifuge the tubes in a tabletop centrifuge at 10,000 x g for 30 min at 4 °C.
NOTE: Optionally, centrifuge the supernatant a second time at 20,000 x g for 30 min at 4 °C to eliminate small fractions of the initial pellet that may have been transferred. This will simplify the subsequent filtration in step 2.3. - Collect the supernatant in a fresh tube and place it on ice. Sequentially filter the supernatant using 0.45 µm and 0.22 µm syringe filter units. Aliquot the supernatant into 10 mL batches and freeze them at -20 °C for short-term storage or at -80 °C for longer storage.
NOTE: Pre-filtering with 0.45 µm removes the majority of the particles before a second filtering step with 0.22 µm removes the finer particles. Using the 0.22 µm filter directly can cause the risk of clogging the filters. - Prepare a 50 µL sample for SDS-PAGE/western blot analysis by adding 10 µL of 5x SDS sample buffer (see Table 1) to 40 µL of the supernatant, and then boiling at 95 °C for 10 min.
- Optionally, resuspend about 100 µL of pellet obtained in step 2.2 in 900 µL of ddH2O, and prepare a sample for SDS-PAGE/western blot analysis as described above.
NOTE: Inclusion of pellet-derived samples in the western blot analysis, in addition to the positive control, will indicate whether the expression of the protein is low, or the antibody is problematic.
- Optionally, resuspend about 100 µL of pellet obtained in step 2.2 in 900 µL of ddH2O, and prepare a sample for SDS-PAGE/western blot analysis as described above.
3. SDS-PAGE and western blot analysis
NOTE: Western blot analysis is required to check for protein solubility. The following describes a protocol for electroblotting, using a Wet/Tank blotting system (see Table of Materials). An alternate protocol for SDS-PAGE may be found elsewhere19.
- Prepare a discontinuous 12.5% polyacrylamide SDS-PAGE gel according to the manufacturer's instructions (i.e., a stacking gel on top of a resolving gel; see Table 1). Run the samples previously prepared during step 2 (similar to steps 4, 5, and 6; see below).
- Load a protein marker ladder into the first well (see Table of Materials). Load 5 ng of hFAHD1 recombinant protein (obtained from bacteria12; see Table 1) as a positive control into the second well.
- Subsequently, load 20 µL of the sample to be analyzed, and fill all the remaining wells with 20 µL of prepared SDS-PAGE 1x sample buffer (i.e., 5x sample buffer diluted with ddH2O). Run the SDS-PAGE gels at 125 V using the SDS-running buffer (see Table 1).
- After SDS-PAGE is complete, perform a western blot analysis and probe the membranes using the available antibody raised against FAHD1 (see Table 1).
NOTE: As the samples are taken from crude tissue homogenate, usually the quality of the SDS-PAGE and the western blot analysis at this point is compromised; however, it is important to check whether the protein to extract is soluble in the supernatant. The following protocol was tested for swine kidney, and different mouse organs, including liver, heart, brain, and kidney.- Prepare the 10x western blot transfer buffer (see Table 1). Prepare the 1x western blot transfer buffer (see Table 1) and cool it to 4 °C.
- Activate a PVDF membrane for 2 min in methanol. Wash the membrane in ddH2O for 2 min. Equilibrate the membrane for 15 min in 1x western blot transfer buffer.
- Wash the SDS-gel with 1x PBS for 10 min while shaking to remove the SDS-running buffer, and then incubate the gel in 1x western blot transfer buffer for 10 min for equilibration. Assemble the electroblotting cassette (i.e., combining the activated PVDF membrane and gels) according to the manufacturer's instructions.
- Run the blot via electroblotting at 300 mA for 1 h in a polystyrene foam box filled with ice or in the cold room (4 °C). Transfer the PVDF membrane into a 50 mL tube with its exposed side facing the inner side of the tube. Incubate the membrane in 20 mL of western blot blocking buffer (see Table 1) overnight at 4 °C while rolling on a tube roller (see Table of Materials).
- The next day, wash the membrane for 5 min with 20 mL of western blot washing buffer (PBS with 0.1% (v/v) Tween 20) in the same tube while rolling. Incubate the membrane in the same tube with the primary antibody2 (targeting FAHD1; see Table 1) diluted 1:500 in western blot blocking buffer for 1 h at room temperature while rolling.
- Wash the membrane in the same tube three times for 10 min each with 20 mL of western blot washing buffer while rolling. Incubate the membrane for 30 min at room temperature with HRP-conjugated secondary antibody (see Table of Materials) diluted 1:3000 in 5 mL of western blot blocking buffer.
- Wash the membrane in the same tube three times for 10 min each with 20 mL of western blot washing buffer and twice for 5 min each with 1x PBS. Dry the membrane by carefully holding it with tweezers on one edge, and by touching a piece of cellulose or a piece of Whatman paper with the opposite (lower) edge of the membrane. Put the membrane (exposed side up) on a cleaned glass plate.
- Carefully cover the whole membrane with 1 mL of prepared ECL western blot substrate using a pipette, taking care not to create any air bubbles. Let the ECL solution incubate for 3 min, and immediately develop the membrane using X-ray film or using an imaging system.
NOTE: If the protein was detected in none of the samples but only in the positive control, this may indicate that the protein is insoluble, or not present in adequate amounts to be detected by the antibody. If only nanograms of the positive control were loaded, the first scenario is more likely. If no protein was detected at all, check the quality of the antibody, and maybe switch to a polyclonal antibody rather than a monoclonal antibody. In rare occasions, i.e., for some hydrophobic proteins, the protein may be detectable after centrifugation, but not after filtration. In such a case, it is recommended to use special filter units for hydrophobic proteins.
- Optionally, stain the PVDF membranes after western blot to control the successful transfer of the protein from the SDS-PAGE gel to the PVDF membrane.
NOTE: Coomassie staining is recommended for troubleshooting, method development, and documentation, but mind that after applying this protocol, membranes are lost to further western blot analysis. Ponceau S staining gives weaker staining but may be used if the membranes are to be re-probed.- Prepare small trays containing the staining (Coomassie or Ponceau S) and de-staining solutions.
- Using tweezers, put the membrane into the staining solution and gently shake until the membrane is stained well (5-10 min).
- Transfer the membrane into the destaining solution and shake until the solution is saturated (5-10 min). Repeat the destaining step until the protein bands can be observed on the membrane; if no bands are observed at all, repeat the staining with a longer incubation time. Dry the membrane by placing it on a glass plate using tweezers.
4. Testing: Ammonium sulfate precipitation
NOTE: Ammonium sulfate precipitation is a method of protein purification by altering the solubility of the protein. In a preliminary experiment, the ammonium sulfate concentration is sequentially increased to a value that precipitates a maximum amount of protein contaminants, while leaving FAHD1 in solution. The solubility of the protein is again probed via western blot analysis.
- Proceed from step 2.3: either thaw an aliquot of the sample or proceed directly after protein extraction (i.e., without freezing the sample). Filter the sample using a 0.22 µm filter unit to exclude possible precipitates after thawing. Prepare six 1.5 mL tubes on ice, and transfer 250 µL of sample into each tube.
- Prepare a dilution series of 5%, 10%, 15%, 20%, 25%, and 30% ammonium sulfate in the tubes prepared above, and make up the final volume to 1000 µL with protein lysis buffer. Incubate the samples at 4 °C overnight on a tube rotator (see Table of Materials).
- Using a tabletop centrifuge, centrifuge at 10,000 x g for 30 min at 4 °C and carefully transfer all of the supernatants into separate tubes. Air-dry the resulting pellets and resuspend each of them in 1000 µL of ddH2O.
- For each pair of resuspended pellet and supernatant from the previous step, mix 40 µL with 10 µL of 5x SDS sample buffer and boil at 95 °C with open lids until most of the liquid has vaporized. Then, resuspend the pellet in a mixture of 50% DMSO in ddH2O.
- Perform SDS-PAGE (step 3) but run the gels at 80 V for 3 h. For each concentration of ammonium sulfate, load the samples derived from the resuspended pellet and supernatant (step 4.3) in pairs. Perform a western blot analysis (step 3).
- Check for the highest concentration of ammonium sulfate, at which the protein to be purified (i.e., FAHD1) remains in the sample derived from the supernatant. Based on the results, define an ammonium sulfate precipitation protocol for the protein of interest, to be used in future experiments.
NOTE: Ammonium sulfate is well known to distort SDS-PAGE and western blot. As the concentration of ammonium sulfate increases, the quality of the western blot analysis will be compromised. However, as with step 3 before, this analysis is used to check for the solubility of the protein of interest at given concentrations of ammonium sulfate. This protocol aims to precipitate other proteins, while the protein to be purified must remain soluble.
5. Testing: ionic exchange chromatography with FPLC
NOTE: Molecules with charged functional groups are bound to a silica particle column for FPLC, enabling the differentiation of proteins according to their surface charge. Perform this step twice, using the cationic exchange column and anionic exchange column (see Table of Materials). The protocol steps are the same for either cationic or anionic exchange chromatography, but the buffers to be used are different (see Table 1); both with "low salt" 15 mM NaCl and "high salt" 1 M NaCl conditions. For the columns used, a flow rate of 1 mL/min is recommended.
- Set up the FPLC system with the anionic or cationic exchange column. Wash the column with 5 column volumes (CVs) of 20% EtOH (in H2O), followed by 5 CVs of ddH2O. Alternately, wash the column with 1 CV of low salt buffer, high salt buffer, and again low salt buffer in the order until no more peaks are observed in the chromatogram, but wash at least once.
- After determining the optimal protocol for ammonium sulfate precipitation on the small scale (step 4), apply the precipitation protocol to 10 mL of original tissue homogenate (step 2). Optionally, dialyze the sample against the low salt buffer.
- Apply the sample onto the column (e.g., by injection or by using a sample pump) and collect the flow-through. Wash the column with 1 CV of the low salt buffer.
- Set up a linear gradient elution from 100% low salt buffer/ 0% high salt buffer to 0% low salt buffer/100% high salt buffer within 3 CVs. Continuously collect 1 mL fractions. After the gradient has finished, continue to run with the high salt buffer until no more protein-associated peaks (UV absorption at 280/255 nm) are detected in the chromatogram over the range of 1 CV.
- Apply 1 mL of 25% SDS dissolved in 0.5 M NaOH (in ddH2O) to clean the column. Consecutively, wash the column with 3 CVs of ddH2O and 3 CVs of 20% EtOH (in ddH2O).
- Collect SDS-PAGE samples of all peak-fractions and the flow-through, and probe them via western blot for the presence of the protein of interest (step 3). Snap-freeze the collected fractions in liquid nitrogen and store them at -80 °C.
- After western blot analysis is complete, thaw and pool the fractions containing the protein of interest and discard the others. Repeat steps 5.1-5.5 with the alternate column (i.e., cationic or anionic exchange column).
- After both columns have been probed, define an FPLC protocol for the protein of interest, to be used in future experiments. Reduce the volume of the protein solution using ultra-centrifugation filter units (10 kDa, see Table of Materials) down to 2 mL.
NOTE: There are two expected outcomes of this series of experiments. Either the protein of interest has attached to one of the columns, and the protein solution is already quite pure after elution, or the protein remained in the flow-through in both cases. In the latter scenario, although the protein is in the flow-through, the cleaning effect of this step might still be significant. In such a case, as for FAHD1 in swine kidney and mouse liver, this step of ionic exchange will still be performed. If neither the cationic or anionic exchange column can provide a proper cleaning effect, one may try to modify the pH of the lysate and buffer, and to dialyze the sample against the running buffer before application to FPLC.
6. Protein extraction using defined sub-protocols for ammonium sulfate-precipitation and FPLC
NOTE: Porous particles in a silica gel column for FPLC (See Table of Materials) enable the differentiation of proteins according to their hydrodynamic radius. The described steps are to be performed with an FPLC system, using size exclusion chromatography (SEC). For the SEC column used (see Table of Materials), a flow rate of 0.3 mL/min is recommended.
- Prepare all the required materials (see step 1), and extract the total protein from the tissue (see step 2). Perform an ammonium sulfate precipitation with all the tissue homogenate that was not used for testing (see step 4). For larger volumes, concentrate the lysate using ultra-centrifugation filter units (10 kDa; see Table of Materials) down to a smaller volume of 50 mL or less.
- Perform a first purification step using ionic exchange chromatography (see step 5).
- Prepare samples for western blot, as described in the previous steps. Perform western blot analysis and pool all FAHD1 containing fractions from ionic exchange chromatography.
- Reduce the volume of the protein solution down to 2 mL using ultra-centrifugation filter units (10 kDa). Sequentially filter the solution with 0.45 µm and 0.22 µm syringe filter units to remove any micro-precipitation.
- Equilibrate the SEC column with 1 CV of SEC running buffer (see Table 1), containing 1 mM DTT. Load the sample onto the column and run the chromatography until all the proteins are eluted (1-2 CV).
- Collect fractions of 1 mL of the flow-through that corresponds to significant peaks in the chromatogram (UV absorption at 280/255 nm) and prepare 50 µL samples of each collected fraction for SDS-PAGE and western blot analysis, as described in the previous steps. Snap-freeze all the fractions using liquid nitrogen, and store them at -80 °C.
- Consecutively wash the SEC column with 1 CV of ddH2O and 1 CV of 20% EtOH (in ddH2O). Perform western-Blot analysis, and pool all FAHD1 containing fractions. Reduce the volume of the protein solution down to 2 mL using ultra-centrifugation filter units (10 kDa, see Table of Materials).
- Assess the protein concentration using a commercial BCA assay kit (see Table of Materials).
NOTE: The pH and salt content of the mobile phase may influence the elution profile of globular proteins20. Acidic or basic conditions may result in peaks being less defined and increased protein-matrix interactions leading to partial retention of protein on the column20. This effect may be exploited for further protein purification. A repetition of step 6 with different flow rates, pH, and salt concentrations may enhance the purity of the protein20.
7. Silver staining
NOTE: Silver staining analysis of SDS-PAGE gels is required to check for protein contaminations that may not be seen with Coomassie staining. The following protocol is one among many versions that can be found in the literature21. Perform all the incubation steps by shaking in a clean glass tray. Collect all silver- and formaldehyde-containing liquids in a special waste container and properly discard them.
- Incubate the SDS-PAGE gels in silver staining fixing solution (see Table 1) overnight in the cold room. Incubate the gels in silver staining incubation solution (see Table 1) for 3 h at room temperature. Optionally, add glutaraldehyde (see Table 1) to improve the detection of faint bands. Wash the gels four times in ddH2O for 10 min each.
- Incubate the gels in silver staining silver solution (see Table 1) for 1 h.
NOTE: Mind that from now on all the liquids and the gel itself contain silver and formaldehyde which are toxic. - Incubate the gels in silver staining developer solution (see Table 1) with vigorous shaking until bands are clearly visible. To stop the reaction, discard the developer solution and immediately incubate the gels in silver staining stop solution (see Table 1) for a minimum of 10 min.
NOTE: Bands stained in steps 7.2 and 7.3 will constantly become more developed. Adding more formaldehyde to the solution than stated may be necessary if the staining is weak.
Results
FAHD1 protein was extracted from swine kidney and mouse liver using the presented protocol. For mouse tissue, multiple organs are required to obtain several µg after the final purification step. For this reason, this article focuses on the extraction of FAHD1 from swine kidneys, which is a much more exemplary experiment. The extraction of FAHD1 from the mouse liver is performed to present the difficulties and possible pitfalls of this protocol. It is generally recommended to use organs that show a high expression level ...
Discussion
Critical steps in the protocol
Following common guidelines for the handling of proteins is essential, such as working on ice and at moderate pH and salt conditions. The use of protease inhibitors is beneficial to the method, while the use of proteasome inhibitors is highly recommended. Freezing and thawing the sample may always result in protein precipitation (at least partially), so any thawed aliquot of initial protein lysate (step 2) should be processed continuously without a break. Centrifugati...
Disclosures
The authors have no competing financial interests.
Acknowledgements
The authors are very thankful for the technical assistance by Ayse Öztürk and Eva Albertini. Mice used for the generation of liver tissue were maintained under the supervision of Univ.-Doz. Dr. Pidder Jansen-Dürr (Institute for Biomedical Aging Research at Innsbruck University, Rennweg 10, 6020 Innsbruck, Austria).
Materials
| Name | Company | Catalog Number | Comments |
| 0.22 µm filter units | MERCK | SLGP033RS | Millex-HP, 0.22 µm, PES 33 mm, not steril |
| 0.45 µm filter units | MERCK | SLHP033NS | Millex-HP, 0.45 µm, PES 33 mm, not steril |
| 15 mL Falcon tubes | VWR | 734-0451 | centrifugal tubes |
| 50 mL Falcon tubes | VWR | 734-0448 | centrifugal tubes |
| 96-Well UV Microplate | Thermo-Fischer | 8404 | UV/VIS transparent flat-bottom 96 well plates |
| Acrylamide/Bis Solution (40%, 29:1 ratio) | BIO-RAD | #1610147 | 40% acrylamide/bis-acrylamide, 29:1 (3.3% crosslinker) solution for casting polyacrylamide gels |
| ÄKTA FPLC system | GE Healthcare Life Sciences / Cytiva | - | using the FPLC system by GE Healthcare; different custom versions exist; this work used the "ÄKTA pure" system |
| Amicon Ultra-15, PLGC Ultracel-PL Membran, 10 kDa | MERCK | UFC901024 | centrifigal filters for protein enrichment; 10 kDa molecular mass filter; 15 mL |
| Amicon Ultra-4, PLGC Ultracel-PL Membran, 10 kDa | MERCK | UFC801024 | centrifigal filters for protein enrichment; 10 kDa molecular mass filter; 4 mL |
| Ammonium sulfate powder | MERCK | A4418 | ammonium sulphate for molecular biology, ≥99.0% |
| Ammoniumpersulfat reagent grade, 98% | MERCK | 215589 | Catalyst for acrylamide gel polymerization. |
| Coomassie Brilliant blue R 250 | MERCK | 1125530025 | Coomassie Brilliant blue R 250 (C.I. 42660) for electrophoresis Trademark of Imperial Chemical Industries PLC. CAS 6104-59-2, pH 6.2 (10 g/l, H2O, 25 °C) |
| Dialysis tubing cellulose membrane | MERCK | D9277 | Cellulose membranes for the exchange of buffers via dialysis. |
| Eppendof tubes 1.5 mL | VWR | 525-1042 | microcentrifugal tubes; autoclaved |
| HiLoad 26/600 Superdex 75 pg | GE Healthcare Life Sciences / Cytiva | 28989334 | HiLoad Superdex 75 pg prepacked columns are for high-resolution size exclusion chromatography of recombinant proteins |
| Immun-Blot PVDF Membrane | BIO-RAD | #1620177 | PVDF membranes are protein blotting membranes optimized for fluorescent and multiplex fluorescent applications. |
| Mini Trans-Blot Electrophoretic Transfer Cell | BIO-RAD | #1703930 | Use the Mini Trans-Blot Cell for rapid blotting of Mini-PROTEAN precast and handcast gels. |
| Mini-PROTEAN Tetra Vertical Electrophoresis Cell for Mini Precast Gels | BIO-RAD | #1658004 | 4-gel vertical electrophoresis system, includes electrode assembly, companion running module, tank, lid with power cables, mini cell buffer dam. |
| Mono Q 10/100 GL | GE Healthcare Life Sciences / Cytiva | 17516701 | Mono Q columns are strong anion exchange chromatography columns for protein analysis or small scale, high resolution polishing of proteins. |
| Mono S 10/100 GL | GE Healthcare Life Sciences / Cytiva | 17516901 | Mono S columns are strong cation exchange chromatography columns for protein analysis or small scale high resolution polishing of proteins. |
| PageRuler Prestained Protein Ladder, 10 to 180 kDa | Thermo-Fischer | 26616 | A mixture of 10 blue-, orange-, and green-stained proteins (10 to 180 kDa) for use as size standards in protein electrophoresis (SDS-PAGE) and western blotting. |
| Pierce BCA Protein Assay Kit | Thermo-Fischer | 23225 | A two-component, high-precision, detergent-compatible protein assay for determination of protein concentration. |
| Sonifier 250; Ultrasonic Cell Disruptor w/ Converter | Branson | - | New models at https://www.emerson.com/documents/automation/brochure-sonifier-sfx250-sfx550-cell-disruptors-homogenizers-branson-en-us-168180.pdf |
| Swine Anti-Rabbit Immunoglobulins/HRP (affinity isolated) | Agilent Dako | P0399 | The antibody used for horseradish peroxidase conjugation reacts with rabbit immunoglobulins of all classes. |
| TEMED, 1,2-Bis(dimethylamino)ethane, TMEDA | MERCK | T9281 | TEMED (N,N,N′,N′-Tetramethylethylenediamine) is molecule which allows rapid polymerization of polyacrylamide gels. |
| Tube Roller | - | - | A general tube rotator roller; e.g. a new model at https://labstac.com/de/Mixer/Roller/c/71 |
| Tube Rotator | - | - | A general tube rotator wheel; e.g. a new model at https://labstac.com/de/Tube-Roller/p/MT123 |
| ULTRA-TURRAX; T 25 digital | IKA | 0003725000 | New models at https://www.ika.com/de/Produkte-Lab-Eq/Dispergierer-Dipergiergeraet-Homogenisierer-Homogenisator-csp-177/T-25-digital-ULTRA-TURRAX-cpdt-3725000/ |
References
- Pircher, H., et al. Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. Journal of Biological Chemistry. 290 (11), 6755-6762 (2015).
- Pircher, H., et al. Identification of human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a novel mitochondrial acylpyruvase. Journal of Biological Chemistry. 286 (42), 36500-36508 (2011).
- Kang, T. -. W., et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 479 (7374), 547-551 (2011).
- Hong, H., Seo, H., Park, W., Kim, K. K. -. J. Sequence, structure and function-based classification of the broadly conserved FAH superfamily reveals two distinct fumarylpyruvate hydrolase subfamilies. Environmental Microbiology. 22 (1), 270-285 (2020).
- Timm, D. E., Mueller, H. A., Bhanumoorthy, P., Harp, J. M., Bunick, G. J. Crystal structure and mechanism of a carbon-carbon bond hydrolase. Structure. 7 (9), 1023-1033 (1999).
- Bateman, R. L., et al. Mechanistic inferences from the crystal structure of Fumarylacetoacetate Hydrolase with a bound phosphorus-based inhibitor. Journal of Biological Chemistry. 276 (18), 15284-15291 (2001).
- Weiss, A. K. H., et al. Structural basis for the bi-functionality of human oxaloacetate decarboxylase FAHD1. Biochemical Journal. 475 (22), 3561-3576 (2018).
- Etemad, S., et al. Oxaloacetate decarboxylase FAHD1 - a new regulator of mitochondrial function and senescence. Mechanisms of Ageing and Development. 177, 22-29 (2019).
- Weiss, A. K. H., et al. Regulation of cellular senescence by eukaryotic members of the FAH superfamily - A role in calcium homeostasis. Mechanisms of Ageing and Development. 190, 111284 (2020).
- Petit, M., Koziel, R., Etemad, S., Pircher, H., Jansen-Dürr, P. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Experimental Gerontology. 92, 7-12 (2017).
- Wiley, C. D., et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metabolism. 23 (2), 303-314 (2016).
- Weiss, A. K. H., et al. Expression, purification, crystallization, and enzyme assays of Fumarylacetoacetate Hydrolase Domain-containing proteins. Journal of Visualized Experiments: JoVE. (148), e59729 (2019).
- Weiss, A. K. H., et al. Inhibitors of Fumarylacetoacetate Hydrolase Domain Containing Protein 1 (FAHD1). Molcules. 26 (16), 5009 (2021).
- Mizutani, H., Kunishima, N. Purification, crystallization and preliminary X-ray analysis of the fumarylacetoacetase family member TTHA0809 from Thermus thermophilus HB8. Acta Crystallographica Section F Structural Biology and Crystallization Communications. 63 (9), 792-794 (2007).
- Lee, C. H. A simple outline of methods for protein isolation and purification. Endocrinology and Metabolism. 32 (1), 18-22 (2017).
- Amer, H. E. A. Purification of proteins: Between meaning and different methods). Proteomics Technologies and Applications. , (2019).
- Niu, L., Yuan, H., Gong, F., Wu, X., Wang, W. Protein extraction methods shape much of the extracted proteomes. Frontiers in Plant Science. 9, 802 (2018).
- Gordon, J. A. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods in Enzymology. 201, 477-482 (1991).
- Gallagher, S. R. SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Current Protocols in Essential Laboratory Techniques. , (2012).
- . Effect of pH on Protein Size Exclusion Chromatography Available from: https://www.agilent.com/cs/library/applications/5990-8138EN.pdf (2011)
- Sørensen, B. K., et al. Silver staining of proteins on electroblotting membranes and intensification of silver staining of proteins separated by polyacrylamide gel electrophoresis. Analytical Biochemistry. 304 (1), 33-41 (2002).
- Fagerberg, L., et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & Cellular Proteomics. 13 (2), 397-406 (2014).
- . Cytiva Life Fundamentals of size exclusion chromatography Available from: https://www.cytivalifesciences.com/en/us/solutions/protein-research/knowledge-center/protein-purification-methods/size-exclusion-chromatography (2022)
- Rosano, G. L., Ceccarelli, E. A. Recombinant protein expression in Escherichia coli: advances and challenges. Frontiers in Microbiology. 5, 172 (2014).
- Rosano, G. L., Morales, E. S., Ceccarelli, E. A. New tools for recombinant protein production in Escherichia coli: A 5-year update. Protein Science: A Publication of the Protein Society. 28 (8), 1412-1422 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved