
Imaging Neutrophil Migration in the Mouse Skin to Investigate Subcellular Membrane Remodeling Under Physiological Conditions
In This Article
Summary
Neutrophil migration relies on the rapid and continuous remodeling of the plasma membrane in response to chemoattractant and its interactions with the extracellular microenvironment. Described herein is a procedure based on Intravital Subcellular Microscopy to investigate the dynamic of membrane remodeling in neutrophils injected in the ear of anesthetized mice.
Abstract
The study of immune cell recruitment and function in tissues has been a very active field over the last two decades. Neutrophils are among the first immune cells to reach the site of inflammation and to participate in the innate immune response during infection or tissue damage. So far, neutrophil migration has been successfully visualized using various in vitro experimental systems based on uniform stimulation, or confined migration under agarose, or micro-fluidic channels. However, these models do not recapitulate the complex microenvironment that neutrophils encounter in vivo. The development of multiphoton microscopy (MPM)-based techniques, such as intravital subcellular microscopy (ISMic), offer a unique tool to visualize and investigate neutrophil dynamics at subcellular resolutions under physiological conditions. In particular, the ear of a live anesthetized mouse provides an experimental advantage to follow neutrophil interstitial migration in real-time due to its ease of accessibility and lack of surgical exposure. ISMic provides the optical resolution, speed, and depth of acquisition necessary to track both cellular and, more importantly, subcellular processes in 3D over time (4D). Moreover, multi-modal imaging of the interstitial microenvironment (i.e., blood vessels, resident cells, extracellular matrix) can be readily accomplished using a combination of transgenic mice expressing select fluorescent markers, exogenous labeling via fluorescent probes, tissue intrinsic fluorescence, and second/third harmonic generated signals. This protocol describes 1) the preparation of neutrophils for adoptive transfer into the mouse ear, 2) different settings for optimal sub-cellular imaging, 3) strategies to minimize motion artifacts while maintaining a physiological response, 4) examples of membrane remodeling observed in neutrophils using ISMic, and 5) a workflow for the quantitative analysis of membrane remodeling in migrating neutrophils in vivo.
Introduction
Directed cell migration is a critical event occurring during different physiological and pathological processes, including development, immune response, tissue repair, and tumor initiation, progression, and dissemination1,2. This process relies on specific extracellular chemotactic signals sensed by their cognate receptors at the plasma membrane and then transduced into intricate intracellular signals. These pathways, in turn, activate cell migration through a series of responses, including local activation of the cytoskeleton, membrane trafficking and remodeling, and cell polarization3. Two distinct types of cell migration have been well characterized: mesenchymal and amoeboid4. Mesenchymal migration is relatively slow (<1 µm/min), relies on strong adhesion between cells and the extracellular matrix (ECM), and depends on metalloproteases-induced matrix degradation. In contrast, ameboid migration, a characteristic of immune cells responding rapidly to inflammatory cues, is faster (>10 µm/min) and utilizes weak adhesions to navigate through the ECM. Regardless of the modality, cell migration requires a constant remodeling of the plasma membrane resulting in the generation of protrusive structures at the leading edge of the cells, which are tightly coordinated with the retraction of the membrane at the rear1.
To unravel the molecular mechanisms underlying these sub-cellular processes, it is fundamental to visualize the dynamics of the membranes in migrating cells at an appropriate temporal and spatial resolution. Moreover, since membrane remodeling is strongly influenced by the properties of the tissue microenvironment surrounding the migrating cells, it is crucial to image this process directly in the native tissue, within live animals. This is accomplished by using intravital microscopy (IVM)5. The very first IVM imaging was performed ~200 years ago, when leukocytes extravasation was visualized using a simple trans-illumination microscope6 and thereafter IVM was used primarily on semi-transparent model organisms, such as Zebrafish and Drosophila7,8. In the last two decades, IVM has been successfully used to investigate cellular processes in mice, rats, and larger mammals such as pigs5,9. This was made possible due to 1) significant developments in confocal and multi-photon (MP) microscopy and 2) the raise of gene-editing technology such as CRISPR-Cas9, which has permitted the rapid engineering of a variety of animal models to express fluorescently tagged proteins and reporters. Besides, the visualization of individual cells and their microenvironment during processes such as tumor initiation, cell migration, and immune response has been made possible by both other forms of exogenous labelings, such as the systemic introduction of dyes to label the vasculature10,11, and the excitation of endogenous molecules, such as collagen through Second Harmonic Generation (SHG)10,12 and nerve fibers through Third Harmonic Generation (THG)11,13. Finally, an improvement in the techniques to minimize the motion artifacts due to heartbeat and respiration has led to the development of intravital subcellular microscopy (ISMic), which has allowed researchers to image and investigate several subcellular events directly in live animals at a level of resolution similar to those observed in vitro14,15,16,17. Examples of ISMic include investigating cytoskeletal dynamics during exocytosis14,15,16,17 and endocytosis15, cytoskeleton dynamic remodeling during cell migration17,18, mitochondrial localization and metabolism19,20, and calcium signaling in the brain21.
This protocol details the different steps and procedures to investigate cell membrane remodeling at a subcellular resolution during neutrophil migration in the ear skin of a live mouse using ISMic. This approach is based on previously described protocols22,23 and adapted to achieve higher spatial and temporal resolution.
Protocol
All animal experiments and procedures were approved by the National Cancer Institute (National Institutes of Health, Bethesda, MD, USA) Animal Care and Use Committee (protocols LCMB-031 and LCMB-035) and were compliant with all relevant ethical regulations. Both male and female mice aged between 2 and 6 months were used for the experiments. mT/mG and wild-type (WT) host mice are in an FVB/NJ background while LyzM-Cre x mTmG mice are in a C57BL/6 background.
1. Materials and preparation of reagents and tools
- Coat tubes, dishes, and needles/syringes with PBS + 1% BSA (without Calcium and Magnesium) overnight under agitation.
- Carefully clean surfaces and tools/instruments to be used for animal work using either 70% ethanol or by autoclaving.
- For neutrophils purification, prepare the following: 3 x 15 mL tubes, 1 x 60 mm dish, 1 x 1.5 mL tube; HBSS containing 10 mM HEPES (pH 7.3); red blood cell lysis solution (ACK); Histopaque 1077; and Histopaque 1119.
- For neutrophil injections, prepare the following: 1 x low volume syringe (10 μL), 1 x 33 G beveled needle, isoflurane, and anesthesia solution (Ketamine, Xylazine, and Acepromazine at 80 mg·kg-1, 2 mg·kg-1 and 4 mg·kg-1, respectively, in saline solution).
2. Purification of neutrophils from a donor mouse
- Euthanize the donor mouse according to the local institutional regulations.
- Collect long bones from the animal (femur, tibia, and humerus), paying attention to preserving bone integrity and avoiding cutting the bones' heads during collection24.
- In a BSA-coated 60 mm dish, remove muscles and fat tissue. Cut the bones' heads to get access to the marrow, and then flush and homogenize the bone marrow using a syringe (27 G needle) filled with HBSS.
- Filter the solution into a 15 mL BSA-coated tube using a 40 µm strainer and centrifuge it at 400 x g for 5 min at room temperature (RT).
- Aspirate the supernatant, resuspend the pellet in 1 mL of ACK for 30 s to lyse the red blood cells, and then add 9 mL of HBSS to stop the lysis. Centrifuge the solution at 400 x g for 5 min at RT.
- Prepare a density step gradient in a 15 mL BSA-coated tube by gently placing 4 mL of 1119 Histopaque at the bottom of the tube, and then gently layering 4 mL of 1077 Histopaque on the top. Take extra care to avoid mixing the two layers.
- Aspirate the supernatant and resuspend the cell pellet in 2 mL of HBSS. Gently layer the solution on top of the step gradient prepared above. Spin at 1,000 x g for 30 min at RT with the lowest possible acceleration and deceleration speed ("no brake" is preferred).
- From the top of the tube, gently aspirate half of the 1077 Histopaque layer. Collect the remaining half of the 1077 Histopaque layer and the top half of the 1119 Histopaque layer into a fresh BSA-coated tube. The cloudy cell suspension between the two density layers contains predominantly neutrophils.
- Add HBSS to reach a final volume of 15 mL and gently mix by inverting the tube. Spin at 400 x g for 5 min at RT.
- After centrifugation, resuspend and wash the cell pellet with 10 mL of HBSS and spin at 400 x g for 5 min at room temperature. Repeat the procedure, and then resuspend the pellet in 15 mL of HBSS to determine the number of cells using a cell counter. Finally, spin and resuspend cells in 1 mL of HBSS and transfer to a 1.5 mL BSA-coated tube.
- After purification, keep the neutrophil suspension in the BSA-coated tube under gentle agitation for 30 min on a tube rotator at RT until labeling and/or injection.
NOTE: A successful purification yields 10-20 x 106 neutrophils per mouse with a 95% purity. Purity is assessed via flow cytometry, using previously described neutrophil markers (Ly6G+, Ly6C-, Gr1+, and CD11b+)25,26.
NOTE: Protocols on neutrophil purification from human peripheral blood and mouse bone marrow are available previously24,27.
3. Neutrophil labeling
NOTE: To visualize the neutrophils, they were purified from LyzM-Cre mT/mG mice, in which myeloid cells express a membrane-targeted peptide fused with the GFP. In addition, purified neutrophils from wild-type (WT) mice were labeled in vitro. The following steps describe the procedure used to label purified neutrophils with a commercial cell-permeable green fluorescent dye and can be adapted to any other MPM compatible fluorescent probe of choice.
- Add the green fluorescent dye (manufacturer's recommended concentration; here, 1 µM) to the neutrophil suspension (purified from WT mouse) in a 1.5 mL BSA-coated tube. Incubate for 30 min at RT under gentle agitation in a tube rotator.
- Wash three times using HBSS, spin at 400 x g for 5 min at RT, and resuspend the pellet in 1 mL of HBSS.
- Keep the neutrophil suspension in the BSA-coated tube under gentle agitation in a tube rotator at RT until the injection procedure.
NOTE: Injection of labeled neutrophils performed right after labeling is highly preferred. However, if necessary, labeled neutrophils can be kept under agitation for an additional 10-15 min post-labeling without compromising the integrity of neutrophil response in vivo. Long-term agitation (more than 60 mins post labeling) will lead to clumping, neutrophil death, and misleading observations in vivo. - Just before the injection, resuspend the cell pellet in saline solution to reach a density of 2-5 x 106 cells per 20 µL.
4. Neutrophil injection
NOTE: Anesthesia must be performed following the local institutional Animal Care and Use Committee guidelines.
- Place the animal in a closed chamber and administer 3%-5% isoflurane (flow rate of 2 L/min) using a calibrated vaporizer connected to an oxygen concentrator.
- Assess whether the animal is unconscious by testing paw withdrawal reflex upon pinching the hind footpad. Then, transfer the animal to a warming pad (37° C) to maintain its body temperature. Continue to administer the isoflurane (1%-2%; flow rate of 0.5 L/min) through a nose cone.
SAFETY NOTE: To limit personnel exposure to toxic isoflurane, the use of a dispenser system equipped with a filtering unit to recapture circulating isoflurane is recommended. - To optimize imaging quality, remove the hairs from the ear using a fine trimmer. This procedure could also be performed a day or two before the experiment.
NOTE: Use of hair-removal cream is discouraged since it is known to exacerbate inflammatory response in the mouse skin and introduce imaging artifacts. - Place the animal on its side. Using surgical tape, hold the edge of the ear, and flatten it along the warming pad. Gently apply the tape to prevent any damage to the ear.
- Fill a syringe equipped with a 33 G needle with the neutrophil suspension (2-5 x 106 cells per 20 µL) and mount it on a micromanipulator.
NOTE: The injection angle between the needle and the ear on a warming pad must be between 10° and 30° to minimize tissue damage and increase the success of cell injection. - Gently move the needle (with the bevel up) toward the ear, and slowly pierce the skin. Once the needle is inside the skin, slowly push the piston and deliver 2-3 µL of cell suspension.
NOTE: Areas with visible blood vessels should be avoided as damaging a blood vessel results in an undesired and complex immune response to injection and issues with the imaging. - Repeat the injection in 2-3 different areas of the ear, preferably 2-3 mm apart. Do not "overfill" the ear with the cell suspension.
NOTE: The injection must be performed in the center of the ear. The periphery of the ear is thin and must be handled with care as it can be easily damaged, whereas the center part is thicker containing larger blood vessels and fewer hair follicles, and can withstand larger injection volumes. - Carefully remove the surgical tape and let the animal regain consciousness under supervision. Allow the animal to rest for 1 h before imaging to let the tissue and the cells recover from the injection procedure.
5. Anesthesia for IVM
NOTE: A deeper anesthesia significantly improves the imaging quality by decreasing the motion artifacts due to heartbeat and respiration. Neutrophil-injected mice are subjected to chemical anesthesia, which provides deeper sedation than gas-induced anesthesia. Anesthesia must be performed following the local institutional Animal Care and Use Committee guidelines.
- One hour after recovery from the neutrophil injection, anesthetize the animals by a subcutaneous injection containing a mixture of Xylazine/Ketamine/Acepromazine (80 mg·kg-1, 2 mg·kg-1, and 4 mg·kg-1, respectively) in saline solution.
NOTE: To ensure reproducibility and reliability of the results, animals must be kept warm from the time of anesthesia to the end of the experiment using a heating pad or water circulating pad connected to a warming pump. - To maintain anesthesia beyond 1 h, inject a mixture of Ketamine/Acepromazine (40 mg·kg-1, and 2 mg·kg-1 respectively) subcutaneously every 40 min. Monitor anesthetized mice periodically for signs of consciousness by testing paw withdrawal reflex.
- Additionally, for imaging procedures lasting more than 1 h, apply a sterile non-medication ophthalmic ointment to the eyes to prevent corneal drying during anesthesia and maintain body hydration by subcutaneous injections of saline solution (200 µL every hour).
NOTE: Alternately, a digitally controlled perfusion-based system can be used to ensure a periodic and smooth injection of the anesthesia solution and/or provide fluids. An automated syringe driver can be set up to deliver the desired volume of anesthetic and provide hydration for longer periods. To this end, a winged infusion needle can be inserted subcutaneously into the dorsal skin of the animal and secured with surgical tape.
6. Imaging
NOTE: The animal setup and the imaging parameters described here are optimized for an inverted multiphoton microscope equipped with a single laser line to excite and simultaneously collect emissions from the different fluorophores.
- Before placing the anesthetized animal on the microscope stage, ensure that the microscope is turned on and that both stage and lenses are pre-heated to 37 °C. Cover the hole on the stage with a glass coverslip (thickness #1 or 1.5) that is aligned with the heated lens.
- Move the animal onto the stage carefully. If imaging is planned for a long duration (>1 h), apply ophthalmic ointment and secure the winged infusion-based perfusion system as described above.
- Place a drop of saline on the center of the coverslip and place the neutrophils-injected ear on top of it. Gently flatten the ear using a sterile cotton swab across the center of the coverslip to remove air pockets.
NOTE: Application of saline (or PBS) between the ear and coverslip reduces light refraction due to air pockets, thereby ensuring even refractive index and superior imaging quality. - Secure the ear by gently pressing a wooden stick (removed from the cotton swab) to the side of the ear closer to the animal's head and locking it using tape (Figure 1A).
NOTE: A secured wooden stick minimizes motion artifacts due to heartbeat and respiration and dramatically increases the imaging quality. Importantly, the wooden stick must be fastened just enough to stabilize the ear without impairing the blood flow. - Using the microscope eyepiece, find an area of interest, and then switch to multiphoton acquisition mode.
- Set the laser excitation wavelength to 900-930 nm to allow simultaneous acquisition of GFP/green fluorescent dye, mTomato as well as collagen-I (via SHG).
- Set appropriate set of mirrors and filter combination on the detectors to collect the emitted light (bandpass filters: Blue = 410-460 nm, Green = 495-540 nm, Red = 580-640 nm).
NOTE: Injected neutrophils, when labeled with a green-fluorescent dye, exhibit a strong green fluorescence making them visible under the microscope eyepiece even if injected into deeper portions of the ear. - Determine the setting most appropriate to the hardware configuration. Even if migrating cells can be tracked with a 30 s to 1 min interval, image subcellular events with a higher speed (at least <10 s interval). In the example below, two different imaging setups are presented to show the difference between classical IVM and ISMic.
- In the classical IVM approach to track cell migration and investigate the host mouse tissue (Figure 1), use a 30x lens and a galvo scanner with an image size of 512 x 512 pixels (pixel size of 0.83 µm), and set z-axis displacement using the motorized stage with a step size of 2 µm, allowing imaging of a 30 µm deep volume every 30 s.
- In the ISMic approach to image highly dynamic membrane remodeling at a higher magnification and resolution (Figure 2), use a 40x lens and a resonant scanner with 3x averaging and image size of 512 x 512 pixels (pixel size of 0.25 µm), and set z-axis displacement using a piezo with a step size of 1 µm, allowing imaging of 20 µm deep volume every 4-5 s.
- Trigger neutrophil migration by inducing a sterile laser injury. Focus the excitation laser at a high power sufficient to reach at least 80 mW22, on a narrow area of the tissue (20 µm x 20 µm) for 10 s.
- Identify the laser-induced injury by its strong auto-fluorescent signals appearing in all the channels and by the resulting alteration of collagen arrangement (Figure 1D).
NOTE: To obtain reliable results, the injury must always be induced as far as possible from the blood vessels; if ruptured, blood cells released into the tissue will impact imaging quality as well as result in a strong overall immune reaction. - Save the data at the end of the experimentation for further analyses.
- Euthanize the animal according to the local institutional guidelines. If needed, tissue can be collected for fixation and further processing.
7. Representative data analysis
NOTE: Data can be visualized and analyzed with either the microscope software, third-party software, or customized programs. The procedure and the tools utilized depend on the specific needs of the investigators. Shown here is one example of a workflow to quantify membrane curvature and local area changes at the leading edge and the rear of the migrating neutrophils.
- Open the imaging files with Imaris or Fiji to visualize the migration of the neutrophils in 4D and determine the quality of the experiment.
NOTE: Imaris and Fiji can read the imaging format of several microscope brands. - Process the data with the following steps by running customized MATLAB code scripts. Read the selected imaging files with the Bio-Formats28 package for MATLAB. Segment each frame of the 3D volume with the Otsu algorithm29 to identify the individual cells.
- Track the identified objects based on the minimum displacement criterion30.
- Determine object's active contours and boundaries.
- Generate a max-projection for each object identified in a time frame.
- Apply the bwboundaries function in MATLAB to the max-projection of each identified object from step 7.2 to generate 100 boundry points.
- Optimize the boundary points identified with the dynamic contour algorithm to achieve subpixel precision31 (Figure 3A) using the functions in the MATLAB package downloaded via http://www.iacl.ece.jhu.edu/static/gvf/.
- Overlay the boundaries for the object and the object trajectory index determined by the tracking in step 7.3 with the max-projection of the original image to generate an output of a 2D overlayed movie for visualization and manual check.
- In the overlayed movie, manually select all the objects identified as migrating cells by excluding non-cell objects, touching objects, and objects with incomplete boundaries. Record the tracking indices, start, and end frames of each cell trajectories in a spread sheet to be used as the input for the following steps implemented by MATLAB code scripts.
- Track the cell boundary points from each frame to the next one according to the minimum displacement criterion. Use the Polyarea function in MATLAB to calculate the area underlined by adjacent boundary points in two consecutive time frames (Figure 3C). If the local cell boundary moves outward from the cell, assign a positive sign to the area change. If the local cell boundary moves inward to the cell, assign a negative sign to the area change.
- Measure the local membrane curvature at each boundary point for each frame by fitting its neighbor boundary points to a circle (5 per side; total 11)32 (Figure 3B).
- Generate kymographs using the imagesc function in MATLAB (Figures 3D,E) to show the changes of the subcellular features over time and relate to the position of the cell.
Representative Results
Here, two different sets of results are presented to illustrate classical IVM and ISMic that provide cellular and sub-cellular resolution, respectively. In the first example, neutrophils were purified from a wild-type (WT) mouse, labeled with Cell Tracker Green to stain the cytoplasm, and injected into a transgenic mouse expressing a fluorescent protein targeting the plasma membrane (mTomato mouse, also known as mT/mG33, Figure 1 and Movie 1 and Movie 2). This recipient mouse enabled the visualization of structural features in the ear tissue such as blood vessels, resident cells, and hair follicles (Figure 1C and Movie 1) via IVM-based approach (step 6.8.1). Collagen fibers, revealed via SHG (detected at half the wavelength of the excitation), were arranged in an intricate network in the dermis, where neutrophils were injected. Along the edge of the hair follicles (HF), a layer of epithelial cells (i.e., keratinocytes) was observed. Occasionally, artifacts from residual hairs resulting in a local depression of the skin were visualized (H). The laser-induced injuries were easily visualized due to their strong auto-fluorescence detected in all the channels and by the alterations of the collagen arrangement (Figure 1D). A more complete view of the 3D architecture of the skin and the localization of the injected neutrophils can be appreciated in Movie 1, which portrays a Z-stack of the skin from the outer to inner layers and a 3D volume rendering. Time-lapse imaging showed the neutrophils sampling the ear skin and interacting with the ECM and host tissue (Movie 2). Imaging at this resolution and acquiring a Z stack every 30 s allows performing cell tracking, and measurement of motility parameters (i.e., speed and directionality), but a precise and detailed analysis of membrane remodeling is challenging at this resolution.
In the second example, membrane remodeling was assessed via the ISMic approach (step 6.8.2) using mGFP-expressing neutrophils purified from LyzM-cre mT/mG mice and injected into WT animals (Figure 2A, experimental flowchart). Using the ISMic protocol (Movie 3 and still images in Figure 2B) and upon laser injury, dynamic remodeling of the plasma membrane is observed during migration, and formation of membrane protrusions at the leading edge and the retraction of the rear of the cells are clearly visualized (Figure 2 and Movie 3). The time-lapse sequences highlighted in Movie 3 reveal the complexity of the interactions with the ECM. Indeed, neutrophils migrated through the interstitial space moving either along the fibers or in the spaces between them. Finally, quantitative aspects such as the changes in curvature and the area changes at the front and the back of the cells were quantified for each time point. Using the cell described in Figure 2B as an example, the local dynamics of the plasma membrane were analyzed using an algorithm pipeline (see step 7) based on the identification of 100 boundary points underlying the cell surface (Figure 3A). The changes in the local curvature (Figure 3B) and in the area underlined by plasma membrane protrusions (Figure 3C) were calculated for each boundary point and reported for each time frame as kymographs (Figure 3D,E). Both the front and the back of the cells maintain higher curvature than the side of the cells (Figure 3D); negative area changes (retractions, blue regions) are more apparent at the back of the cells than at the leading edge where the positive area changes are more prominent (protrusions, red regions) (Figure 3E).

Figure 1: IVM of neutrophils in the mouse ear skin. (A) Schematic drawing of the ear set-up on the microscope stage. (B) Experimental flowchart. (C) Representative image of the ear skin of a mTomato mouse injected with fluorescently labeled neutrophils, with different projections. Hair follicle (HF), Blood Vessel (BV), Epidermis (E), Dermis (D), Basal Cell Layer (BCL), and Hair artifact (due to a residual hair at the skin surface, H). (D) Representative image of mouse skin after a sterile laser injury: blue channel (right), green and red channels (middle), merged image (left). Injury is visible on the left of the image by a strong autofluorescence emission in both green and red channels as well as a disruption of the ECM observed by the formation of a hole in the collagen-I fibers. In C and D, Green: neutrophils; Red: mouse host tissue; Cyan: Collagen-I SHG. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Time-lapse of mGFP neutrophil migrating in a WT mouse ear. (A) Experimental flowchart. (B) Representative still images from Movie 3. (C) Volume rendering of the same cell to visualize membrane 3D organization. The area of high membrane dynamics is illustrated by an arrow (red for retraction and blue for protrusion). (D) Close-up and tilted visualization of the rendered cell volume. Please click here to view a larger version of this figure.
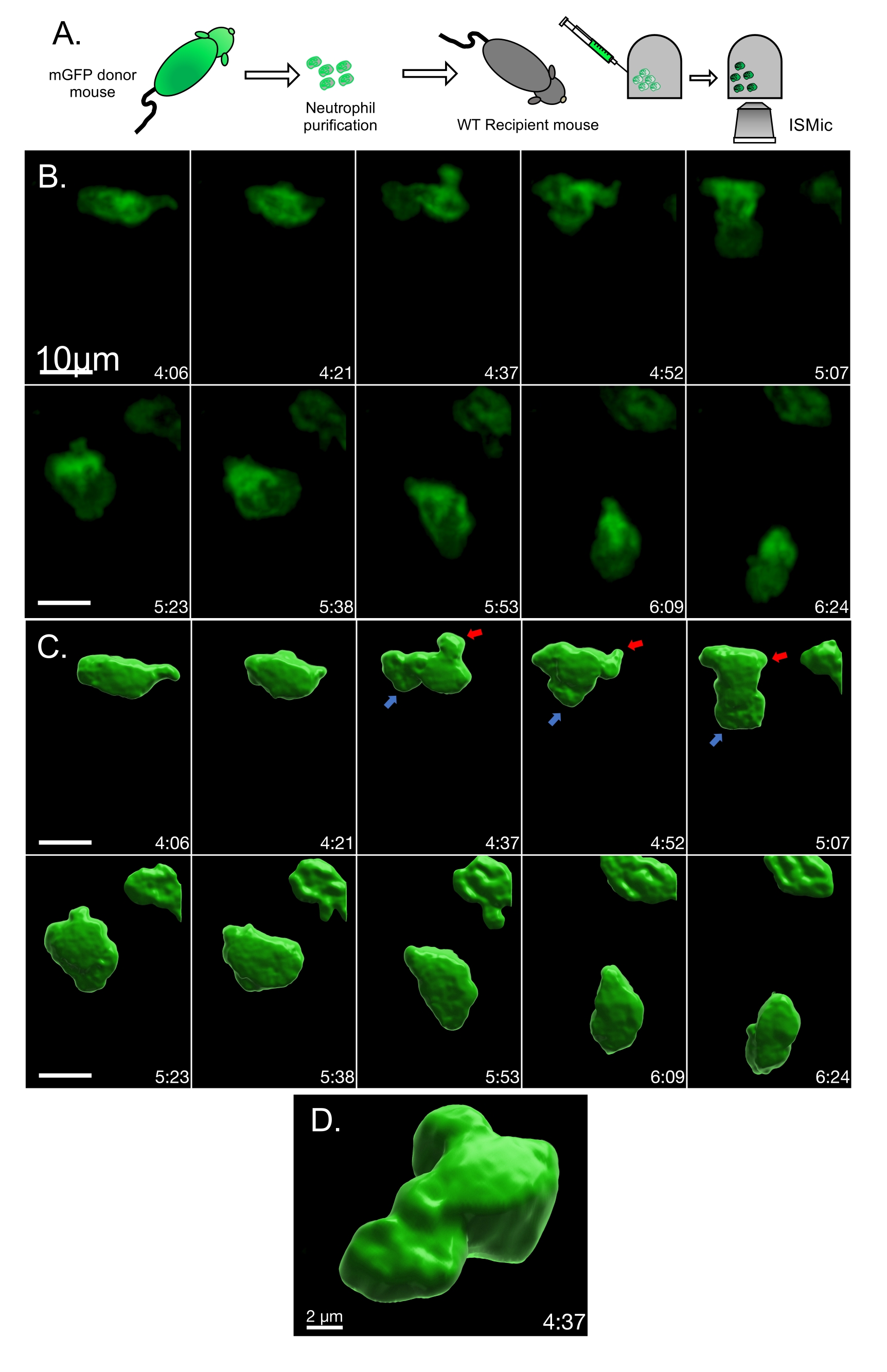{kind=link}

Figure 3: Analysis pipeline for quantification of membrane dynamics collected using ISMic. (A) Cell contour determination and boundary points repartition (100 boundary points) between two consecutive frames for mGFP neutrophil described in Figure 2B. (B) Colored boundaries represent local membrane curvature determined for every boundary point. (C) Local area changes between two consecutive frames (current: blue, and next: red). The green areas represent the tracking results of the local membrane motion. Yellow arrows indicate the general direction of the membrane displacement. (D) Boundary curvature kymograph of the cell over time. The vertical axis represents the boundary points indices where 1 and 100 represent the rear of the cell according to its migration direction. (E) Local area change kymograph reflecting the membrane protrusion (red) and the membrane retraction (blue) of the cell over time. Please click here to view a larger version of this figure.
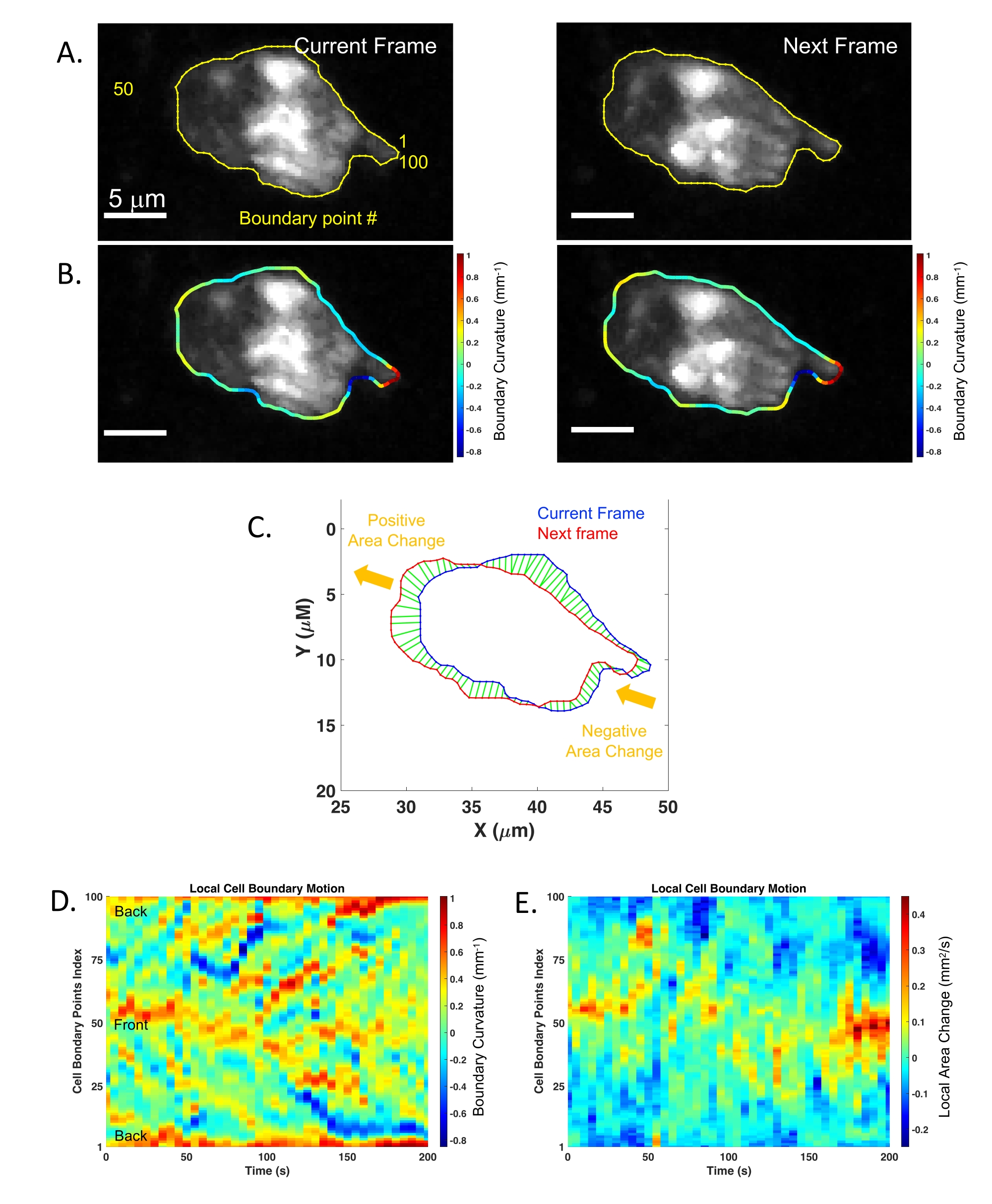{kind=link}
Movie 1: Green labeled neutrophils visualized in a mTomato mouse ear using IVM. Red: mTomato host mouse tissue. Cyan: Collagen-I SHG; Green: neutrophil. Please click here to download this Movie.
Movie 2: IVM of neutrophils migrating in a mTomato mouse ear skin. Videos are maximum-intensity projections of an image stack acquired for ῀27 min with a frame rate of 5 frames/s. Red: mTomato host mouse tissue. Cyan: Collagen-I SHG; Green: neutrophil. Please click here to download this Movie.
Movie 3: ISMic of membrane remodeling during neutrophil migration in a WT mouse ear. Videos are maximum-intensity projections of an image stack acquired for ῀8 min with a frame rate of 10 frames/s. Red: mTomato host mouse tissue. Cyan: Collagen-I SHG; Green: mGFP neutrophil; Light green: rendered neutrophil. Please click here to download this Movie.
Discussion
Despite decades of advances in the field of cell migration and membrane remodeling, very few studies have employed IVM to visualize sub-cellular features in live animals. The procedures described in this protocol provide a powerful tool to gain novel insights into neutrophil migration in live animals and more specifically on plasma membrane remodeling during this process. This approach makes it possible to investigate neutrophil migration under physiological conditions considering the inherent complexity of the tissue microenvironment. Indeed, the use of MPM enables the visualization of multiple features within the host tissue by using a combination of mouse strains expressing selected fluorescent markers, exogenous tissue labeling, excitation of endogenous fluorescence, and signals generated through SHG or THG12,13.
One potential issue to consider in this procedure is photodamage. Even though MPM is generally safer than confocal microscopy with respect to photobleaching and phototoxicity, care must be taken when imaging live tissues34. Phototoxicity can drastically hamper the results by either creating imaging artifacts ranging from small bright speckles to larger areas of damaged tissue or inhibiting/stimulating a variety of intracellular pathways. To prevent phototoxicity, laser power should be kept at a minimum and the appropriate controls have to be designed to verify that physiological conditions are maintained (e.g., measuring blood flow, probes for oxidative stress)35,36. Moreover, in this specific procedure, which involves the skin, albino strains are highly recommended as the melanin present in dark-coated animals is more sensitive to phototoxicity36,37,38.
Among the main limitations of the procedure are the microscope system used and the nature of neutrophils. Although using MPM significantly increases the depth of tissue imaging when compared with other light microscopy techniques (e.g., confocal, spinning disk), the ability to visualize subcellular dynamics in the ear is restricted to the outmost layers of the tissue (80-100 µm). This is due to the intense light scattering produced by the thick ECM layer making it difficult to investigate migration in the deeper layers. Another limitation is the fact that neutrophils are very short-lived and cannot be kept in culture long enough to perform gene-editing techniques. This can be overcome by engineering mice to produce neutrophils lacking specific genes or harboring selected mutations, which of course increases the research costs and duration.
The procedures described here, although designed to investigate membrane dynamics, can be tailored to address any cell biological question not only in neutrophils but also in other immune cell types and migratory cells. The information gathered through low magnification IVM on cellular behaviors (e.g., migratory phenotype, cell speed, and directionality22) can be complemented and correlated with mechanistic information acquired through ISMic on organelle repositioning, protein secretion, endocytosis, nuclear dynamics, calcium dynamics, cytoskeleton organization, and netosis. The use of pharmacological and/or genetic manipulations can highlight the role of specific molecular pathways in the process of interest making this a unique and very powerful approach.
Disclosures
The authors declare no competing financial interests.
Acknowledgements
This research was supported by the intramural research program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL tubes | USA Scientific | 4036-3204 | |
| 15 mL tubes | Corning | 430766 | |
| 1 mL syringe | Covidien | 8881501400 | |
| 27 G needle | Kendall | 827112 | |
| 27 G winged infusion set | Terumo | SV*27EL | |
| 30x objective | Olympus | UPLSAPO30XS | 1.05 NA, silicon oil immersion |
| 40x objective | Olympus | UPLSAPO40XS | 1.23 NA, silicon oil immersion |
| 60 mm dishes | Falcon | 353002 | |
| 6 mL syringe | Kendall | 8881516937 | |
| Acepromazine (10 mg/mL) | Vet one | 13985-587-50 | |
| ACK lysis buffer | Quality Biological | 118-156-101 | |
| Balance | AND | EK-1200A | |
| BSA | Sigma Aldrich | A9647 | |
| Cell strainer 40 µm | Sigma Aldrich | CLS431750 | |
| Fiji | ImageJ | N/A | Image visualization/analysis software |
| Fluoview Software | Olympus | N/A | Acquisition software |
| FVB mouse strain | Jackson | N/A | FVB background |
| Gas Anesthesia system | Patterson veterinary | 07-8915712 | Link 7 model |
| Green Cell tracker | Thermo | C2925 | Solubilized in cell culture grade DMSO to reach 1 mM concentration (1000x) |
| Hair removal cream | Nair | N/A | |
| HBSS (w/o Ca2+, Mg2+) | Gibco | 14175-095 | |
| HEPES 1 M pH 7.3 | Quality Biological | 118-089-721 | |
| Histopaque 1077 | Sigma Aldrich | 10771-100ML | |
| Histopaque 1991 | Sigma Aldrich | 11191-100ML | |
| Imaris | Bitplane | N/A | Image visualization/analysis software |
| Isoflurane | Vet one | 13985-528-40 | |
| Ketamine (100 mg/mL) | Vet one | 13985-584-10 | |
| LyzM-cre x mT/mG | generated in the lab | N/A | C57BL/6J background |
| Manual micromanipulator | WPI | M3301R | |
| MATLAB | MatWorks | N/A | Analysis software |
| mtomato mouse strain | generated in the lab | N/A | mT/mG, FVB background |
| Multiphoton laser | Spectra Physics | Insight DS+ | |
| Multiphoton Microscope | Olympus | MPE-RS | |
| Nanofil 10 µL syringe | WPI | NANOFIL | |
| Nanofil 33 G needle | WPI | NF33BV-2 | |
| Objective heater | Bioptechs | N/A | |
| Objective heater controller | Bioptechs | 150803 | |
| Ophtalmic ointment | Major | NDC 0904-6488-38 | |
| Oxygen concentrator | Caire | VisionAire 5 | |
| PBS (w/o Ca2+, Mg2+) | Quality Biological | 114-058-131 | |
| Saline | Quality Biological | 114-055-101 | |
| Stage heater | Okolab | N/A | |
| Stage heater controller | Okolab | H401-T | |
| Surgical tape | 3M | 1538-1 | Hypoallergenic |
| Syringe driver | Harvard Apparatus | PHD Ultra | |
| Warming Pads | Parkland Scientific | A2789B | |
| Warming Pump | Parkland Scientific | TP-700 | |
| Xylazine (100 mg/mL) | Vet one | 13985-704-10 |
References
- Trepat, X., Chen, Z., Jacobson, K. Cell migration. Comprehensive Physiology. 2 (4), 2369-2392 (2012).
- Oudin, M. J., Weaver, V. M. Physical and chemical gradients in the tumor microenvironment regulate tumor cell invasion, migration, and metastasis. Cold Spring Harbor Symposia on Quantitative Biology. 81, 189-205 (2016).
- Devreotes, P., Horwitz, A. R. Signaling networks that regulate cell migration. Cold Spring Harbor Perspectives in Biology. 7 (8), 005959 (2015).
- Alexandrova, A. Y., Chikina, A. S., Svitkina, T. M. Actin cytoskeleton in mesenchymal-to-amoeboid transition of cancer cells. International Review of Cell and Molecular Biology. 356, 197-256 (2020).
- Masedunskas, A., et al. Intravital microscopy. Bioarchitecture. 2 (5), 143-157 (2012).
- Wagner, R. . Explanatory panels on physiology and development history 1839. , (1839).
- Yaniv, K., et al. Live imaging of lymphatic development in the zebrafish. Nature Medicine. 12 (6), 711-716 (2006).
- Vinegoni, C., et al. Mesoscopic fluorescence tomography for in-vivo imaging of developing Drosophila. Journal of Visualized Experiments. (30), e1510 (2009).
- Sack, F. -. U. Intravital microscopy of pulmonary microcirculation after single lung transplantation in pigs. Transplantation Proceedings. 38 (3), 737-740 (2006).
- Rehberg, M., Krombach, F., Pohl, U., Dietzel, S. Label-free 3D visualization of cellular and tissue structures in intact muscle with second and third harmonic generation microscopy. PloS One. 6 (11), 28237 (2011).
- Weigelin, B., Bakker, G. -. J., Friedl, P. Intravital third harmonic generation microscopy of collective melanoma cell invasion: Principles of interface guidance and microvesicle dynamics. Intravital. 1, 32-43 (2012).
- Poole, J. J. A., Mostaço-Guidolin, L. B. Optical microscopy and the extracellular matrix structure: A review. Cells. 10 (7), 1760 (2021).
- Weigelin, B., Bakker, G. -. J., Friedl, P. Third harmonic generation microscopy of cells and tissue organization. Journal of Cell Science. 129 (2), 245-255 (2016).
- Ebrahim, S., et al. Dynamic polyhedral actomyosin lattices remodel micron-scale curved membranes during exocytosis in live mice. Nature Cell Biology. 21 (8), 933-939 (2019).
- Shitara, A., et al. Cdc42 negatively regulates endocytosis during apical membrane maintenance in live animals. Molecular Biology of the Cell. 30 (3), 324-332 (2019).
- Masedunskas, A., et al. Role for the actomyosin complex in regulated exocytosis revealed by intravital microscopy. Proceedings of the National Academy of Sciences of the United States of America. 108 (33), 13552-13557 (2011).
- Subramanian, B. C., et al. The LTB4-BLT1 axis regulates actomyosin and β2-integrin dynamics during neutrophil extravasation. The Journal of Cell Biology. 219 (10), 201910215 (2020).
- Yan, S. L. S., Hwang, I. -. Y., Kamenyeva, O., Kehrl, J. H. In vivo F-Actin filament organization during lymphocyte transendothelial and interstitial migration revealed by intravital microscopy. iScience. 16, 283-297 (2019).
- Porat-Shliom, N., et al. In vivo tissue-wide synchronization of mitochondrial metabolic oscillations. Cell Reports. 9 (2), 514-521 (2014).
- Takihara, Y., et al. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10515-10520 (2015).
- Calvo-Rodriguez, M., et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer's disease. Nature Communications. 11, 2146 (2020).
- Lämmermann, T., et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 498 (7454), 371-375 (2013).
- Li, J. L., et al. Intravital multiphoton imaging of immune responses in the mouse ear skin. Nature Protocols. 7 (2), 221-234 (2012).
- Swamydas, M., Lionakis, M. S. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. Journal of Visualized Experiments:JoVE. (77), e50586 (2013).
- Rose, S., Misharin, A., Perlman, H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytometry. Part A: The Journal of the International Society for Analytical Cytology. 81 (4), 343-350 (2012).
- Lakschevitz, F. S. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Experimental Cell Research. 342 (2), 200-209 (2016).
- Oh, H., Siano, B., Diamond, S. Neutrophil Isolation Protocol. Journal of Visualized Experiments:JoVE. (17), e745 (2008).
- . MATLAB - Bio-Formats 6.1.0 documentation Available from: https://docs.openmicroscopy.org/bio-formats/6.1.0/users/matlab/index.html (2022)
- Otsu, N. A Threshold selection method from gray-level histograms. IEEE Transactions on Systems, Man, and Cybernetics. 9, 62-66 (1979).
- Crocker, J. C., Grier, D. G. Methods of digital video microscopy for colloidal studies. Journal of Colloid and Interface Science. 179, 298-310 (1996).
- Xu, C., Prince, J. L. Snakes, shapes, and gradient vector flow. IEEE Transactions on Image Processing: A Publication of the IEEE Signal Processing Society. 7 (3), 359-369 (1998).
- Driscoll, M. K., et al. Automated image analysis of nuclear shape: what can we learn from a prematurely aged cell. Aging. 4 (2), 119-132 (2012).
- Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L., Luo, L. A global double-fluorescent Cre reporter mouse. Genesis. 45 (9), 593-605 (2007).
- Tauer, U. Advantages and risks of multiphoton microscopy in physiology. Experimental Physiology. 87 (6), 709-714 (2002).
- Débarre, D., Olivier, N., Supatto, W., Beaurepaire, E. Mitigating phototoxicity during multiphoton microscopy of live Drosophila embryos in the 1.0-1.2 µm wavelength range. PloS One. 9 (8), 104250 (2014).
- Masedunskas, A., et al. Intravital microscopy: a practical guide on imaging intracellular structures in live animals. Bioarchitecture. 2 (5), 143-157 (2012).
- Ng, L. G., et al. Visualizing the neutrophil response to sterile tissue injury in mouse dermis reveals a three-phase cascade of events. The Journal of Investigative Dermatology. 131 (10), 2058-2068 (2011).
- Wu, X. S., et al. Melanoregulin regulates a shedding mechanism that drives melanosome transfer from melanocytes to keratinocytes. Proceedings of the National Academy of Sciences of the United States of America. 109 (31), 2101-2109 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved