
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Neuroscience
Génération robuste et hautement reproductible d’organoïdes cérébraux corticaux pour la modélisation de la sénescence neuronale cérébrale in vitro
Dans cette étude, nous fournissons une technique détaillée pour un système de culture organoïde corticale simple mais robuste utilisant des cultures hPSC standard sans mangeoire. Il s’agit d’un protocole rapide, efficace et reproductible pour générer des organoïdes qui modélisent les aspects de la sénescence cérébrale in vitro.
Les organoïdes cérébraux sont des modèles tridimensionnels du cerveau humain en développement et fournissent une plate-forme convaincante et de pointe pour la modélisation des maladies et le dépistage génomique et médicamenteux à grande échelle. En raison de la nature auto-organisée des cellules dans les organoïdes cérébraux et de la gamme croissante de protocoles disponibles pour leur génération, des problèmes d’hétérogénéité et de variabilité entre les organoïdes ont été identifiés. Dans cet article de protocole, nous décrivons un protocole robuste et reproductible qui surmonte en grande partie ces problèmes et génère des organoïdes corticaux à partir de progéniteurs neuroectodermiques en 1 mois, et qui peut être maintenu pendant plus de 1 an. Ce protocole hautement reproductible peut être facilement réalisé dans une salle de culture tissulaire standard et donne des organoïdes avec une riche diversité de types de cellules que l’on trouve généralement dans le cortex humain en développement. Malgré leur composition développementale précoce, les neurones et autres types de cellules cérébrales humaines commenceront à présenter les signes typiques de sénescence dans les cellules neuronales après une culture in vitro prolongée, ce qui en fait une plate-forme précieuse et utile pour l’étude des processus neuronaux liés au vieillissement. Ce protocole décrit également une méthode pour détecter de telles cellules sénescentes dans les organoïdes cérébraux corticaux en utilisant la coloration à la bêta-galactosidase associée à la sénescence.
Nos connaissances actuelles sur le cerveau humain ont été largement basées sur des modèles animaux et des spécimens de cerveau post-mortem. La biologie des cellules souches est un domaine en évolution rapide qui fournit de nouvelles informations sur la biologie de base du développement du cerveau humain et les moteurs pathologiques des troubles cérébraux humains. Les cellules souches pluripotentes humaines (CSEh) sont un outil inestimable pour modéliser le cerveau humain via la génération d’organoïdes, de tissus tridimensionnels (3D) semblables à des organes qui récapitulent généralement les trajectoires de développement, la composition cellulaire et l’architecture du cerveau humain en développement. Les organoïdes cérébraux sont auto-assemblés et composés de cellules souches neurales, de progéniteurs neuronaux spécifiés, de neurones matures et de types de cellules gliales. Les organoïdes offrent donc une occasion unique d’étudier le cerveau humain primitif, qui est souvent inaccessible pour l’expérimentation directe, mais qui présente également des limites intrinsèques telles que l’absence de système vasculaire et immunitaire.
Les méthodologies pour générer des organoïdes cérébraux ont été poursuivies de deux manières différentes: la différenciation non guidée et la différenciation guidée. Les méthodes organoïdes cérébrales non guidées reposent sur les capacités de différenciation intrinsèque spontanée des cellules souches qui déterminent la morphogenèsetissulaire 1,2 et permettent l’émergence d’une variété d’identités de lignée cellulaire allant du cerveau antérieur, du mésencéphale et du cerveau postérieur au plexus choroïde, à la rétine et au mésoderme. En revanche, les méthodes d’organoïdes cérébraux guidés nécessitent une utilisation substantielle de facteurs externes pour conduire les CSEh vers le modèle souhaité de lignées neuronales représentant un type de région cérébrale, telles que l’éminence ganglionnaire médiale3, le cerveau antérieur4, le mésencéphale5, l’hypothalamus6, le cervelet7 et le plexus choroïde8. Cette capacité à générer différentes régions du cerveau avec différentes lignées cellulaires, et le potentiel de les fusionner à volonté, font des organoïdes cérébraux un excellent modèle pour étudier le développement du cerveau humain et déchiffrer les mécanismes sous-jacents des maladies liées au cerveau. Bien que ces méthodes de génération d’organoïdes cérébraux offrent une percée dans la modélisation des régions du cerveau humain, la variabilité et l’hétérogénéité entre les organoïdes restent une limitation importante pour les études systématiques et quantitatives, telles que le dépistage des drogues.
Le protocole actuel est basé sur une méthode développée dans notre récent article9 et implique la différenciation sélective des colonies de hPSC vers l’identité neuroectoderme (NEct) avec des inhibiteurs SMAD doubles (SB-431542 et LDN 193189), qui ont ensuite la capacité de s’auto-organiser en 4 jours en sphéroïdes 3D neuroépithélium sous l’influence de la signalisation FGF2. Ces sphéroïdes neuroépithélium génèrent de manière fiable des organoïdes corticaux homogènes avec une composition cellulaire de type in vivo dans les 4 semaines suivant la différenciation. Le protocole décrit ici est construit sur nos résultats précédents montrant que l’inhibition de la signalisation double SMAD (Suppressor of Mothers Against Decapentaplegic) favorise la différenciation des hPSC vers les cellules souches neurales rostrales dérivées des progéniteurs neuroectodermiques10 en inhibant, entre autres, le choix du devenir des cellules endodermiques, mésodermiques et trophectodermiques11 . De plus, l’incorporation des sphéroïdes du neuroépithélie dans la matrice de membrane basale qualifiée hESC déclenche un bourgeonnement important de la neuroépithélie, formant des ventricules à polarité apicobasale. La culture à grande échelle a montré la reproductibilité et l’homogénéité des organoïdes corticaux indépendamment des lignées cellulaires, des clones ou des lots, et représente donc un système de cellules souches fiable et stable pour imiter le développement cortical humain précoce dans la santé et la maladie in vitro. Nous décrivons en outre un protocole pour détecter les marqueurs de cellules neuronales sénescentes dans les organoïdes cérébraux corticaux dérivés des HSPc qui ont été cultivés pendant de longues périodes.
Après avoir plaqué des CSEh à une densité d’ensemencement de 20% à 30%, les cellules sont traitées avec des inhibiteurs doubles de SMAD pendant 3 jours pour différencier les colonies de hPSC en colonies neuroectodermiques. Ces colonies sont ensuite doucement soulevées avec de la dispase et ensemencées dans des plaques à 6 puits à fixation ultra-faible complétées par du FGF2. Les colonies flottantes 2D s’auto-organisent en sphéroïdes neuroectodermiques 3D pendant la nuit et sont maintenues pendant 4 jours dans un milieu N2 complété quotidiennement par FGF2. Une fois que les sphéroïdes ont établi la couche neuroépithéliale, ils peuvent être intégrés dans la matrice de la membrane basale. En ajoutant régulièrement un nouveau milieu de différenciation terminal, les chercheurs observeront une expansion progressive et un bourgeonnement de neuroépithélie dans les organoïdes corticaux. Les chercheurs pourraient souhaiter dissocier ces organoïdes pour effectuer un profilage transcriptionnel et protéomique. De plus, l’imagerie en champ clair est recommandée pour surveiller la qualité des organoïdes corticaux. L’analyse peut être effectuée par fixation, cryosection et immunocoloration. Des descriptions et des méthodes pour ces techniques ont déjà été décrites12. En fin de compte, ce protocole permet aux chercheurs de générer rapidement et de manière robuste des organoïdes cérébraux corticaux homogènes pour modéliser le cerveau cortical humain en développement, avec un équipement peu coûteux et limité, et pour étudier les aspects de la sénescence neuronale cellulaire, comme indiqué dans cet article.
1. Génération d’organoïdes cérébraux corticaux
REMARQUE : Toutes les étapes de cette section du protocole se dérouleront dans une hotte de biosécurité de classe 2, sauf indication contraire.
- Induction de colonies neuroectodermiques 2D à partir de la culture hPSC 2D (jours -1 à 3)
- Avant l’induction, plaquez les colonies de hPSC sur une matrice de membrane basale qualifiée hESC dans une plaque de 6 puits à une densité de 20% à 30%. Atteindre cette densité en faisant passer les colonies de CSEh d’un puits d’une plaque de 6 puits à 60 % de confluence dans trois puits d’une plaque de 6 puits.
- Pour le revêtement de matrice de membrane basale, diluer la matrice de membrane basale à un rapport de 1:50 dans un milieu basal uni. Déposer uniformément 1 mL/puits d’une plaque de 6 puits, incuber pendant 1 h à température ambiante (RT), puis aspirer.
- Maintenir les colonies de CSEh pendant 1 jour dans 2 mL d’un milieu de culture cellulaire sans sérum avant la différenciation.
- Le jour de la différenciation hPSC, inspectez les colonies hPSC à l’aide de la microscopie à fond clair à un grossissement de 4x à 10x pour assurer des colonies saines sans différenciation détectable.
REMARQUE: Les CSEh sains formeront des colonies de bord serrées avec des cellules qui ont un gros noyau, un très petit cytoplasme et des nucléoles proéminents. Les colonies iPSC différenciées présenteront des différences morphologiques claires par rapport à celles des colonies hPSC décrites ci-dessus, en particulier autour des bords extérieurs des colonies ou au centre. - Ajouter les réactifs énumérés dans le tableau 1 pour constituer le milieu N2 requis pour la différenciation. Apportez ce support à RT avant utilisation.
- Une fois à TA, aspirer le milieu de culture cellulaire sans sérum de chaque puits de la plaque de 6 puits et remplacer par 2 mL de milieu N2 ajouté doucement avec une pipette sérologique de 5 mL.
- Ajouter les inhibiteurs doubles de SMAD, SB-431542 (10 μM) et LDN 193189 (100 nM).
REMARQUE: Les inhibiteurs de SMAD peuvent être ajoutés au milieu N2 après que le milieu a été placé dans chaque puits, ou à la quantité requise de milieu N2 avant de remplacer le milieu de culture cellulaire sans sérum. Les inhibiteurs peuvent être incorporés uniformément dans le milieu en faisant doucement tourbillonner la plaque ou en inversant le tube contenant le milieu et les inhibiteurs 3 à 4 fois. - Ajouter des milieux N2 frais complétés par sb-431542 (10 μM) et LDN 193189 (100 nM) par jour à chaque puits pendant les 2 prochains jours.
REMARQUE: Un milieu N2 frais est ajouté pour réduire l’exposition prolongée des cellules au DMSO qui est utilisé pour dissoudre SB-431542 et LDN 193189 composés et prévenir la cytotoxicité.
- Génération de sphéroïdes neuroectodermiques 3D à partir de colonies neuroectodermiques 2D induites (jours 3 à 7)
- Soulever les colonies neuroectodermiques induites à l’aide de dispase en suivant les étapes 1.2.2-1.2.8.
- Tout d’abord, retirez 2 mL de milieu N2 de la plaque à 6 puits et lavez 1x avec HBSS pour vous assurer que tout le milieu N2 est retiré.
REMARQUE: Le milieu N2 peut interférer avec l’activité enzymatique de la dispase, empêchant un détachement adéquat des colonies neuroectodermiques du puits. - Ajouter 1 mL de dispase de 2,4 unités/mL à chaque puits contenant une colonie.
- Incuber le puits pendant 20-25 min (maximum 30 min) à 37 °C. Vérifiez régulièrement le détachement de la colonie.
REMARQUE: Les petites colonies peuvent se détacher dans les 20 minutes. Toutes les colonies qui restent coincées après 30 minutes doivent être ignorées. - Après l’incubation, ajouter 1 mL de milieu N2 au puits pour arrêter l’activité de l’enzyme dispase et transférer les colonies dans un tube de 15 mL à l’aide d’une pointe de pipette P1000 à large alésage ou d’une pointe de pipette P1000 modifiée coupée avec des ciseaux stériles (ce qui en fait une pointe P1000 à large alésage).
- Laissez les touffes de la colonie s’enfoncer au fond du tube avec gravité.
REMARQUE: Ce processus prendra environ 1 min. - Une fois les touffes enfoncées, retirez soigneusement le surnageant avec une pointe de pipette P1000 standard et remplacez-le par 1 mL de milieu N2 frais. Répétez cette étape de lavage trois fois pour assurer l’élimination complète de la dispase.
REMARQUE: Toute dispase restante empêchera une formation uniforme de sphéroïdes neuroectodermiques et induira la mort cellulaire. - Après le lavage, remettre en suspension les amas cellulaires dans 3 mL de milieu N2 et transférer dans un puits d’une plaque de 6 puits et ajouter 40 ng/mL de bFGF.
REMARQUE: Si un grand nombre de colonies neuroectodermiques étaient détachées, ces colonies pourraient être plaquées sur deux puits ou plus d’une plaque de 6 puits pour empêcher la fusion des sphéroïdes. 24 h après le détachement des colonies, vérifiez si les sphéroïdes se sont formés. - Maintenir les sphéroïdes dans le même milieu pendant les 3-4 prochains jours, mais ajouter du bFGF frais (40 ng / mL) à chaque puits tous les jours pour favoriser la prolifération des cellules neuroectodermiques, l’auto-organisation et induire et développer la neuroépithélie.
REMARQUE: Si les sphéroïdes ont été plaqués à une densité plus élevée, le milieu est susceptible de jaunir et de devoir être remplacé tous les 2 jours par du bFGF frais (40 ng / mL). Cependant, cela n’est pas recommandé, et au lieu de cela, un nombre inférieur de sphéroïdes doit être maintenu dans chaque puits pour éviter ce problème. Les sphéroïdes peuvent être incorporés dans la matrice de la membrane basale après 3 jours si les neuroépithélies sont évidentes. Si les neuroépithélies ne sont pas évidentes ou ne semblent pas fortes, maintenez les sphéroïdes pour un autre jour et vérifiez à nouveau.
- Différenciation et entretien des organoïdes corticaux du cerveau (Jour 8)
- Préparer le support de différenciation terminal (DM) à l’aide des réactifs répertoriés dans le tableau 2. Apportez ce média à RT.
- Décongeler la matrice de membrane basale 100 % qualifiée hESC sur la glace.
- Préparez une feuille de parafilm avec des fossettes stérilisées à 70% d’éthanol dans une boîte de Petri de 10 cm et placez-la sur un stéréomicroscope sous une hotte.
REMARQUE: Un plateau vide de 200 μL d’embouts de pipette peut être utilisé pour générer une grille de fossettes. - Coupez l’extrémité d’une pointe de pipette de 100 μL (pour la rendre à large alésage). Cela sera utilisé pour intégrer des sphéroïdes neuronaux dans la matrice du sous-sol sans les casser.
- À l’aide d’un stéréomicroscope, sélectionnez des sphéroïdes neuronaux de taille similaire (500 μm) dans la plaque de 6 puits et transférez-les dans les fossettes du parafilm à l’aide de la pointe de pipette de 100 μL à large alésage, en plaçant un seul sphéroïde neural par fossette.
- Retirez doucement tout excès de support, en laissant juste assez pour couvrir les sphéroïdes.
REMARQUE: Il s’agit de maintenir la qualité des sphéroïdes avant d’ajouter la matrice du sous-sol et de s’assurer qu’ils ne sèchent pas. - Ajoutez doucement 18 μL de matrice de sous-sol sur le sphéroïde, en positionnant le sphéroïde au centre de la goutte de matrice. Utilisez l’extrémité d’une pointe de pipette de 10 μL pour centrer les sphéroïdes dans la matrice.
REMARQUE: Essayez d’ajouter la matrice du sous-sol le plus rapidement possible pour éviter que l’excès de média autour des sphéroïdes ne se dessèche. - Couvrir la boîte de Petri de 10 cm et transférer dans l’incubateur et incuber la parabole avec des sphéroïdes incorporés dans la matrice du sous-sol à 37 °C pendant 25 min.
- Après l’incubation, rincez les sphéroïdes incorporés dans une plaque à 24 puits à faible fixation à l’aide d’une pointe P1000 avec 0,5 mL de support DM, en vous assurant que les sphéroïdes intégrés individuels sont placés dans un puits chacun.
REMARQUE: Si plus d’un sphéroïde incorporé tombe dans un puits, utilisez une pointe P1000 à large alésage pour transférer l’autre sphéroïde dans un nouveau puits. - Maintenez les organoïdes cérébraux corticaux différenciés dans le milieu DM pendant de longues périodes, avec des changements de média se produisant tous les 2 jours lorsque les organoïdes deviennent plus gros et plus âgés.
REMARQUE: Au cours de la première semaine de différenciation, le support peut être changé tous les 3 jours.
2. Caractérisation du vieillissement neuronal chez les organoïdes corticaux
- Organoïdes corticaux de procédé pour cryosections:
REMARQUE : Les étapes ont été effectuées dans une hotte de biosécurité de classe 2.- Préparer des tubes de 2 mL, chacun rempli de 1,5 mL de paraformaldéhyde (PFA) à 4 %.
- Coupez l’extrémité d’une pointe de pipette P1000 (pour en faire un alésage large) et transférez doucement chaque organoïde dans l’un des tubes de 2 mL préparés ci-dessus (un organoïde par tube).
REMARQUE: Pour éviter que l’excès de milieu DM ne se mélange avec le PFA, laissez l’organoïde s’enfoncer vers l’ouverture de l’embout de la pipette P1000 et placez la pointe juste au-dessus du sommet du PFA dans le tube avant de pipeter l’organoïde. Cela permettra au chercheur de transférer uniquement le milieu organoïde et très minimal. - Laisser le processus de fixation se dérouler à 4°C pendant 1 h.
- À l’aide d’une pointe de pipette P1000 non coupée, aspirez soigneusement l’excès de PFA et ajoutez 1,5 mL de PBS froid 1x.
- Transférer les tubes dans un agitateur orbital réglé à 70 tr/min pendant 10 min à RT.
- Répétez le processus de lavage avec 1x PBS froid trois fois pour vous assurer que tout PFA a été soigneusement éliminé.
REMARQUE : Ne pas jeter les PFA dans des contenants de déchets ordinaires; préparez plutôt un conteneur d’élimination des déchets chimiques spécifique pour cela, car le PFA est un danger. - Immerger les organoïdes dans 1x PBS contenant 30% de saccharose et incuber à 4 °C jusqu’à ce que tous les organoïdes aient coulé au fond du tube.
REMARQUE: Le temps nécessaire pour permettre aux organoïdes de couler dépend de la taille / âge des organoïdes. Les organoïdes âgés de 3 mois peuvent prendre jusqu’à 5 h. - À l’aide d’une pointe de pipette P1000 coupée et à large alésage, transférez doucement trois à cinq organoïdes dans un moule de montage contenant une solution de montage composée de 30 % de saccharose et d’un milieu de température de coupe optimale (OCT) à 100 %, dans un rapport de 3:2.
- Utilisez une pointe de pipette de 10 μL à l’aide d’un stéréomicroscope pour orienter et positionner les organoïdes dans un motif en forme de grille.
- Placez le moule sur de la glace carbonique pour solidifier la solution OCT de saccharose avant de procéder à la cryo-section (16-20 μm) à l’aide d’un cryostat.
REMARQUE: Pour la bêta-galactosidase associée à la sénescence, tous les tissus doivent être traités pour le sectionnement une fois qu’ils ont coulé. Pour l’immunofluorescence, les tissus peuvent être traités pour le sectionnement le lendemain. Toutes les lames contenant des sections doivent être conservées à -20 °C avant l’immunofluorescence ultérieure ou la bêta-galactosidase, si elles ne sont pas colorées immédiatement.
- Processus d’analyse de la sénescence dans les organoïdes cérébraux corticaux:
REMARQUE: Les étapes suivantes peuvent être effectuées sur un banc de laboratoire ordinaire.- Transférez les lames dans un récipient de coloration à lames de microscope avec un couvercle et lavez le tissu organoïde sectionné trois fois avec 1x PBS pendant 10 minutes à TA pour éliminer tout excès de solution de montage.
- Ensuite, incuber le tissu lavé avec une solution de coloration à la bêta-galactosidase fraîchement préparée pendant la nuit à 37 ° C.
NOTE: La solution de coloration à la bêta-galactosidase est constituée de tampon phosphate (pour 10 mL de tampon phosphate: 8,15 mL de 1 M NaH2PO4, 1,85 mL de 1M Na2HPO4) pH ajusté = 6,100 mM d’hexacyanoferrate de potassium (III), 100 mM d’hexacyanoferrate de potassium (II) trihydraté, 5 M de NaCl, 1 M de MgCl2, 20 mg/mL de X-Gal. Évitez d’utiliser un incubateur de culture cellulaire standard contenant du CO2 , car le CO2 modifiera le pH de la solution de coloration à la bêta-galactosidase. - Lavez les tissus colorés avec 1x PBS trois fois pendant 10 minutes chacun à TA pour enlever la solution de bêta-galactosidase.
- Montez les tissus lavés avec un support antifade en verre et laissez la solution de montage se solidifier pendant 30 min à TA avant de la visualiser au microscope.
Nous avons décrit ici un protocole robuste qui permet aux chercheurs de générer des organoïdes cérébraux corticaux homogènes dérivés de hPSC qui imitent la région cérébrale corticale humaine in vivo dans les 1 à 3 mois suivant la culture. Les colonies de cSEh sont d’abord cultivées dans des milieux de différenciation pour générer des colonies neuroectodermiques, qui peuvent ensuite être utilisées pour former des sphéroïdes neuronaux. Ces sphéroïdes sont ensuite incorporés dans une matrice de membrane basale et maintenus pendant de longues périodes pour produire des organoïdes qui peuvent être utilisés pour modéliser le vieillissement neuronal (voir la figure 1 pour un aperçu du protocole). Il est à noter que la culture de ces organoïdes dans des plaques ultra-non revêtues de 24 puits provoque un stress cellulaire et favorise les phénotypes associés à la sénescence sur 13 semaines de culture in vitro . Les organoïdes dérivés de ce protocole peuvent également être maintenus dans des bioréacteurs agités pour une croissance et une différenciation optimales des cellules neurales de la plaque corticale ou à l’interface air-liquide.
Pour commencer, les colonies de CSEh sont cultivées pendant 1 jour avant la différenciation neuroectodermique. Il est essentiel que ces colonies de CSEh soient cultivées à seulement 20 % à 30 % de confluence et qu’elles soient de la plus haute qualité possible : une monocouche plate et serrée sans cellules différenciées contaminant les colonies (Figure 2A, B). La pluripotence des colonies de cSEh doit être confirmée par l’expression de marqueurs tels que NANOG (Figure 2C). Les colonies de CSEh validées sont ensuite exposées au milieu de différenciation neuroectodermique N2 avec sb-431542 et LDN 193189. Après 3 jours d’entretien dans ce milieu, les colonies de CSEh devraient s’être différenciées en colonies neuroectodermiques et ne plus présenter la même morphologie monocouche plate serrée des CSEh (Figure 2B), mais plutôt devenir des cellules plus longues en forme de colonne (Figure 2B). Ces cellules seront également négatives pour les marqueurs de pluripotence tels que NANOG (Figure 2C).
C’est à ce stade que les colonies neuroectodermiques sont détachées enzymatiquement avec la dispase, et chaque colonie saine et détachée avec succès est autorisée à s’auto-organiser et à former un jeune sphéroïde neural (Figure 2D, Vidéo supplémentaire 1). Seules des colonies neuroectodermiques saines et propres se détacheront dans le délai spécifié pour l’activité de dispase; toutes les autres colonies doivent être ignorées car elles entraîneront une qualité de sphéroïde inférieure. Avec l’exposition quotidienne au FGF2 dans le milieu N2, les cellules souches neurales (SOX2+) dans ces sphéroïdes (Figure 2D, jour 1) vont proliférer et former un nombre important de rosettes neurales (Figure 2D, jour 4). Ces rosettes exprimeront la jonction serrée et le marqueur épithélial ZO1 dans les cellules situées au centre des rosettes et le long du bord externe du sphéroïde, démontrant la polarité apicale-basale du sphéroïde (Figure 2D, jour 4). La méthode d’imagerie 3D complète des sphéroïdes a été décrite avant13. L’inspection quotidienne des sphéroïdes devrait élucider la formation d’un bord extérieur étroit et sombre et d’une périphérie brillante des sphéroïdes, à savoir la couche neuroépithéliale. Cette couche doit être suffisamment formée après 3-4 jours avec un diamètre approximatif de 500 μm, moment auquel les sphéroïdes peuvent être incorporés dans la matrice du sous-sol. Si cette couche n’est pas présente ou n’est que faiblement formée, les sphéroïdes ne sont pas suffisamment développés pour avancer. Il est recommandé d’attendre un autre jour pour observer tout changement, mais si cela n’est pas observé, ne tenez pas compte de ces sphéroïdes.
Une image représentative en champ lumineux des sphéroïdes après 3 jours de culture peut être vue sur la figure 2D. Les sphéroïdes avec une couche neuroépithéliale serrée qui n’ont pas fusionné avec d’autres sphéroïdes voisins, ont un tissu semi-transparent et démontrent la formation de rosette neurale, sont choisis pour être intégrés dans la matrice du sous-sol. Une fois intégré, le sphéroïde proliférera rapidement et commencera à bourgeonner: des nœuds de tissu compact apparaîtront, s’étendant vers l’extérieur du corps principal du sphéroïde. Ceci est évident entre 1 et 3 semaines dans la matrice du sous-sol et peut être observé à travers plusieurs lignées cellulaires (Figure 3A). L’analyse quantitative des sphéroïdes incorporés confirme la présence de cellules épithéliales dans jusqu’à 100 % des sphéroïdes sur trois lignées cellulaires différentes, affirmant l’homogénéité et la reproductibilité attendues de ce protocole (Figure 3B). La quantification du diamètre des organoïdes lors de la différenciation in vitro confirme en outre la reproductibilité entre différentes lignées de cSEh (Figure 3C). Si le bourgeonnement ne se produit pas, les sphéroïdes ne se développent pas de manière appropriée et doivent être jetés. Une fois que les sphéroïdes ont été intégrés dans une matrice, leur développement progresse, et les sphéroïdes sont maintenant appelés organoïdes. La coloration par immunofluorescence confirme également la présence de cellules progénitrices neurales (PAX6) ainsi que de marqueurs de couche corticale colorés avec CTIP2 et SATB2 dans les organoïdes à couche claire (Figure 3D, E). Cette superposition est observable à travers différents points temporels de maintenance organoïde (Figure 3D,E). La méthode d’immunohistochimie des tissus a été décrite avant14.
Une application possible de ces organoïdes est d’étudier comment les processus liés au vieillissement neuronal affectent le cerveau. Pour étudier cela, des organoïdes générés avec succès sont prélevés à plusieurs points temporels différents pour la section et la coloration pour les biomarqueurs moléculaires standard de la sénescence tels que la bêta-galactosidase associée à la sénescence et p21. La figure 4A montre une image représentative de la coloration à la bêta-galactosidase associée à la sénescence des organoïdes 4 et 13 semaines après leur incorporation dans la matrice de la membrane basale. Entre les semaines 4 et 13, il y a une augmentation marquée de la présence de bêta-galactosidase associée à la sénescence, ce qui suggère que la sénescence cellulaire, un moteur reconnu du vieillissement de l’organisme, s’est produite au cours de cette période en culture. La coloration par immunofluorescence des organoïdes à la semaine 13 a confirmé la présence d’un autre marqueur de sénescence, p21, co-marqué avec le marqueur neuronal cortical mature (CTIP2) et peut être vu à la figure 4B. Il convient toutefois de noter que la présence de p21 est un marqueur de l’arrêt du cycle cellulaire et n’est pas en soi un marqueur définitif de la sénescence, et la détection d’autres marqueurs de sénescence tels que p16 et les facteurs SASP (phénotype sécrétoire associé à la sénescence) est recommandée pour identifier définitivement les cellules comme sénescentes.
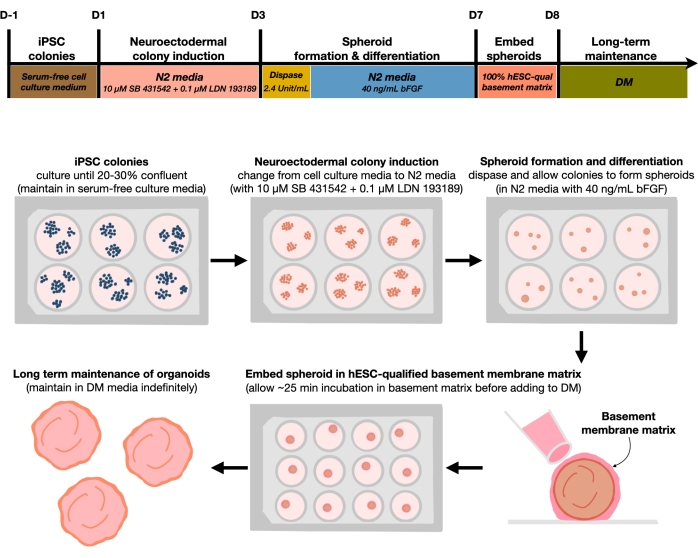
Figure 1 : Diagramme schématique pour la génération d’organoïdes cérébraux corticaux reproductibles. Flux de travail schématique de la procédure expérimentale pour la génération d’organoïdes cérébraux corticaux à partir de CSEh maintenus dans le milieu sans mangeoire. Le flux de travail fournit une vue d’ensemble des six étapes impliquées pour différencier les hPSC 2D en tissus humains à plaque corticale à motifs 3D dans des organoïdes. Veuillez cliquer ici pour voir une version agrandie de cette figure.
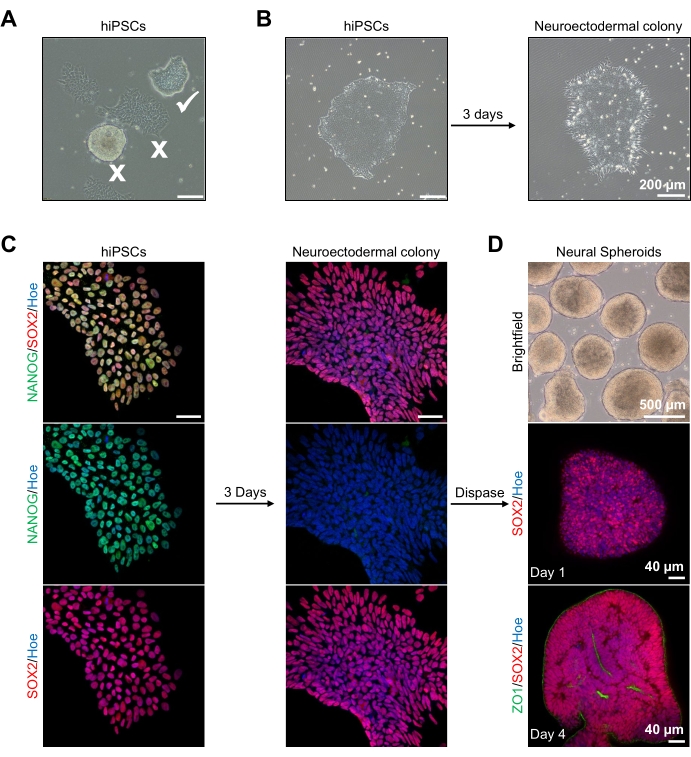
Figure 2 : Génération de sphéroïdes neuronaux dérivés de colonies neuroectodermiques-CSEh. (A) Images représentatives de CSP humaines présentant des colonies optimales (tique blanche) et différenciées (croix blanche). Barre d’échelle: 200 μm, grossissement 4x. (B) Image représentative de la colonie neuroectodermique dérivée des CSEh après 3 jours de traitements à double inhibiteur de la SMAD. Barre d’échelle: 200 μm, grossissement 4x. (C) Les colonies humaines de CSP ont été différenciées en colonies neuroectodermiques. Les images représentent la coloration des colonies de PSC (au jour 1) et neuroectodermiques (au jour 3) avec SOX2 (rouge), NANOG (vert), tous les noyaux ont été contre-colorés avec Hoechst 33342 (bleu). Barre d’échelle: 40 μm, grossissement 100x. (D) Images montrant les stades de développement des sphéroïdes corticaux du cerveau au fil du temps en culture in vitro sous fond clair, et immunocolorés en quantité entière avec SOX2 (rouge) au jour 1, et doublement immunocolorés avec SOX2 (rouge) et ZO1 (vert) au jour 4. La barre d’échelle de l’image en fond clair est de 500 μm, le grossissement 4x, les barres d’échelle des images du bas sont de 40 μm, le grossissement 20x. Veuillez cliquer ici pour voir une version agrandie de cette figure.
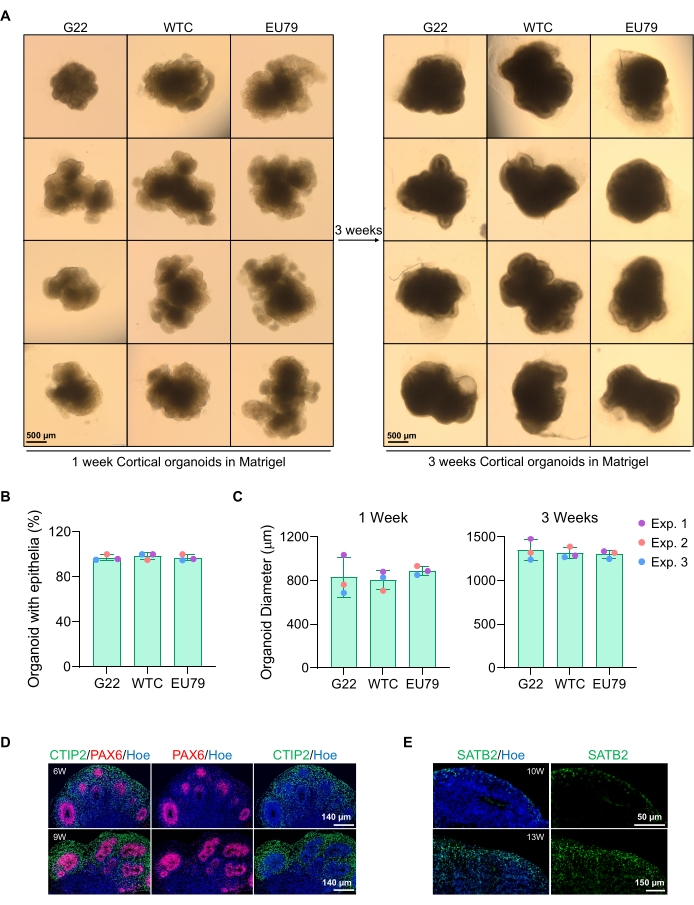
Figure 3 : Caractérisation d’organoïdes cérébraux corticaux dérivés de différentes lignées de cSEh. (A) Images représentatives d’organoïdes cérébraux corticaux dérivés de lignées iPSC humaines G22, WTC et EU79 cultivées pendant 3 semaines in vitro. La barre d’échelle de toutes les images est de 500 μm, grossissement 2x. (B) Pourcentages de la génération réussie d’organoïdes cérébraux corticaux à 3 semaines de différenciation in vitro dans différentes lignées hPSC (G22, WTC et EU79). N = 3. Les données sont présentées comme une moyenne ± un écart-type. (C) Graphiques à barres montrant la croissance des organoïdes corticaux du cerveau (sur la base du diamètre moyen) aux semaines 1 et 3 de différenciation in vitro dans différentes lignées de lignées de cellules souches pluripotentes humaines (G22, WTC et EU79). N = 3. Les données sont présentées comme une moyenne ± un écart-type. (D) Images représentatives de sections d’organoïdes cérébraux corticaux âgés de 6 semaines et de 9 semaines dérivés de CSEh G22, immunocolorés pour la zone ventriculaire PAX6 (rouge) et la plaque corticale CTIP2 (vert). Toutes les sections ont été contre-colorées avec Hoechst 33342 (bleu). Barre d’échelle = 140 μm, grossissement 20x. W est la semaine. (E) Images représentatives de sections d’organoïdes cérébraux corticaux âgés de 10 et 13 semaines dérivés de csthp wtc, immunocolorés pour la couche corticale IV SATB2 (vert). Toutes les sections ont été contre-colorées avec Hoechst 33342 (bleu). 10 semaines d’image Barre d’échelle = 50 μm, grossissement 40x. 13 semaines d’image Barre d’échelle = 150 μm, grossissement 40x. W est la semaine. Veuillez cliquer ici pour voir une version agrandie de cette figure.
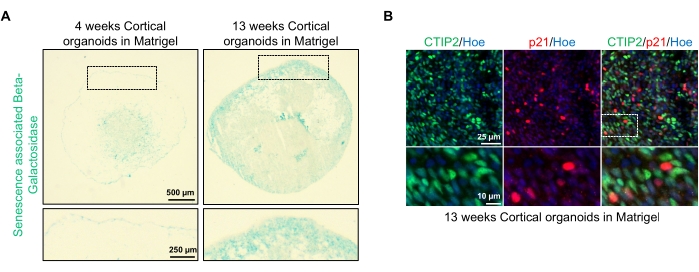
Figure 4 : Caractérisation de la sénescence dans les organoïdes cérébraux corticaux dérivés des CSEh. (A) Images représentatives de sections d’organoïdes cérébraux corticaux humains dérivés de CSEh du WTC cultivés pendant 4 et 13 semaines in vitro et colorés avec du SA-β-gal. Barre d’échelle = 500 μm, barre d’échelle des images zoomées = 250 μm, grossissement 4x. La zone en pointillés indique une image agrandie. (B) Images représentatives de coupes d’organoïdes cérébraux corticaux âgés de 13 semaines dérivés de CSEh humains eu79, immunocolorés pour les neurones corticaux CTIP2 (vert) et p21 (rouge). Toutes les sections ont été contre-colorées avec Hoechst 33342 (bleu). Barre d’échelle = 25 μm, barre d’échelle des images zoomées = 10 μm, grossissement 40x. Veuillez cliquer ici pour voir une version agrandie de cette figure.
| Composants multimédias | Concentration |
| DMEM Mélange nutritif F12 10x 500 mL (DMEM/F-12) | |
| Supplément N2 5 mL (100x) | Supplmenté à 1% |
| B 27 Supplément 10 mL | Supplémenté à 2% |
| Solution d’acides aminés non essentiels MEM (100x) | Supplémenté à 1% |
| Pénicilline-streptomycine (10 000 U/mL) | Supplémenté à 1% |
| 2-Mercaptoéthanol 50 mL (1000x ) | Supplémenté à 0,1 % |
Tableau 1 : Milieu N2. Le tableau répertorie les réactifs nécessaires à la préparation du milieu N2.
| Composants multimédias | Concentration |
| DMEM Mélange nutritif F12 10x 500 mL (DMEM/F-12) | Les médias DM sont fabriqués avec un rapport 1: 1 de DMEM / F12 et de milieux neurobasaux |
| Milieu neurobasal | |
| Supplément N2 5 mL (100x) | Augmenté à 0,5 % |
| B 27 Supplément 10 mL | Supplémenté à 1% |
| Solution d’acides aminés non essentiels MEM (100x) | Supplémenté à 1% |
| GlutaMAX Supplément 100x | Supplémenté à 1% |
| Pénicilline-streptomycine (10 000 U/mL) | Supplémenté à 1% |
| Solution d’insuline humaine recombinante | 12,5 μL pour 50 mL de milieu |
| 2-Mercaptoéthanol 50 mL (1000x ) | 17,5 μL pour 50 mL de milieu |
Tableau 2 : Milieu de différenciation (MS). Le tableau répertorie les réactifs nécessaires à la préparation du milieu de différenciation.
Vidéo supplémentaire 1. Imagerie en direct de la conversion induite de la feuille/colonie hNEct 2D en 3D sous le traitement du bFGF. Les colonies induites de hNEct ont été doucement détachées du plat avec de la dispase comme indiqué ci-dessus et transférées dans une plaque de culture à 6 puits à faible fixation. Les colonies 2D hNEct ont été converties en sphéroïdes hNEct 3D en 12 h. Des images en série ont été capturées toutes les 5 minutes. Barre d’échelle = 100 μm. Veuillez cliquer ici pour télécharger cette vidéo.
Pour permettre l’utilisation d’organoïdes cérébraux dérivés de la hPSC dans le dépistage des médicaments et la modélisation des maladies, il est crucial de fabriquer des organoïdes selon un protocole reproductible et fiable15. Les organoïdes cérébraux sont généralement générés à partir de corps embryoïdes dérivés de CSEh, qui sont ensuite intégrés dans une matrice extracellulaire qui favorise l’expansion tissulaire et la différenciation neuronale. Par rapport à des protocoles tels que Lancaster 1,16,17 et Velasco 18, qui partent de corps embryidés et permettent de suivre une voie de différenciation par défaut par les organoïdes en développement, nous avons constaté que le début de la création d’organoïdes cérébraux corticaux avec des cellules NEct humaines plutôt qu’avec des corps embryoïdes améliore la cohérence de la formation d’organoïdes cérébraux corticaux. Cela permet donc également la mise à l’échelle requise pour le dépistage médicamenteux et phénotypique. Étant donné que les cellules NEct humaines peuvent non seulement être étendues en quantités considérables, mais peuvent également être facilement cryoconservées, cette approche améliore également la reproductibilité entre les expériences. Il convient également de noter que, par rapport à d’autres protocoles qui ont adopté l’utilisation de bioréacteurs et de technologies similaires, aucun équipement spécialisé n’est requis pour ce protocole, ce qui le rend adapté à tout laboratoire6. Enfin, le temps nécessaire pour générer des organoïdes matures positifs pour les marqueurs de la couche corticale tels que SATB2 est réduit par rapport aux protocoles Lancaster1 et bioréacteur 6,19, ce qui le rend plus approprié pour étudier la trajectoire de développement du développement cortical humain dans la santé et les maladies 1,6,16.
En outre, étant donné l’impact croissant des maladies liées au vieillissement telles que la démence sur les soins de santé à l’échelle mondiale, qui sont associées à une augmentation des types de cellules sénescentes dans le cerveau qui contribue à la pathogenèse, la capacité d’identifier et de tester des composés capables d’améliorer le vieillissement du cerveau est d’un intérêt énorme. Bien que les CSEh soient connus pour être rajeunis épigénétiquement au cours du processus de reprogrammation20, nous constatons des augmentations robustes des cellules sénescentes dans les organoïdes cérébraux corticaux cultivés pendant de longues périodes. Il s’agit d’un développement prometteur qui permet maintenant le dépistage de médicaments qui éliminent ces cellules sénescentes du cerveau (sénolytiques) ou qui ralentissent ce processus (sénostatiques)21. Étant donné que les organoïdes cérébraux corticaux humains dérivés de NEct sont d’origine humaine, cette approche raccourcira probablement la voie traditionnelle vers la commercialisation de ces nouveaux traitements.
Ce protocole comporte deux étapes critiques. Le premier est le niveau correct de confluence des colonies de cSEh au moment de la différenciation. Les colonies de cSEh doivent être confluentes à 30 % au maximum pour s’assurer que les colonies de NEct générées ne fusionnent pas avec les colonies voisines et que les organoïdes individuels sont clonés. La deuxième étape critique implique l’utilisation correcte de dispase pour soulever les colonies NEct et produire les sphéroïdes neuronaux. Le moment de l’incubation avec dispase est essentiel à la qualité éventuelle des sphéroïdes neuronaux générés. En effet, la surexposition des colonies avec dispase est toxique pour les cellules22 et finit par affecter la qualité des organoïdes générés. La limite de ce protocole est qu’il est difficile de contrôler la taille des sphéroïdes neuronaux car il dépend de la taille des colonies initiales qui sont levées avec dispase. Cependant, ce problème peut être surmonté en sélectionnant des sphéroïdes neuronaux de taille similaire lors de l’étape d’intégration.
Enfin, les applications futures pourraient s’étendre à l’utilisation de ces organoïdes corticaux reproductibles dans l’analyse robotique et les approches de dépistage biopharmaceutique généralement utilisées dans cette industrie. Ceci est soutenu par les données préliminaires de notre laboratoire indiquant que la génération d’organoïdes cérébraux corticaux à partir de cellules NEct humaines peut être facilement automatisée, ce qui la rend compatible avec ces approches.
Les auteurs n’ont rien à divulguer.
Ce travail est soutenu par le Medical Research Future Fund-Accelerated Research, le fleuron de la leucodystrophie Massimo’s Mission (EPCD000034), medical Research Future Fund-Stem Cell Mission (APP2007653). Les auteurs tiennent à remercier le Dr Ju-Hyun Lee (Université de Corée) d’avoir généré des données dans la vidéo supplémentaire 1.
| Name | Company | Catalog Number | Comments |
| 16% Formaldehyde (W/V) Methanol-free | Thermo Fisher Scientific | 28908 | 4% of PFA are diluted in 1x PBS |
| 2-Mercaptoethanol 50 mL(1000x) | Life Technologies Australia (TFS) | 21985023 | Used in NM and DM media |
| B 27 Supplement 10 mL | Life Technologies Australia (TFS) | 17504044 | Used in NM and DM media |
| CKX53 microscope with SC50 camera | Olympus | ||
| Corning Costar 6 well cell culture plates | Sigma Aldrich Pty Ltd | CLS3516-50EA | |
| Dispase II powder | Thermo Fisher Scientific | 17105041 | Powder is dissolve in HBSS, filtered through 0.22 µm filter, aliquote at 10 mL and store at -20 °C |
| DMEM Nutrient Mix F12 10x 500 mL (DMEM/F-12) | Thermofisher | 11320082 | Used in NM and DM media |
| DMSO Dimethyl Sulfoxide | Sigma Aldrich Pty Ltd | D2650-100ML | |
| Dulbecco's Phosphate Buffered Saline | Sigma Aldrich Pty Ltd | D1408-500ML | |
| Falcon Matrigel hESC-qualified Matrix | In Vitro Technologies Pty Ltd | FAL354277 | Make aliquotes of 100 µL and stored at -20 °C |
| GlutaMAX Supplement 100x | Thermo Fisher Scientific | 35050061 | Used in NM and DM media |
| Hanks Balanced Salt Solution | Sigma Aldrich Pty Ltd | H8264 | |
| Human induced pluripotent stem cells (EU79) | In-house reporogrammed from skin fibroblast | ||
| Human induced pluripotent stem cells (G22) | Genea Biocells | Obtained from Genea Biocells (San Diego, United States) | |
| Human induced pluripotent stem cells (WTC) | Gift from Professor Bruce Conklin | ||
| InSolution TGF-Β RI Kinase Inhibitor VI, SB431542 | Merck | US1616464-5MG | |
| Insulin Solution Human Recombinant | Sigma Aldrich Pty Ltd | I9278 | Used in NM and DM media |
| LDN193189 Dihydrochloride | Sigma Aldrich Pty Ltd | SML0559-5MG | Used during differentiation |
| MEM Non-Essential Amino Acids Solution (100x) | Thermo Fisher Scientific | 11140050 | Used in NM and DM media |
| mTeSR Plus | STEMCELL TECHNOLOGIES | 100-0276 | Used to maintain hiPSC colonies prior to differentiation with NM media |
| N2 Supplement 5 mL (100x) | Life Technologies Australia Pty Ltd | 17502048 | Used in NM and DM media |
| Neurobasal Medium | Thermo Fisher Scientific | 21103049 | Used in DM media |
| OCT Embedding Compound Sakura Clear (118 mL/Bottle) | Tissue Tek | 4583 | |
| Parafilm M Roll Size 4 in. x 125 Ft | Sigma Aldrich Pty Ltd | P7793 | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 | Used in NM and DM media |
| Potassium Hexacyanoferrate (II) Trihydrate | Sigma Aldrich Pty Ltd | CP1087 | |
| Potassium hexacyanoferrate(III) | Sigma Aldrich Pty Ltd | 455946 | |
| Prolong Glass Antifade Mountant | Life Technologies Australia (TFS) | P36980 | |
| Recombinant Human FGF basic | R&D Systems | 233-FB-01M | Aliquotes are made at 20 µg/mL and stored at -20 °C |
| SB431542 | Tocris | 1614 | Used during differentiation |
| Sucrose | Sigma Aldrich Pty Ltd | PHR1001-1G | 30% of sucrose are diluted in 1x PBS |
| Ultra-Low attachment multiwell plates , 24 well plate, polystyrene | Sigma Aldrich Pty Ltd | CLS3473-24EA | |
| X-GAL EA | Life Technologies Australia (TFS) | R0404 | Make aliquotes of 20 mg/mL and storde at -80 °C |
- Lancaster, M. A., Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nature Protocols. 9 (10), 2329-2340 (2014).
- Mansour, A. A., et al. An in vivo model of functional and vascularized human brain organoids. Nature Biotechnology. 36 (5), 432-441 (2018).
- Xiang, Y., et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell. 21 (3), 383-398 (2017).
- Bagley, J. A., Reumann, D., Bian, S., Lévi-Strauss, J., Knoblich, J. A. Fused cerebral organoids model interactions between brain regions. Nature Methods. 14 (7), 743 (2017).
- Kwak, T. H., et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson's disease modeling. Stem Cells. 38 (6), 727-740 (2020).
- Qian, X., et al. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nature Protocols. 13 (3), 565-580 (2018).
- Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Reports. 10 (4), 537-550 (2015).
- Shaker, M. R., Cooper-White, J., Wolvetang, E. J. Self-organizing 3D human choroid plexus-ventricle-cortical organoids. BioRxiv. , (2020).
- Shaker, M. R., Aguado, J., Chaggar, H. K., Wolvetang, E. J. Klotho inhibits neuronal senescence in human brain organoids. npj Aging and Mechanisms of Disease. 7 (1), 1-12 (2021).
- Shaker, M. R., et al. Anteroposterior Wnt-RA gradient defines adhesion and migration properties of neural progenitors in developing spinal cord. Stem Cell Reports. 15 (4), 898-911 (2020).
- Chambers, S. M., et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology. 27 (3), 275-280 (2009).
- Shaker, M. R., et al. Rapid and efficient generation of myelinating human oligodendrocytes in organoids. Frontiers in Cellular Neuroscience. 15, 631548 (2021).
- Lee, J. -. H., Shaker, M. R., Lee, E., Lee, B., Sun, W. NeuroCore formation during differentiation of neurospheres of mouse embryonic neural stem cells. Stem Cell Research. 34, 101691 (2020).
- Shaker, M. R., et al. Spatiotemporal contribution of neuromesodermal progenitor-derived neural cells in the elongation of developing mouse spinal cord. Life Sciences. 282, 119393 (2021).
- Shaker, M. R., et al. Neural epidermal growth factor-like like protein 2 Is expressed in human oligodendroglial cell types. Frontiers in Cell and Developmental Biology. 10, 803061 (2022).
- Lancaster, M. A., et al. Cerebral organoids model human brain development and microcephaly. Nature. 501 (7467), 373-379 (2013).
- Giandomenico, S. L., Sutcliffe, M., Lancaster, M. A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nature Protocols. 16 (2), 579-602 (2021).
- Velasco, S., et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 570 (7762), 523-527 (2019).
- Qian, X., et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 165 (5), 1238-1254 (2016).
- Hunter, Z. L., Leeson, H. C., Shaker, M. R., Wolvetang, E. J., Vadlamudi, L. Human induced pluripotent stem cells generated from epilepsy patients for use as in vitro models for drug screening. Stem Cell Research. 60, 102673 (2022).
- Kaur, A., Macip, S., Stover, C. M. An appraisal on the value of using nutraceutical based senolytics and senostatics in aging. Frontiers in Cell and Developmental Biology. 8, 218 (2020).
- Wang, F., et al. Safety and efficacy of dispase and plasmin in pharmacologic vitreolysis. Investigative Ophthalmology & Visual Science. 45 (9), 3286-3290 (2004).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved