
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Neuroscience
Robuste und hochreproduzierbare Erzeugung kortikaler Hirnorganoide zur Modellierung der neuronalen Seneszenz des Gehirns in vitro
In dieser Studie stellen wir eine detaillierte Technik für ein einfaches, aber robustes kortikales Organoid-Kultursystem unter Verwendung von standardmäßigen feederfreien hPSC-Kulturen zur Verfügung. Dies ist ein schnelles, effizientes und reproduzierbares Protokoll zur Erzeugung von Organoiden, die Aspekte der Hirnseneszenz in vitro modellieren.
Gehirnorganoide sind dreidimensionale Modelle des sich entwickelnden menschlichen Gehirns und bieten eine überzeugende, hochmoderne Plattform für die Krankheitsmodellierung und das groß angelegte genomische und medikamentöse Screening. Aufgrund der selbstorganisierenden Natur von Zellen in Gehirnorganoiden und der wachsenden Bandbreite verfügbarer Protokolle für ihre Erzeugung wurden Probleme mit der Heterogenität und Variabilität zwischen Organoiden identifiziert. In diesem Protokollpapier beschreiben wir ein robustes und replizierbares Protokoll, das diese Probleme weitgehend überwindet und innerhalb von 1 Monat kortikale Organoide aus neuroektodermalen Vorläufern erzeugt und länger als 1 Jahr aufrechterhalten werden kann. Dieses hochgradig reproduzierbare Protokoll kann leicht in einem Standard-Gewebekulturraum durchgeführt werden und führt zu Organoiden mit einer reichen Vielfalt von Zelltypen, die typischerweise im sich entwickelnden menschlichen Kortex vorkommen. Trotz ihrer frühen Entwicklungsstruktur werden Neuronen und andere menschliche Gehirnzelltypen nach längerer In-vitro-Kultur die typischen Anzeichen von Seneszenz in neuronalen Zellen zeigen, was sie zu einer wertvollen und nützlichen Plattform für die Untersuchung altersbedingter neuronaler Prozesse macht. Dieses Protokoll beschreibt auch eine Methode zum Nachweis solcher seneszenten Zellen in kortikalen Gehirnorganoiden unter Verwendung der seneszenzassoziierten Beta-Galactosidase-Färbung.
Unser derzeitiges Wissen über das menschliche Gehirn basiert weitgehend auf Tiermodellen und postmortalen Hirnproben. Die Stammzellbiologie ist ein sich schnell entwickelndes Feld, das neue Einblicke in die grundlegende Biologie der Entwicklung des menschlichen Gehirns und die pathologischen Treiber menschlicher Gehirnstörungen liefert. Menschliche pluripotente Stammzellen (hPSCs) sind ein unschätzbares Werkzeug für die Modellierung des menschlichen Gehirns durch die Erzeugung von Organoiden, organähnlichem dreidimensionalem (3D) Gewebe, das typischerweise die Entwicklungsverläufe, die zelluläre Zusammensetzung und die Architektur des sich entwickelnden menschlichen Gehirns rekapituliert. Gehirnorganoide sind selbstorganisiert und bestehen aus neuralen Stammzellen, spezifizierten neuralen Vorläufern, reifen Neuronen und Gliazelltypen. Organoide bieten daher eine einzigartige Gelegenheit, das frühe menschliche Gehirn zu untersuchen, das oft für direkte Experimente unzugänglich ist, aber auch intrinsische Einschränkungen wie das Fehlen von Gefäßen und einem Immunsystem aufweist.
Methoden zur Erzeugung von Gehirnorganoiden wurden auf zwei verschiedene Arten verfolgt: ungeführte und geführte Differenzierung. Ungelenkte Organoidmethoden des Gehirns beruhen auf den spontanen intrinsischen Differenzierungskapazitäten der Stammzellen, die die Gewebemorphogenese1,2 antreiben und die Entstehung einer Vielzahl von Zelllinienidentitäten ermöglichen, die von Vorderhirn, Mittelhirn und Hinterhirn bis hin zu Plexus choroideus, Netzhaut und Mesoderm reichen. Im Gegensatz dazu erfordern geführte Hirnorganoidmethoden eine erhebliche Verwendung externer Faktoren, um hPS-Zellen in Richtung der gewünschten Musterung neuronaler Linien zu treiben, die einen Gehirnregionentyp darstellen, wie mediale ganglionäre Eminenz3, Vorderhirn4, Mittelhirn5, Hypothalamus6, Kleinhirn7 und Aderhautplexus8. Diese Fähigkeit, verschiedene Gehirnregionen mit unterschiedlichen Zelllinien zu erzeugen, und das Potenzial, diese nach Belieben zu verschmelzen, macht Gehirnorganoide zu einem ausgezeichneten Modell, um die Entwicklung des menschlichen Gehirns zu untersuchen und die zugrunde liegenden Mechanismen von Erkrankungen des Gehirns zu entschlüsseln. Obwohl diese Methoden zur Erzeugung von Gehirnorganoiden einen Durchbruch bei der Modellierung menschlicher Hirnregionen bieten, bleiben die Variabilität und Heterogenität zwischen Organoiden eine signifikante Einschränkung für systematische und quantitative Studien wie das Arzneimittelscreening.
Das aktuelle Protokoll basiert auf einer Methode, die in unserer kürzlich erschienenen Arbeit9 entwickelt wurde, und beinhaltet die selektive Differenzierung von hPSC-Kolonien in Richtung Neuroektoderm (NEct) -Identität mit dualen SMAD-Inhibitoren (SB-431542 und LDN 193189), die dann die Fähigkeit haben, sich innerhalb von 4 Tagen in 3D-Neuroepithel-Sphäroide unter dem Einfluss der FGF2-Signalgebung selbst zu organisieren. Diese Neuroepithel-Sphäroide erzeugen innerhalb von 4 Wochen nach der Differenzierung zuverlässig homogene kortikale Organoide mit einer in vivo-ähnlichen zellulären Zusammensetzung. Das hier beschriebene Protokoll baut auf unseren früheren Ergebnissen auf, die zeigen, dass die Hemmung der dualen SMAD-Signalgebung (Suppressor of Mothers Against Decapentaplegic) die Differenzierung von hPS-Zellen gegenüber rostralen neuralen Stammzellen fördert, die von neuroektodermalen Vorläuferzellen abgeleitet werden10 unter anderem durch Hemmung der endodermalen, mesodermalen und trophektodermen Zellschicksalswahl11 . Darüber hinaus löst die Einbettung der Neuroepithel-Sphäroide in die hESC-qualifizierte Basalmembranmatrix ein signifikantes Austreiben der Neuroepithele aus und bildet Ventrikel mit apikobasaler Polarität. Großkultur zeigte Reproduzierbarkeit und Homogenität von kortikalen Organoiden unabhängig von Zelllinien, Klonen oder Chargen und stellt somit ein zuverlässiges und stabiles Stammzellsystem dar, um die frühe kortikale Entwicklung des Menschen in Gesundheit und Krankheit in vitro nachzuahmen. Wir skizzieren ferner ein Protokoll zum Nachweis seneszenter neuronaler Zellmarker in hPSCs-abgeleiteten kortikalen Gehirnorganoiden, die über längere Zeiträume kultiviert wurden.
Nach der Beschichtung von hPS-Zellen mit einer Aussaatdichte von 20%-30% werden die Zellen 3 Tage lang mit dualen SMAD-Inhibitoren behandelt, um hPSC-Kolonien in Richtung neuroektodermaler Kolonien zu differenzieren. Diese Kolonien werden dann vorsichtig mit Dispase angehoben und in extrem niedrige 6-Well-Platten gesät, die mit FGF2 ergänzt werden. Die schwimmenden 2D-Kolonien organisieren sich über Nacht in 3D-neuroektodermale Sphäroide und werden 4 Tage lang in N2-Medium gehalten, das täglich mit FGF2 ergänzt wird. Sobald die Sphäroide die neuroepitheliale Schicht aufgebaut haben, können sie in die Basalmembranmatrix eingebettet werden. Durch die routinemäßige Zugabe von frischem terminalem Differenzierungsmedium werden die Forscher eine fortschreitende Expansion und Knospektik der Neuroepithel in kortikalen Organoiden beobachten. Forscher möchten diese Organoide möglicherweise dissoziieren, um transkriptionelle und proteomische Profilerstellung durchzuführen. Darüber hinaus wird die Hellfeldbildgebung zur Überwachung der Qualität der kortikalen Organoide empfohlen. Die Analyse kann durch Fixierung, Kryosektion und Immunfärbung durchgeführt werden. Beschreibungen und Methoden für diese Techniken wurden zuvorbeschrieben 12. Letztendlich ermöglicht dieses Protokoll den Forschern, schnell und robust homogene kortikale Gehirnorganoide zu erzeugen, um das sich entwickelnde menschliche kortikale Gehirn mit niedrigen Kosten und begrenzter Ausrüstung zu modellieren und Aspekte der zellulären neuronalen Seneszenz zu untersuchen, wie in diesem Artikel beschrieben.
1. Erzeugung kortikaler Gehirnorganoide
HINWEIS: Alle Schritte in diesem Abschnitt des Protokolls werden in einer Biosicherheitshaube der Klasse 2 ausgeführt, sofern nicht anders angegeben.
- Induktion von 2D-neuroektodermalen Kolonien aus der hPSC-2D-Kultur (Tage -1 bis 3)
- Vor der Induktion die hPSC-Kolonien auf einer hESC-qualifizierten Basalmembranmatrix in einer 6-Well-Platte mit einer Dichte von 20% -30% platten. Erreichen Sie diese Dichte, indem Sie hPSC-Kolonien von einem Bohrloch einer 6-Well-Platte mit 60% Konfluenz in drei Wells einer 6-Well-Platte leiten.
- Für die Basalmembranmatrixbeschichtung verdünnen Sie die Basalmembranmatrix in einem Verhältnis von 1:50 in einem einfachen Basalmedium. Schichten Sie 1 ml/Well einer 6-Well-Platte gleichmäßig ab, inkubieren Sie 1 h bei Raumtemperatur (RT) und saugen Sie dann ab.
- Halten Sie hPSC-Kolonien für 1 Tag in 2 ml eines serumfreien Zellkulturmediums vor der Differenzierung aufrecht.
- Untersuchen Sie am Tag der hPSC-Differenzierung die hPSC-Kolonien mittels Hellfeldmikroskopie bei 4- bis 10-facher Vergrößerung, um gesunde Kolonien ohne nachweisbare Differenzierung zu gewährleisten.
HINWEIS: Gesunde hPS-Zellen bilden enge Kantenkolonien mit Zellen, die einen großen Kern, ein sehr kleines Zytoplasma und prominente Nukleolen haben. Differenzierte iPSC-Kolonien zeigen deutliche morphologische Unterschiede zu den oben beschriebenen hPSC-Kolonien, insbesondere an den äußeren Rändern der Kolonien oder in der Mitte. - Fügen Sie die in Tabelle 1 aufgeführten Reagenzien hinzu, um das für die Differenzierung erforderliche N2-Medium zu bilden. Bringen Sie dieses Medium vor Gebrauch zu RT.
- Sobald Sie bei RT angekommen sind, aspirieren Sie das serumfreie Zellkulturmedium aus jeder Vertiefung der 6-Well-Platte und ersetzen Sie es durch 2 ml N2-Medium, das vorsichtig mit einer 5 ml serologischen Pipette hinzugefügt wird.
- Fügen Sie die beiden SMAD-Inhibitoren SB-431542 (10 μM) und LDN 193189 (100 nM) hinzu.
HINWEIS: Die SMAD-Inhibitoren können dem N2-Medium hinzugefügt werden, nachdem das Medium in jedes Vertiefungsfeld gegeben wurde, oder in die erforderliche Menge an N2-Medium, bevor das serumfreie Zellkulturmedium ersetzt wird. Inhibitoren können gleichmäßig in das Medium eingearbeitet werden, indem die Platte vorsichtig gewirbelt oder das Röhrchen, das das Medium und die Inhibitoren enthält, 3-4 Mal invertiert wird. - Fügen Sie in den nächsten 2 Tagen täglich frisches N2-Medium hinzu, das mit SB-431542 (10 μM) und LDN 193189 (100 nM) ergänzt wird.
HINWEIS: Frisches N2-Medium wird hinzugefügt, um die längere Exposition von Zellen gegenüber dem DMSO zu reduzieren, das zum Auflösen von SB-431542 und LDN 193189 Verbindungen verwendet wird, und um Zytotoxizität zu verhindern.
- Generierung von 3D-neuroektodermalen Sphäroiden aus induzierten 2D-neuroektodermalen Kolonien (Tage 3 bis 7)
- Heben Sie die induzierten neuroektodermalen Kolonien mit Dispase an, indem Sie die Schritte 1.2.2-1.2.8 ausführen.
- Entfernen Sie zuerst 2 ml N2-Medium von der 6-Well-Platte und waschen Sie 1x mit HBSS, um sicherzustellen, dass das gesamte N2-Medium entfernt wird.
HINWEIS: N2-Medium kann die Enzymaktivität der Dispase stören und eine ausreichende Ablösung neuroektodermaler Kolonien aus dem Brunnen verhindern. - Fügen Sie 1 ml 2,4 Einheit / ml Dispase zu jeder koloniehaltigen Vertiefung hinzu.
- Inkubieren Sie den Brunnen für 20-25 min (maximal 30 min) bei 37 °C. Überprüfen Sie regelmäßig auf Kolonieablösung.
HINWEIS: Kleine Kolonien können sich innerhalb von 20 Minuten ablösen. Alle Kolonien, die nach 30 Minuten stecken bleiben, sollten ignoriert werden. - Nach der Inkubation fügen Sie 1 ml N2-Medium in die Vertiefung hinzu, um die Aktivität des Dispase-Enzyms zu stoppen, und übertragen Sie die Kolonien in ein 15-ml-Rohr mit einer P1000-Pipettenspitze mit breiter Bohrung oder einer modifizierten P1000-Pipettenspitze, die mit einer sterilen Schere geschnitten wird (was sie zu einer P1000-Spitze mit breiter Bohrung macht).
- Lassen Sie die Kolonieklumpen mit Schwerkraft auf den Boden der Röhre sinken.
HINWEIS: Dieser Vorgang dauert ca. 1 Minute. - Sobald die Klumpen gesunken sind, entfernen Sie den Überstand vorsichtig mit einer Standard-P1000-Pipettenspitze und ersetzen Sie ihn durch 1 ml frisches N2-Medium. Wiederholen Sie diesen Waschschritt dreimal, um eine vollständige Entfernung der Dispase sicherzustellen.
HINWEIS: Jede verbleibende Dispase verhindert eine gleichmäßige Bildung von neuroektodermalen Sphäroiden und induziert den Zelltod. - Nach dem Waschen suspendieren Sie die Zellklumpen in 3 ml N2-Medium und übertragen Sie sie auf eine Vertiefung einer 6-Well-Platte und fügen Sie 40 ng / ml bFGF hinzu.
HINWEIS: Wenn eine hohe Anzahl von neuroektodermalen Kolonien abgelöst würde, könnten diese Kolonien über zwei oder mehr Vertiefungen einer 6-Well-Platte plattiert werden, um die Fusion von Sphäroiden zu verhindern. Überprüfen Sie 24 h nach der Entfernung der Kolonien, ob sich die Sphäroide gebildet haben. - Halten Sie die Sphäroide in den nächsten 3-4 Tagen im selben Medium, fügen Sie jedoch täglich frisches bFGF (40 ng / ml) zu jedem Brunnen hinzu, um die Proliferation neuroektodermaler Zellen zu fördern, sich selbst zu organisieren und Neuroepithele zu induzieren und zu erweitern.
HINWEIS: Wenn Sphäroide mit einer höheren Dichte plattiert wurden, wird das Medium wahrscheinlich gelb und muss alle 2 Tage durch frisches bFGF (40 ng / ml) ersetzt werden. Dies wird jedoch nicht empfohlen, und stattdessen sollte in jedem Bohrloch eine geringere Anzahl von Sphäroiden beibehalten werden, um dieses Problem zu vermeiden. Sphäroide können nach 3 Tagen in die Basalmembranmatrix eingebettet werden, wenn Neuroepithele offensichtlich sind. Wenn Neuroepithele nicht offensichtlich sind oder nicht stark aussehen, dann halten Sie die Sphäroide für einen weiteren Tag und überprüfen Sie es erneut.
- Kortikale Organoiddifferenzierung und -erhaltung des Gehirns (Tag 8)
- Bereiten Sie die Klemmendifferenzierungsmedien (DM) mit den in Tabelle 2 aufgeführten Reagenzien vor. Bringen Sie dieses Medium zu RT.
- Tauen Sie 100% hESC-qualifizierte Basalmembranmatrix auf Eis auf.
- Bereiten Sie ein Blatt Parafilm mit Grübchen vor, das mit 70% Ethanol in einer 10 cm großen Petrischale sterilisiert ist, und legen Sie es auf ein Stereomikroskop unter einer Haube.
HINWEIS: Ein leeres Tablett mit 200 μL Pipettenspitzen kann verwendet werden, um ein Raster von Grübchen zu erzeugen. - Schneiden Sie das Ende von einer 100 μL Pipettenspitze ab (um es breit zu bohren). Dies wird verwendet, um neuronale Sphäroide in die Kellermatrix einzubetten, ohne sie zu brechen.
- Wählen Sie mit einem Stereomikroskop neuronale Sphäroide ähnlicher Größe (500 μm) aus der 6-Well-Platte aus und übertragen Sie sie mit der 100-μL-Pipettenspitze mit breiter Bohrung auf die Parafilm-Grübchen, wobei Sie pro Grübchen ein einzelnes neuronales Sphäroid platzieren.
- Entfernen Sie vorsichtig überschüssige Medien und lassen Sie gerade genug übrig, um die Sphäroide zu bedecken.
HINWEIS: Dies dient dazu, die Qualität der Sphäroide vor dem Hinzufügen der Kellermatrix zu erhalten und sicherzustellen, dass sie nicht austrocknen. - Fügen Sie vorsichtig 18 μL Kellermatrix über das Sphäroid hinzu und positionieren Sie das Sphäroid in der Mitte des Matrixtropfens. Verwenden Sie das Ende einer 10 μL Pipettenspitze, um die Sphäroide in der Matrix zu zentrieren.
HINWEIS: Versuchen Sie, die Kellermatrix so schnell wie möglich hinzuzufügen, um zu vermeiden, dass die überschüssigen Medien um die Sphäroide herum austrocknen. - Die 10 cm Petrischale abdecken und in den Inkubator geben und die Parafilmschale mit in die Kellermatrix eingebetteten Sphäroiden bei 37 °C für 25 min inkubieren.
- Spülen Sie nach der Inkubation die eingebetteten Sphäroide mit einer P1000-Spitze mit 0,5 ml des DM-Mediums in eine niedrig angebrachte 24-Well-Platte ab, wobei Sie sicherstellen, dass einzelne eingebettete Sphäroide in jeweils einer Vertiefung platziert werden.
HINWEIS: Wenn mehr als ein eingebettetes Sphäroid in ein Bohrloch fällt, verwenden Sie eine P1000-Spitze mit breiter Bohrung, um das andere Sphäroid in ein neues Bohrloch zu übertragen. - Halten Sie die differenzierten kortikalen Gehirnorganoide in den DM-Medien für längere Zeit aufrecht, wobei Medienveränderungen alle 2 Tage auftreten, wenn die Organoide größer und älter werden.
HINWEIS: Während der ersten Woche der Differenzierung kann das Medium alle 3 Tage gewechselt werden.
2. Charakterisierung des neuronalen Alterns in kortikalen Organoiden
- Prozesskortikale Organoide für Kryosektionen:
HINWEIS: Die Schritte wurden in einer Biosicherheitshaube der Klasse 2 durchgeführt.- Bereiten Sie 2 ml-Röhrchen vor, die jeweils mit 1,5 ml 4% Paraformaldehyd (PFA) gefüllt sind.
- Schneiden Sie das Ende einer P1000-Pipettenspitze ab (um sie zu einer breiten Bohrung zu machen) und übertragen Sie jedes Organoid vorsichtig auf eine der oben vorbereiteten 2-ml-Röhren (ein Organoid pro Röhrchen).
HINWEIS: Um zu verhindern, dass sich überschüssige DM-Medien mit dem PFA vermischen, lassen Sie das Organoid in Richtung der Öffnung der P1000-Pipettenspitze sinken und legen Sie die Spitze direkt über der Oberseite der PFA in die Röhre, bevor Sie das Organoid herauspipettieren. Dies ermöglicht es dem Forscher, nur das Organoid und sehr minimale Medien zu übertragen. - Lassen Sie den Fixationsprozess bei 4 ° C für 1 h stattfinden.
- Mit einer ungeschnittenen P1000-Pipettenspitze überschüssiges PFA vorsichtig absaugen und 1,5 ml kaltes 1x PBS hinzufügen.
- Übertragen Sie die Röhren auf einen Orbitalschüttler, der bei 70 U / min für 10 min bei RT eingestellt ist.
- Wiederholen Sie den Waschvorgang mit kaltem 1x PBS dreimal, um sicherzustellen, dass alle PFA gründlich entfernt wurden.
HINWEIS: Entsorgen Sie PFA nicht in normalen Abfallbehältern; Bereiten Sie stattdessen einen speziellen Entsorgungsbehälter für chemische Abfälle dafür vor, da PFA eine Gefahr darstellt. - Tauchen Sie die Organoide in 1x PBS mit 30% Saccharose und inkubieren Sie bei 4 °C, bis alle Organoide auf den Boden des Schlauches gesunken sind.
HINWEIS: Die Zeit, die benötigt wird, um die Organoide absinken zu lassen, hängt von der Größe / dem Alter der Organoide ab. 3 Monate alte Organoide können bis zu 5 Stunden dauern. - Mit einer geschnittenen, breit gebohrten P1000-Pipettenspitze werden drei bis fünf Organoide vorsichtig in eine Montageform mit einer Montagelösung aus 30% Saccharose und 100% optimalem Schnitttemperaturmedium (OCT) im Verhältnis 3:2 übertragen.
- Verwenden Sie eine 10 μL Pipettenspitze mit Hilfe eines Stereomikroskops, um Organoide in einem gitterartigen Muster zu orientieren und zu positionieren.
- Legen Sie die Form auf Trockeneis, um die Soccharose-OCT-Lösung zu verfestigen, bevor Sie mit der Kryosektion (16-20 μm) unter Verwendung eines Kryostaten fortfahren.
HINWEIS: Für seneszenzassoziierte Beta-Galactosidase müssen alle Gewebe für die Schnittaufnahme verarbeitet werden, sobald sie gesunken sind. Für die Immunfluoreszenz können Gewebe für die Schnittaufnahme am nächsten Tag verarbeitet werden. Alle Objektträger, die Abschnitte enthalten, müssen vor der anschließenden Immunfluoreszenz oder Beta-Galactosidase bei -20 °C gelagert werden, wenn sie nicht sofort verfärbt werden.
- Verfahren zur Analyse der Seneszenz in den kortikalen Hirnorganoiden:
HINWEIS: Die folgenden Schritte können auf einem normalen Labortisch ausgeführt werden.- Übertragen Sie die Objektträger in einen Objektträger-Färbebehälter mit einem Deckel und waschen Sie das geschnittene Organoidgewebe dreimal mit 1x PBS für 10 Minuten bei RT, um überschüssige Montagelösungen zu entfernen.
- Anschließend das gewaschene Gewebe mit frisch hergestellter Beta-Galactosidase-Färbelösung über Nacht bei 37 °C inkubieren.
HINWEIS: Die Beta-Galactosidase-Färbelösung besteht aus Phosphatpuffer (für 10 ml Phosphatpuffer: 8,15 ml 1 M NaH2PO 4, 1,85 ml 1M Na2 HPO4) eingestellter pH-Wert = 6, 100 mM Kaliumhexacyanoferrat (III), 100 mM Kaliumhexacyanoferrat(II)-trihydrat, 5 M NaCl, 1 M MgCl2, 20 mg/ml X-Gal. Vermeiden Sie die Verwendung eines Standard-Zellkultur-Inkubators, der CO 2 enthält, da das CO2 den pH-Wert der Beta-Galactosidase-Färbelösung verändert. - Waschen Sie die gefärbten Taschentücher dreimal mit 1x PBS für jeweils 10 Minuten bei RT, um die Beta-Galactosidase-Lösung zu entfernen.
- Montieren Sie die gewaschenen Taschentücher mit einem Antifade-Mountant aus Glas und lassen Sie die Montagelösung 30 Minuten lang bei RT erstarren, bevor sie unter dem Mikroskop betrachtet wird.
Hier haben wir ein robustes Protokoll beschrieben, das es Forschern ermöglicht, homogene hPSC-abgeleitete kortikale Gehirnorganoide zu erzeugen, die die in vivo menschliche kortikale Gehirnregion innerhalb von 1-3 Monaten nach der Kultur nachahmen. hPSC-Kolonien werden zunächst in Differenzierungsmedien kultiviert, um neuroektodermale Kolonien zu erzeugen, die dann zur Bildung neuronaler Sphäroide verwendet werden können. Diese Sphäroide werden anschließend in eine Basalmembranmatrix eingebettet und über einen längeren Zeitraum aufrechterhalten, um Organoide zu erzeugen, die zur Modellierung des neuronalen Alterns verwendet werden können (siehe Abbildung 1 für einen Überblick über das Protokoll). Es ist erwähnenswert, dass die Kultivierung dieser Organoide in ultra-unbeschichteten 24-Well-Platten zellulären Stress verursacht und seneszenzassoziierte Phänotypen über 13 Wochen In-vitro-Kultur fördert. Von diesem Protokoll abgeleitete Organoide können auch in gerührten Bioreaktoren für ein optimales Wachstum und eine optimale Differenzierung von neuralen Zellen der kortikalen Platte oder an der Luft-Flüssigkeits-Grenzfläche aufrechterhalten werden.
Zu Beginn werden hPSC-Kolonien 1 Tag lang vor der neuroektodermalen Differenzierung kultiviert. Es ist von entscheidender Bedeutung, dass diese hPSC-Kolonien nur zu 20% -30% Konfluenz kultiviert werden und von höchstmöglicher Qualität sind: eine enge flache Monoschicht ohne differenzierte Zellen, die die Kolonien kontaminieren (Abbildung 2A, B). Die Pluripotenz der hPSC-Kolonien sollte durch die Expression von Markern wie NANOG bestätigt werden (Abbildung 2C). Die validierten hPSC-Kolonien werden dann den neuroektodermalen N2-Differenzierungsmedien mit SB-431542 und LDN-193189 ausgesetzt. Nach 3 Tagen der Erhaltung in diesem Medium sollten sich die hPSC-Kolonien in neuroektodermale Kolonien differenziert haben und nicht mehr die gleiche enge flache Monoschichtmorphologie der hPSCs aufweisen (Abbildung 2B), sondern sie werden zu längeren säulenförmigen Zellen (Abbildung 2B). Diese Zellen werden auch negativ für Pluripotenzmarker wie NANOG sein (Abbildung 2C).
In diesem Stadium werden die neuroektodermalen Kolonien enzymatisch mit Dispase abgelöst, und jede gesunde und erfolgreich abgelöste Kolonie darf sich selbst organisieren und ein junges neuronales Sphäroid bilden (Abbildung 2D, Ergänzendes Video 1). Nur gesunde, saubere neuroektodermale Kolonien lösen sich in dem für die Dispase-Aktivität angegebenen Zeitrahmen; Alle anderen Kolonien sollten ignoriert werden, da sie zu einer schlechteren Qualität des Sphäroides führen. Bei täglicher Exposition gegenüber FGF2 in den N2-Medien vermehren sich die neuralen Stammzellen (SOX2+) in diesen Sphäroiden (Abbildung 2D, Tag 1) und bilden eine signifikante Anzahl neuronaler Rosetten (Abbildung 2D, Tag 4). Diese Rosetten exprimieren die Tight Junction und den Epithelmarker ZO1 in Zellen, die sich im Zentrum der Rosetten und entlang des äußeren Randes des Sphärooids befinden, und demonstrieren die apikal-basale Polarität des Sphäroids (Abbildung 2D, Tag 4). Das Verfahren zur 3D-Bildgebung von Sphäroiden im Großraum wurde vordem 13. Die tägliche Inspektion der Sphäroide sollte die Bildung eines engen, dunklen äußeren Randes und einer hellen Peripherie der Sphäroide, der neuroepithelialen Schicht, aufklären. Diese Schicht sollte nach 3-4 Tagen mit einem ungefähren Durchmesser von 500 μm ausreichend gebildet sein, wobei die Sphäroide in die Grundmatrix eingebettet werden können. Ist diese Schicht nicht vorhanden oder nur schwach ausgebildet, sind die Sphäroide nicht ausreichend entwickelt, um voranzukommen. Es wird empfohlen, einen weiteren Tag zu warten, um eine Veränderung zu beobachten, aber wenn dies nicht beobachtet wird, ignorieren Sie diese Sphäroide.
Ein repräsentatives Hellfeldbild der Sphäroide nach 3 Tagen Kultur ist in Abbildung 2D zu sehen. Sphäroide mit einer engen neuroepithelialen Schicht, die nicht mit anderen benachbarten Sphäroiden verschmolzen sind, halbtransparentes Gewebe haben und eine neuronale Rosettenbildung aufweisen, werden so gewählt, dass sie in die Kellermatrix eingebettet werden. Einmal eingebettet, wird sich das Sphäroid schnell vermehren und anfangen zu knospen: Knoten von kompaktem Gewebe werden erscheinen, die sich vom Hauptkörper des Sphäroides nach außen ausdehnen. Dies zeigt sich zwischen 1-3 Wochen in der Kellermatrix und kann über mehrere Zelllinien beobachtet werden (Abbildung 3A). Die quantitative Analyse der eingebetteten Sphäroide bestätigt das Vorhandensein von Epithelzellen in bis zu 100% der Sphäroide über drei verschiedene Zelllinien hinweg, was die von diesem Protokoll erwartete Homogenität und Reproduzierbarkeit bestätigt (Abbildung 3B). Die Quantifizierung des Durchmessers von Organoiden während der In-vitro-Differenzierung bestätigt die Reproduzierbarkeit über verschiedene hPSC-Linien hinweg (Abbildung 3C). Wenn keine Knospen auftreten, entwickeln sich die Sphäroide nicht angemessen und sollten verworfen werden. Sobald die Sphäroide in eine Matrix eingebettet wurden, schreitet ihre Entwicklung voran, und die Sphäroide werden nun als Organoide bezeichnet. Die Immunfluoreszenzfärbung bestätigt auch das Vorhandensein von neuralen Vorläuferzellen (PAX6) sowie kortikalen Schichtmarkern, die mit CTIP2 und SATB2 in den Organoiden mit klarer Schichtung angefärbt sind (Abbildung 3D, E). Diese Schichtung ist über verschiedene Zeitpunkte der Organoiderhaltung hinweg beobachtbar (Abbildung 3D,E). Die Methode der Immunhistochemie von Geweben wurde vor14 beschrieben.
Eine mögliche Anwendung dieser Organoide besteht darin, zu untersuchen, wie sich neuronale Alterungsprozesse auf das Gehirn auswirken. Um dies zu untersuchen, werden erfolgreich erzeugte Organoide aus mehreren verschiedenen Zeitpunkten für die Schnittung und Färbung für molekulare Standard-Biomarker der Seneszenz wie Seneszenz-assoziierte Beta-Galactosidase und p21 geerntet. Abbildung 4A zeigt ein repräsentatives Bild der seneszenzassoziierten Beta-Galactosidase-Färbung von Organoiden 4 und 13 Wochen nach der Einbettung in die Basalmembranmatrix. Zwischen den Wochen 4 und 13 gibt es einen deutlichen Anstieg der Präsenz von Seneszenz-assoziierter Beta-Galactosidase, was darauf hindeutet, dass zelluläre Seneszenz, ein anerkannter Treiber der organismischen Alterung, in dieser Zeit in der Kultur aufgetreten ist. Die Immunfluoreszenzfärbung von Organoiden in Woche 13 bestätigte das Vorhandensein eines anderen Seneszenzmarkers, p21, der mit dem reifen kortikalen neuronalen Marker (CTIP2) ko-markiert wurde und in Abbildung 4B zu sehen ist. Es sollte jedoch beachtet werden, dass das Vorhandensein von p21 ein Marker für den Stillstand des Zellzyklus ist und an sich kein definitiver Marker der Seneszenz ist, und der Nachweis anderer Seneszenzmarker wie p16 und SASP (Seneszenz-assoziierter sekretorischer Phänotyp) wird empfohlen, um Zellen definitiv als seneszent zu identifizieren.
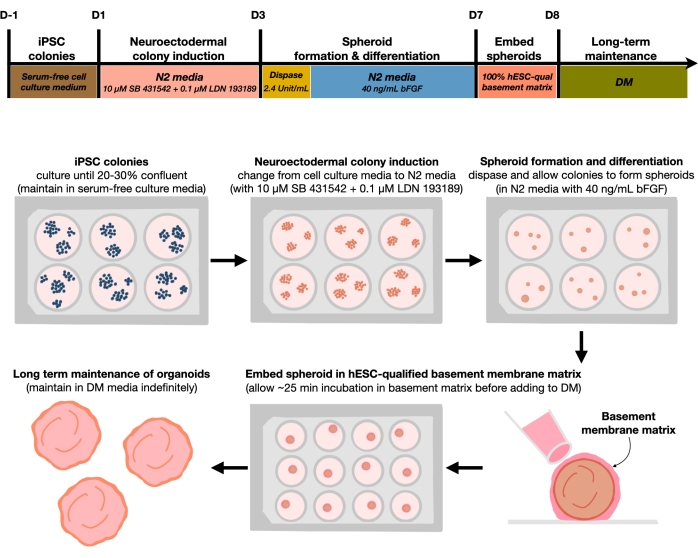
Abbildung 1: Schematische Darstellung zur Erzeugung reproduzierbarer kortikaler Hirnorganoide. Schematischer Ablauf des experimentellen Verfahrens zur Erzeugung kortikaler Hirnorganoide aus hPS-Zellen, die im feederfreien Medium gehalten werden. Der Workflow bietet einen Überblick über sechs Schritte, die erforderlich sind, um die 2D-hPSCs in 3D-gemusterte kortikale Plattenmenschliche Gewebe in Organoiden zu unterscheiden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
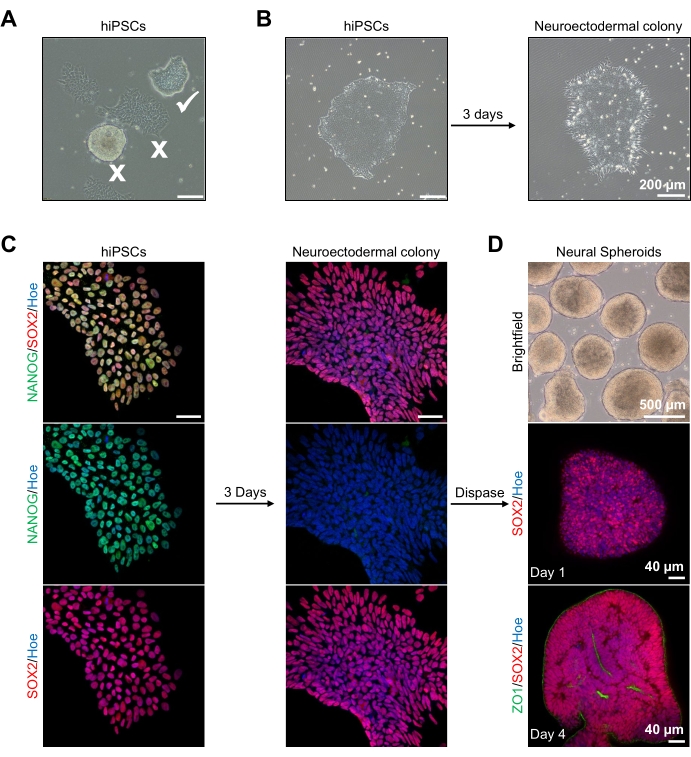
Abbildung 2: Erzeugung neuronaler Sphäroide aus neuroektodermalen Kolonien-hPS-Kolonien . (A) Repräsentative Bilder von menschlichem PSK, die optimale (weiße Zecke) und differenzierte Kolonien (weißes Kreuz) aufweisen. Skalenbalken: 200 μm, 4-fache Vergrößerung. (B) Repräsentatives Bild der neuroektodermalen Kolonie, abgeleitet von hPS-Zellen nach 3 Tagen dualer SMAD-Inhibitor-Behandlungen. Skalenbalken: 200 μm, 4-fache Vergrößerung. (C) Menschliche PSC-Kolonien wurden in neuroektodermale Kolonien unterschieden. Die Bilder zeigen die Färbung von PSC- (an Tag 1) und neuroektodermalen (an Tag 3) Kolonien mit SOX2 (Rot), NANOG (Grün), alle Kerne wurden mit Hoechst 33342 (blau) konterkariert. Skalenbalken: 40 μm, 100-fache Vergrößerung. (D) Bilder, die die Entwicklungsstadien von kortikalen Hirnsphäroiden im Laufe der Zeit in Kultur in vitro unter Hellfeld zeigen, und Wholemount-Immunen, die an Tag 1 mit SOX2 (rot) und doppelt immunisiert mit SOX2 (rot) und ZO1 (grün) an Tag 4 gefärbt sind. Die Skalierungsleiste des Hellfeldbildes beträgt 500 μm, 4-fache Vergrößerung, Skalierungsbalken der unteren Bilder sind 40 μm, 20-fache Vergrößerung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
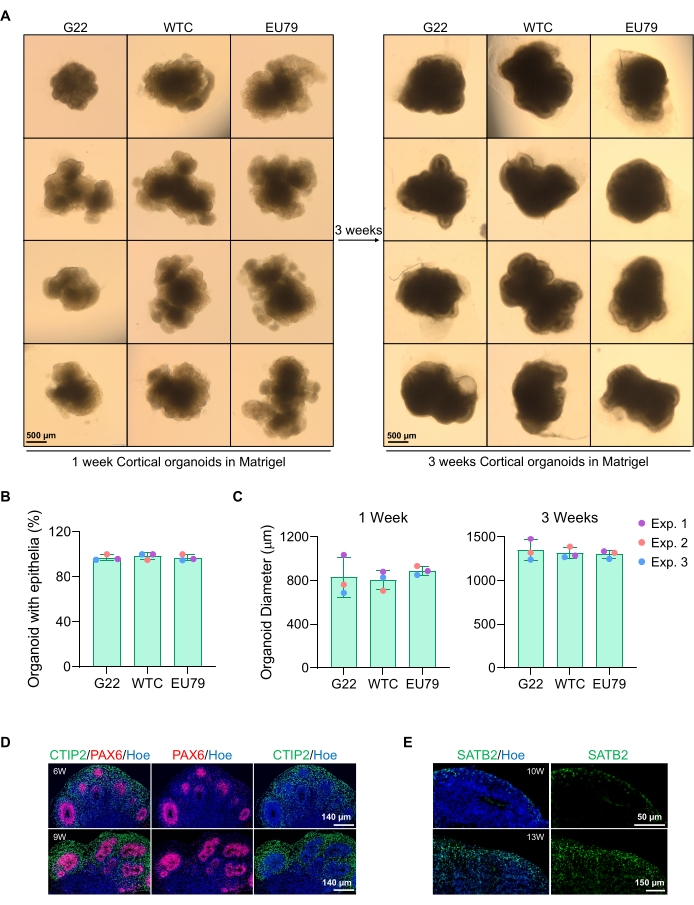
Abbildung 3: Charakterisierung kortikaler Gehirnorganoide, abgeleitet von verschiedenen hPSC-Linien. (A) Repräsentative Bilder von kortikalen Gehirnorganoiden, abgeleitet von G22-, WTC- und EU79-iPSC-Linien für Menschen, die über 3 Wochen in vitro kultiviert wurden. Der Maßstabsbalken aller Bilder beträgt 500 μm, 2-fache Vergrößerung. (B) Prozentsätze der erfolgreichen Erzeugung kortikaler Gehirnorganoide nach 3 Wochen In-vitro-Differenzierung in verschiedenen hPSC-Linien (G22, WTC und EU79). N = 3. Die Daten werden als mittlere ± Standardabweichung dargestellt. (C) Balkendiagramme, die das Wachstum kortikaler Gehirnorganoide (basierend auf dem durchschnittlichen Durchmesser) in den Wochen 1 und 3 der In-vitro-Differenzierung in verschiedenen Linien humaner pluripotenter Stammzelllinien (G22, WTC und EU79) zeigen. N = 3. Die Daten werden als mittlere ± Standardabweichung dargestellt. (D) Repräsentative Bilder von Abschnitten von 6 Wochen und 9 Wochen alten kortikalen Gehirnorganoiden, die von G22-hPS-Zellen abgeleitet sind, immungefärbt für die Ventrikelzone PAX6 (rot) und die kortikale Platte CTIP2 (grün). Alle Abschnitte wurden mit Hoechst 33342 (blau) gegengebeizt. Maßstabsleiste = 140 μm, 20-fache Vergrößerung. W ist Woche. (E) Repräsentative Bilder von Abschnitten von 10 Wochen und 13 Wochen alten kortikalen Gehirnorganoiden, die von WTC-hPS-Zellen abgeleitet sind, immungefärbt für kortikale Schicht IV SATB2 (grün). Alle Abschnitte wurden mit Hoechst 33342 (blau) gegengebeizt. 10 Wochen Bildmaßstabsleiste = 50 μm, 40-fache Vergrößerung. 13 Wochen Bild Maßstabsleiste = 150 μm, 40-fache Vergrößerung. W ist Woche. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
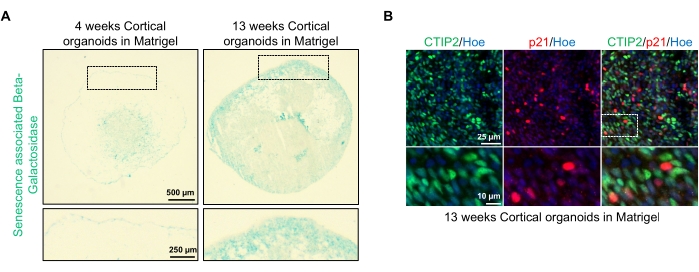
Abbildung 4: Charakterisierung der Seneszenz in kortikalen Gehirnorganoiden aus hPSCs. (A) Repräsentative Bilder von Abschnitten menschlicher kortikaler Gehirnorganoide, abgeleitet von WTC-hPSCs, die 4 und 13 Wochen lang in vitro kultiviert und mit SA-β-gal gefärbt wurden. Maßstabsleiste = 500 μm, Maßstabsleiste von vergrößerten Bildern = 250 μm, 4-fache Vergrößerung. Das gepunktete Feld zeigt ein vergrößertes Bild an. (B) Repräsentative Bilder von Schnitten von 13 Wochen alten kortikalen Gehirnorganoiden, die aus humanen EU79-hPS-Zellen gewonnen wurden, immunfärbt für kortikale Neuronen CTIP2 (grün) und p21 (rot). Alle Abschnitte wurden mit Hoechst 33342 (blau) gegengebeizt. Maßstabsleiste = 25 μm, Maßstabsleiste für gezoomte Bilder = 10 μm, 40-fache Vergrößerung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
| Medienkomponenten | Konzentration |
| DMEM Nährstoffmischung F12 10x 500 ml (DMEM/F-12) | |
| N2 Ergänzung 5 ml (100x) | Supplmentiert zu 1% |
| B 27 Zuschlag 10 ml | Ergänzt um 2% |
| MEM-Lösung für nicht-essentielle Aminosäuren (100x) | Ergänzt um 1% |
| Penicillin-Streptomycin (10.000 U/ml) | Ergänzt um 1% |
| 2-Mercaptoethanol 50 ml (1000x) | Ergänzt um 0,1% |
Tabelle 1: N2 Mittel. In der Tabelle sind die Reagenzien aufgeführt, die zur Herstellung des N2-Mediums erforderlich sind.
| Medienkomponenten | Konzentration |
| DMEM Nährstoffmischung F12 10x 500 ml (DMEM/F-12) | DM-Medien werden im Verhältnis 1:1 von DMEM/F12 und neurobasalen Medien hergestellt |
| Neurobasales Medium | |
| N2 Ergänzung 5 ml (100x) | Supplmented bei 0,5% |
| B 27 Zuschlag 10 ml | Ergänzt um 1% |
| MEM-Lösung für nicht-essentielle Aminosäuren (100x) | Ergänzt um 1% |
| GlutaMAX Ergänzung 100x | Ergänzt um 1% |
| Penicillin-Streptomycin (10.000 U/ml) | Ergänzt um 1% |
| Insulinlösung Humanes Rekombinantes | 12,5 μL für 50 mL Medien |
| 2-Mercaptoethanol 50 ml (1000x) | 17,5 μL für 50 mL Medien |
Tabelle 2: Differenzierungsmedium (DM). In der Tabelle sind die Reagenzien aufgeführt, die zur Herstellung des Differenzierungsmediums erforderlich sind.
Ergänzendes Video 1. Live-Bildgebung der induzierten hNEct 2D-Blatt-/Koloniekonvertierung in 3D unter der Behandlung von bFGF. Induzierte Kolonien von hNEct wurden vorsichtig von der Schale mit Dispase wie oben beschrieben gelöst und auf eine 6-Well-Kulturplatte mit niedriger Befestigung übertragen. 2D-hNEct-Kolonien wurden innerhalb von 12 h in 3D-hNEct-Sphäroide umgewandelt. Serienbilder wurden alle 5 Minuten aufgenommen. Maßstabsleiste = 100 μm. Bitte klicken Sie hier, um dieses Video herunterzuladen.
Um die Verwendung von hPSC-abgeleiteten Gehirnorganoiden im Arzneimittel-Screening und in der Krankheitsmodellierung zu ermöglichen, ist es von entscheidender Bedeutung, Organoide nach einem replizierbaren und zuverlässigen Protokollherzustellen 15. Gehirnorganoide werden üblicherweise aus embryonalen Körpern erzeugt, die aus hPS-Zellen gewonnen werden, die dann in eine extrazelluläre Matrix eingebettet werden, die die Gewebeexpansion und neuronale Differenzierung fördert. Im Vergleich zu Protokollen wie Lancasters 1,16,17 und Velasco 18, die von embryonalen Körpern ausgehen und einen Standard-Differenzierungspfad ermöglichen, dem die sich entwickelnden Organoide folgen, haben wir festgestellt, dass der Beginn der kortikalen Gehirnorganoidbildung mit menschlichen NEct-Zellen und nicht mit embryonalen Körpern die Konsistenz der kortikalen Gehirnorganoidbildung verbessert. Dies ermöglicht folglich auch die für das medikamentöse und phänotypische Screening erforderliche Skalierung. Da humane NEct-Zellen nicht nur in beträchtliche Mengen erweitert werden können, sondern auch leicht kryokonserviert werden können, verbessert dieser Ansatz auch die Replizierbarkeit zwischen den Experimenten. Es sollte auch beachtet werden, dass im Vergleich zu anderen Protokollen, die die Verwendung von Bioreaktoren und ähnlichen Technologien übernommen haben, keine spezielle Ausrüstung für dieses Protokoll erforderlich ist, so dass es für jedes Laborgeeignet ist 6. Schließlich wird die Zeit, die benötigt wird, um reife Organoide zu erzeugen, die positiv für kortikale Schichtmarker wie SATB2 sind, im Vergleich zu Lancaster 1 und Bioreaktorprotokollen 6,19 reduziert, wodurch es besser geeignet ist, den Entwicklungsverlauf der menschlichen kortikalen Entwicklung in Gesundheit und Krankheiten zu untersuchen 1,6,16.
Angesichts der kontinuierlich wachsenden globalen Auswirkungen alterungsbedingter Krankheiten wie Demenz, die mit einer Zunahme seneszenter Zelltypen im Gehirn verbunden sind, die zur Pathogenese beitragen, ist die Fähigkeit, Verbindungen zu identifizieren und zu testen, die die Alterung des Gehirns verbessern können, von enormem Interesse. Obwohl bekannt ist, dass hPS-Zellen während des Reprogrammierungsprozesses epigenetisch verjüngtwerden 20, finden wir einen robusten Anstieg der seneszenten Zellen in kortikalen Gehirnorganoiden, die über einen längeren Zeitraum kultiviert wurden. Dies ist eine vielversprechende Entwicklung, die nun das Screening von Medikamenten ermöglicht, die solche seneszenten Zellen aus dem Gehirn eliminieren (Senolytika) oder diesen Prozess verlangsamen (Senostatika)21. Da humane NEct-abgeleitete kortikale Gehirnorganoide menschlichen Ursprungs sind, wird dieser Ansatz wahrscheinlich den traditionellen Weg zur Vermarktung solcher neuartigen Therapeutika verkürzen.
Es gibt zwei kritische Schritte in diesem Protokoll. Der erste ist der korrekte Grad der Konfluenz der hPSC-Kolonien zum Zeitpunkt der Differenzierung. hPSC-Kolonien müssen zu höchstens 30% konfluent sein, um sicherzustellen, dass erzeugte NEct-Kolonien nicht mit benachbarten Kolonien verschmelzen und dass einzelne Organoide klongetrieben werden. Der zweite kritische Schritt beinhaltet die korrekte Verwendung von Dispase, um die NEct-Kolonien zu heben und die neuronalen Sphäroide zu produzieren. Der Zeitpunkt der Inkubation mit Dispase ist entscheidend für die letztendliche Qualität der erzeugten neuronalen Sphäroide. Dies liegt daran, dass eine übermäßige Exposition von Kolonien mit Dispase für die Zellentoxisch ist 22 und schließlich die Qualität der erzeugten Organoide beeinflusst. Die Einschränkung dieses Protokolls besteht darin, dass es schwierig ist, die Größe der neuronalen Sphäroide zu kontrollieren, da sie von der Größe der anfänglichen Kolonien abhängt, die mit Dispase angehoben werden. Dieses Problem kann jedoch überwunden werden, indem neuronale Sphäroide mit ähnlicher Größe ausgewählt werden, wenn mit der Einbettungsphase fortgefahren wird.
Schließlich könnten sich zukünftige Anwendungen auf die Verwendung dieser reproduzierbaren kortikalen Organoide in der Roboteranalyse und bei biopharmazeutischen Screening-Ansätzen erstrecken, die typischerweise in dieser Branche verwendet werden. Dies wird durch vorläufige Daten aus unserem Labor gestützt, die darauf hindeuten, dass die Erzeugung von kortikalen Gehirnorganoiden aus menschlichen NEct-Zellen leicht automatisiert werden kann, was sie mit diesen Ansätzen kompatibel macht.
Die Autoren haben nichts offenzulegen.
Diese Arbeit wird vom Medical Research Future Fund-Accelerated Research, dem Leukodystrophie-Flaggschiff Massimo's Mission (EPCD000034), dem Medical Research Future Fund-Stem Cell Mission (APP2007653) unterstützt. Die Autoren danken Dr. Ju-Hyun Lee (Korea University) für die Generierung von Daten in Supplementary Video 1.
| Name | Company | Catalog Number | Comments |
| 16% Formaldehyde (W/V) Methanol-free | Thermo Fisher Scientific | 28908 | 4% of PFA are diluted in 1x PBS |
| 2-Mercaptoethanol 50 mL(1000x) | Life Technologies Australia (TFS) | 21985023 | Used in NM and DM media |
| B 27 Supplement 10 mL | Life Technologies Australia (TFS) | 17504044 | Used in NM and DM media |
| CKX53 microscope with SC50 camera | Olympus | ||
| Corning Costar 6 well cell culture plates | Sigma Aldrich Pty Ltd | CLS3516-50EA | |
| Dispase II powder | Thermo Fisher Scientific | 17105041 | Powder is dissolve in HBSS, filtered through 0.22 µm filter, aliquote at 10 mL and store at -20 °C |
| DMEM Nutrient Mix F12 10x 500 mL (DMEM/F-12) | Thermofisher | 11320082 | Used in NM and DM media |
| DMSO Dimethyl Sulfoxide | Sigma Aldrich Pty Ltd | D2650-100ML | |
| Dulbecco's Phosphate Buffered Saline | Sigma Aldrich Pty Ltd | D1408-500ML | |
| Falcon Matrigel hESC-qualified Matrix | In Vitro Technologies Pty Ltd | FAL354277 | Make aliquotes of 100 µL and stored at -20 °C |
| GlutaMAX Supplement 100x | Thermo Fisher Scientific | 35050061 | Used in NM and DM media |
| Hanks Balanced Salt Solution | Sigma Aldrich Pty Ltd | H8264 | |
| Human induced pluripotent stem cells (EU79) | In-house reporogrammed from skin fibroblast | ||
| Human induced pluripotent stem cells (G22) | Genea Biocells | Obtained from Genea Biocells (San Diego, United States) | |
| Human induced pluripotent stem cells (WTC) | Gift from Professor Bruce Conklin | ||
| InSolution TGF-Β RI Kinase Inhibitor VI, SB431542 | Merck | US1616464-5MG | |
| Insulin Solution Human Recombinant | Sigma Aldrich Pty Ltd | I9278 | Used in NM and DM media |
| LDN193189 Dihydrochloride | Sigma Aldrich Pty Ltd | SML0559-5MG | Used during differentiation |
| MEM Non-Essential Amino Acids Solution (100x) | Thermo Fisher Scientific | 11140050 | Used in NM and DM media |
| mTeSR Plus | STEMCELL TECHNOLOGIES | 100-0276 | Used to maintain hiPSC colonies prior to differentiation with NM media |
| N2 Supplement 5 mL (100x) | Life Technologies Australia Pty Ltd | 17502048 | Used in NM and DM media |
| Neurobasal Medium | Thermo Fisher Scientific | 21103049 | Used in DM media |
| OCT Embedding Compound Sakura Clear (118 mL/Bottle) | Tissue Tek | 4583 | |
| Parafilm M Roll Size 4 in. x 125 Ft | Sigma Aldrich Pty Ltd | P7793 | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 | Used in NM and DM media |
| Potassium Hexacyanoferrate (II) Trihydrate | Sigma Aldrich Pty Ltd | CP1087 | |
| Potassium hexacyanoferrate(III) | Sigma Aldrich Pty Ltd | 455946 | |
| Prolong Glass Antifade Mountant | Life Technologies Australia (TFS) | P36980 | |
| Recombinant Human FGF basic | R&D Systems | 233-FB-01M | Aliquotes are made at 20 µg/mL and stored at -20 °C |
| SB431542 | Tocris | 1614 | Used during differentiation |
| Sucrose | Sigma Aldrich Pty Ltd | PHR1001-1G | 30% of sucrose are diluted in 1x PBS |
| Ultra-Low attachment multiwell plates , 24 well plate, polystyrene | Sigma Aldrich Pty Ltd | CLS3473-24EA | |
| X-GAL EA | Life Technologies Australia (TFS) | R0404 | Make aliquotes of 20 mg/mL and storde at -80 °C |
- Lancaster, M. A., Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nature Protocols. 9 (10), 2329-2340 (2014).
- Mansour, A. A., et al. An in vivo model of functional and vascularized human brain organoids. Nature Biotechnology. 36 (5), 432-441 (2018).
- Xiang, Y., et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell. 21 (3), 383-398 (2017).
- Bagley, J. A., Reumann, D., Bian, S., Lévi-Strauss, J., Knoblich, J. A. Fused cerebral organoids model interactions between brain regions. Nature Methods. 14 (7), 743 (2017).
- Kwak, T. H., et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson's disease modeling. Stem Cells. 38 (6), 727-740 (2020).
- Qian, X., et al. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nature Protocols. 13 (3), 565-580 (2018).
- Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Reports. 10 (4), 537-550 (2015).
- Shaker, M. R., Cooper-White, J., Wolvetang, E. J. Self-organizing 3D human choroid plexus-ventricle-cortical organoids. BioRxiv. , (2020).
- Shaker, M. R., Aguado, J., Chaggar, H. K., Wolvetang, E. J. Klotho inhibits neuronal senescence in human brain organoids. npj Aging and Mechanisms of Disease. 7 (1), 1-12 (2021).
- Shaker, M. R., et al. Anteroposterior Wnt-RA gradient defines adhesion and migration properties of neural progenitors in developing spinal cord. Stem Cell Reports. 15 (4), 898-911 (2020).
- Chambers, S. M., et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology. 27 (3), 275-280 (2009).
- Shaker, M. R., et al. Rapid and efficient generation of myelinating human oligodendrocytes in organoids. Frontiers in Cellular Neuroscience. 15, 631548 (2021).
- Lee, J. -. H., Shaker, M. R., Lee, E., Lee, B., Sun, W. NeuroCore formation during differentiation of neurospheres of mouse embryonic neural stem cells. Stem Cell Research. 34, 101691 (2020).
- Shaker, M. R., et al. Spatiotemporal contribution of neuromesodermal progenitor-derived neural cells in the elongation of developing mouse spinal cord. Life Sciences. 282, 119393 (2021).
- Shaker, M. R., et al. Neural epidermal growth factor-like like protein 2 Is expressed in human oligodendroglial cell types. Frontiers in Cell and Developmental Biology. 10, 803061 (2022).
- Lancaster, M. A., et al. Cerebral organoids model human brain development and microcephaly. Nature. 501 (7467), 373-379 (2013).
- Giandomenico, S. L., Sutcliffe, M., Lancaster, M. A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nature Protocols. 16 (2), 579-602 (2021).
- Velasco, S., et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 570 (7762), 523-527 (2019).
- Qian, X., et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 165 (5), 1238-1254 (2016).
- Hunter, Z. L., Leeson, H. C., Shaker, M. R., Wolvetang, E. J., Vadlamudi, L. Human induced pluripotent stem cells generated from epilepsy patients for use as in vitro models for drug screening. Stem Cell Research. 60, 102673 (2022).
- Kaur, A., Macip, S., Stover, C. M. An appraisal on the value of using nutraceutical based senolytics and senostatics in aging. Frontiers in Cell and Developmental Biology. 8, 218 (2020).
- Wang, F., et al. Safety and efficacy of dispase and plasmin in pharmacologic vitreolysis. Investigative Ophthalmology & Visual Science. 45 (9), 3286-3290 (2004).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved