
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Neuroscience
Generazione robusta e altamente riproducibile di organoidi cerebrali corticali per la modellazione della senescenza neuronale cerebrale in vitro
In questo studio, forniamo una tecnica dettagliata per un sistema di coltura organoide corticale semplice ma robusto che utilizza colture hPSC standard prive di alimentatore. Questo è un protocollo rapido, efficiente e riproducibile per la generazione di organoidi che modellano aspetti della senescenza cerebrale in vitro.
Gli organoidi cerebrali sono modelli tridimensionali del cervello umano in via di sviluppo e forniscono una piattaforma convincente e all'avanguardia per la modellazione delle malattie e lo screening genomico e farmacologico su larga scala. A causa della natura auto-organizzante delle cellule negli organoidi cerebrali e della crescente gamma di protocolli disponibili per la loro generazione, sono stati identificati problemi di eterogeneità e variabilità tra gli organoidi. In questo documento di protocollo, descriviamo un protocollo robusto e replicabile che supera in gran parte questi problemi e genera organoidi corticali da progenitori neuroectodermici entro 1 mese e che può essere mantenuto per più di 1 anno. Questo protocollo altamente riproducibile può essere facilmente eseguito in una sala di coltura tissutale standard e si traduce in organoidi con una ricca diversità di tipi di cellule che si trovano tipicamente nella corteccia umana in via di sviluppo. Nonostante la loro composizione precoce dello sviluppo, i neuroni e altri tipi di cellule cerebrali umane inizieranno a mostrare i tipici segni di senescenza nelle cellule neuronali dopo una prolungata coltura in vitro , rendendoli una piattaforma preziosa e utile per studiare i processi neuronali legati all'invecchiamento. Questo protocollo delinea anche un metodo per rilevare tali cellule senescenti negli organoidi cerebrali corticali utilizzando la colorazione beta-galattosidasi associata alla senescenza.
La nostra attuale conoscenza del cervello umano è stata in gran parte basata su modelli animali e campioni di cervello post-mortem. La biologia delle cellule staminali è un campo in rapida evoluzione che fornisce nuove intuizioni sulla biologia di base dello sviluppo del cervello umano e sui driver patologici dei disturbi del cervello umano. Le cellule staminali pluripotenti umane (hPSC) sono uno strumento inestimabile per modellare il cervello umano attraverso la generazione di organoidi, tessuto tridimensionale (3D) simile a un organo che in genere ricapitola le traiettorie di sviluppo, la composizione cellulare e l'architettura del cervello umano in via di sviluppo. Gli organoidi cerebrali sono auto-assemblati e composti da cellule staminali neurali, progenitori neurali specificati, neuroni maturi e tipi di cellule gliali. Gli organoidi, quindi, offrono un'opportunità unica per studiare il cervello umano precoce, che è spesso inaccessibile per la sperimentazione diretta ma ha anche limitazioni intrinseche come l'assenza di vascolarizzazione e un sistema immunitario.
Le metodologie per generare organoidi cerebrali sono state perseguite in due modi diversi: differenziazione non guidata e guidata. I metodi organoidi cerebrali non guidati si basano sulle capacità di differenziazione intrinseca spontanea delle cellule staminali che guidano la morfogenesi tissutale 1,2 e consentono l'emergere di una varietà di identità di lignaggio cellulare che vanno dal proencefalo, al mesencefalo e al cervello posteriore, al plesso coroideo, alla retina e al mesoderma. Al contrario, i metodi organoidi cerebrali guidati richiedono un uso sostanziale di fattori esterni per guidare le hPSC verso il modello desiderato di linee neuronali che rappresentano un tipo di regione cerebrale, come l'eminenza gangliare mediale3, il proencefalo4, il mesencefalo5, l'ipotalamo6, il cervelletto7 e il plesso coroideo8. Questa capacità di generare diverse regioni del cervello con diverse linee cellulari e il potenziale di fonderle a piacimento, rende gli organoidi cerebrali un modello eccellente per studiare lo sviluppo del cervello umano e decifrare i meccanismi alla base delle malattie legate al cervello. Sebbene questi metodi per generare organoidi cerebrali offrano una svolta nella modellazione delle regioni del cervello umano, la variabilità e l'eterogeneità tra gli organoidi rimangono una limitazione significativa per gli studi sistematici e quantitativi, come lo screening farmacologico.
L'attuale protocollo si basa su un metodo sviluppato nel nostro recente articolo9 e prevede la differenziazione selettiva delle colonie di hPSC verso l'identità del neuroectoderma (NEct) con doppi inibitori SMAD (SB-431542 e LDN 193189), che quindi hanno la capacità di auto-organizzarsi entro 4 giorni in sferoidi 3D neuroepitelio sotto l'influenza della segnalazione FGF2. Questi sferoidi del neuroepitelio generano in modo affidabile organoidi corticali omogenei con una composizione cellulare simile a quella in vivo entro 4 settimane dalla differenziazione. Il protocollo qui descritto si basa sui nostri precedenti risultati che mostrano che l'inibizione della doppia segnalazione SMAD (Suppressor of Mothers Against Decapentaplegic) promuove la differenziazione delle hPSC verso cellule staminali neurali rostrali derivate da progenitori neuroectodermici10 , tra gli altri, inibendo la scelta del destino delle cellule endodermiche, mesodermiche e trofoectodermiche11 . Inoltre, l'incorporazione degli sferoidi del neuroepitelio nella matrice della membrana basale qualificata hESC innesca un significativo germogliamento della neuroepitelia, formando ventricoli con polarità apicobasale. La coltura su larga scala ha mostrato riproducibilità e omogeneità degli organoidi corticali indipendenti da linee cellulari, cloni o lotti, e quindi rappresenta un sistema di cellule staminali affidabile e stabile per imitare lo sviluppo corticale umano precoce in salute e malattia in vitro. Delineiamo inoltre un protocollo per rilevare marcatori di cellule neuronali senescenti in organoidi cerebrali corticali derivati da hPSC che sono stati coltivati per periodi di tempo prolungati.
Dopo aver placcato le hPSC ad una densità di semina del 20%-30%, le cellule vengono trattate con doppi inibitori SMAD per 3 giorni per differenziare le colonie di hPSC verso colonie neuroectodermiche. Queste colonie vengono quindi sollevate delicatamente con dispase e seminate in piastre a 6 pozzetti di attacco ultra-basso integrate con FGF2. Le colonie 2D galleggianti si auto-organizzano in sferoidi neuroectodermici 3D durante la notte e vengono mantenute per 4 giorni in N2 medio integrato quotidianamente con FGF2. Una volta che gli sferoidi hanno stabilito lo strato neuroepiteliale, possono essere incorporati nella matrice della membrana basale. Aggiungendo di routine un nuovo mezzo di differenziazione terminale, i ricercatori osserveranno una progressiva espansione e germogliamento della neuroepitelia negli organoidi corticali. I ricercatori potrebbero voler dissociare questi organoidi per condurre la profilazione trascrizionale e proteomica. Inoltre, l'imaging a campo luminoso è raccomandato per monitorare la qualità degli organoidi corticali. L'analisi può essere eseguita mediante fissazione, criosezione e immunocolorazione. Descrizioni e metodi per queste tecniche sono stati precedentemente descritti12. In definitiva, questo protocollo consente ai ricercatori di generare rapidamente e robustamente organoidi cerebrali corticali omogenei per modellare il cervello corticale umano in via di sviluppo, con attrezzature a basso costo e limitate, e per studiare aspetti della senescenza neuronale cellulare, come delineato in questo articolo.
1. Generazione di organoidi cerebrali corticali
NOTA: Tutti i passaggi in questa sezione del protocollo si verificheranno in una cappa di biosicurezza di Classe 2, se non diversamente specificato.
- Induzione di colonie neuroectodermiche 2D da coltura hPSC 2D (Giorni da -1 a 3)
- Prima dell'induzione, placcare le colonie di hPSC su una matrice di membrana basale qualificata hESC in una piastra a 6 pozzetti con una densità del 20%-30%. Raggiungi questa densità passando colonie di hPSC da un pozzetto di una piastra a 6 pozzetti con una confluenza del 60% in tre pozzetti di una piastra a 6 pozzetti.
- Per il rivestimento della matrice della membrana basale, diluire la matrice della membrana basale con un rapporto di 1:50 in un mezzo basale semplice. Depositare uniformemente 1 mL/pozzetto di una piastra a 6 pozzetti, incubare per 1 ora a temperatura ambiente (RT) e quindi aspirare.
- Mantenere le colonie di hPSC per 1 giorno in 2 mL di un terreno di coltura cellulare privo di siero prima della differenziazione.
- Il giorno della differenziazione hPSC, ispezionare le colonie di hPSC utilizzando la microscopia a campo luminoso con ingrandimento da 4x a 10x per garantire colonie sane senza differenziazione rilevabile.
NOTA: le hPSC sane formeranno colonie di bordi stretti con cellule che hanno un nucleo grande, un citoplasma molto piccolo e nucleoli prominenti. Le colonie di iPSC differenziate mostreranno chiare differenze morfologiche rispetto a quelle delle colonie hPSC descritte sopra, in particolare intorno ai bordi esterni delle colonie o al centro. - Aggiungere i reagenti elencati nella Tabella 1 per costituire il mezzo N2 necessario per la differenziazione. Portare questo mezzo a RT prima dell'uso.
- Una volta a RT, aspirare il terreno di coltura cellulare privo di siero da ciascun pozzetto della piastra a 6 pozzetti e sostituirlo con 2 ml di mezzo N2 delicatamente aggiunto con una pipetta sierologica da 5 ml.
- Aggiungere i doppi inibitori SMAD, SB-431542 (10 μM) e LDN 193189 (100 nM).
NOTA: Gli inibitori SMAD possono essere aggiunti al mezzo N2 dopo che il mezzo è stato inserito in ciascun pozzetto o alla quantità richiesta di terreno N2 prima di sostituire il terreno di coltura cellulare privo di siero. Gli inibitori possono essere incorporati uniformemente nel mezzo facendo roteare delicatamente la piastra o invertendo il tubo contenente il mezzo e gli inibitori 3-4 volte. - Aggiungere nuovi supporti N2 integrati con SB-431542 (10 μM) e LDN 193189 (100 nM) al giorno a ciascun pozzetto per i successivi 2 giorni.
NOTA: Il mezzo N2 fresco viene aggiunto per ridurre l'esposizione prolungata delle cellule al DMSO che viene utilizzato per sciogliere i composti 193189 SB-431542 e LDN e prevenire la citotossicità.
- Generazione di sferoidi neuroectodermici 3D da colonie neuroectodermiche 2D indotte (giorni da 3 a 7)
- Sollevare le colonie neuroectodermiche indotte usando la dispasi seguendo i passaggi 1.2.2-1.2.8.
- In primo luogo, rimuovere 2 mL di mezzo N2 dalla piastra a 6 pozzetti e lavare 1x con HBSS per assicurarsi che tutto il mezzo N2 venga rimosso.
NOTA: Il mezzo N2 può interferire con l'attività enzimatica della dispasi, impedendo un adeguato distacco delle colonie neuroectodermiche dal pozzo. - Aggiungere 1 mL di 2,4 unità/mL di dispase a ciascun pozzo contenente colonie.
- Incubare il pozzetto per 20-25 min (massimo 30 min) a 37 °C. Controllare regolarmente il distacco della colonia.
NOTA: Piccole colonie possono staccarsi entro 20 min. Tutte le colonie che rimangono bloccate dopo 30 minuti dovrebbero essere ignorate. - Dopo l'incubazione, aggiungere 1 mL di mezzo N2 al pozzo per fermare l'attività dell'enzima dispasi e trasferire le colonie in un tubo da 15 ml utilizzando una punta della pipetta P1000 a foro largo o una punta della pipetta P1000 modificata tagliata con forbici sterili (rendendola una punta P1000 a foro largo).
- Lascia che i ciuffi della colonia affondino sul fondo del tubo con gravità.
NOTA: questo processo richiederà circa 1 minuto. - Una volta che i ciuffi sono affondati, rimuovere con cura il surnatante con una punta della pipetta P1000 standard e sostituirlo con 1 mL di mezzo N2 fresco. Ripetere questa fase di lavaggio tre volte per garantire la completa rimozione della spedizione.
NOTA: Qualsiasi dispacciamento residuo impedirà una formazione uniforme di sferoidi neuroectodermici e indurrà la morte cellulare. - Dopo il lavaggio, risospesere i grumi cellulari in 3 mL di mezzo N2 e trasferirli in un pozzetto di una piastra a 6 pozzetti e aggiungere 40 ng / mL di bFGF.
NOTA: Se un numero elevato di colonie neuroectodermiche fosse staccato, queste colonie potrebbero essere placcate su due o più pozzetti di una piastra a 6 pozzetti per prevenire la fusione degli sferoidi. 24 ore dopo il distacco delle colonie, verificare se si sono formati gli sferoidi. - Mantenere gli sferoidi nello stesso mezzo per i prossimi 3-4 giorni, ma aggiungere bFGF fresco (40 ng / mL) a ciascun pozzo ogni giorno per promuovere la proliferazione delle cellule neuroectodermiche, auto-organizzarsi e indurre ed espandere la neuroepitelia.
NOTA: se gli sferoidi sono stati placcati a una densità più elevata, è probabile che il fluido diventi giallo e richieda la sostituzione ogni 2 giorni con bFGF fresco (40 ng / mL). Tuttavia, questo non è raccomandato e, invece, un numero inferiore di sferoidi dovrebbe essere mantenuto in ciascun pozzetto per evitare questo problema. Gli sferoidi possono essere incorporati nella matrice della membrana basale dopo 3 giorni se i neuroepitelia sono evidenti. Se i neuroepitelia non sono evidenti o non sembrano forti, mantenere gli sferoidi per un altro giorno e controllare di nuovo.
- Differenziazione e mantenimento degli organoidi cerebrali corticali (Giorno 8)
- Preparare il mezzo di differenziazione terminale (DM) utilizzando i reagenti elencati nella Tabella 2. Porta questi media su RT.
- Scongelare la matrice della membrana basale qualificata hESC al 100% su ghiaccio.
- Preparare un foglio di parafilm con fossette sterilizzate con etanolo al 70% in una capsula di Petri da 10 cm e posizionarlo su uno stereomicroscopio sotto un cappuccio.
NOTA: un vassoio vuoto di punte di pipetta da 200 μL può essere utilizzato per generare una griglia di fossette. - Tagliare l'estremità di una punta della pipetta da 100 μL (per renderla a foro largo). Questo verrà utilizzato per incorporare sferoidi neurali nella matrice del seminterrato senza romperli.
- Utilizzando uno stereomicroscopio, selezionare sferoidi neurali di dimensioni simili (500 μm) dalla piastra a 6 pozzetti e trasferirli alle fossette parafilm utilizzando la punta della pipetta da 100 μL a foro largo, posizionando un singolo sferoide neurale per fossetta.
- Rimuovere delicatamente eventuali mezzi in eccesso, lasciando quel tanto che basta per coprire gli sferoidi.
NOTA: Questo per mantenere la qualità degli sferoidi prima di aggiungere la matrice del seminterrato e per assicurarsi che non si asciughino. - Aggiungere delicatamente 18 μL di matrice basale sopra lo sferoide, posizionando lo sferoide all'interno del centro della caduta della matrice. Utilizzare l'estremità di una punta della pipetta da 10 μL per centrare gli sferoidi nella matrice.
NOTA: Cercare di aggiungere la matrice del seminterrato il più rapidamente possibile per evitare che il fluido in eccesso intorno agli sferoidi si secchi. - Coprire la capsula di Petri da 10 cm e trasferirla nell'incubatrice e incubare la parafilm con sferoidi incorporati a matrice basale a 37 °C per 25 min.
- Dopo l'incubazione, risciacquare gli sferoidi incorporati in una piastra a 24 pozzetti ad attacco basso utilizzando una punta P1000 con 0,5 ml di supporto DM, assicurandosi che i singoli sferoidi incorporati siano collocati in un pozzetto ciascuno.
NOTA: se più di uno sferoide incorporato cade in un pozzetto, utilizzare una punta P1000 a foro largo per trasferire l'altro sferoide in un nuovo pozzo. - Mantenere gli organoidi cerebrali corticali differenziati nei mezzi DM per periodi di tempo prolungati, con cambiamenti dei media che si verificano ogni 2 giorni quando gli organoidi diventano sempre più grandi.
NOTA: Durante la prima settimana di differenziazione, il mezzo può essere cambiato ogni 3 giorni.
2. Caratterizzazione dell'invecchiamento neuronale negli organoidi corticali
- Organoidi corticali di processo per criosezioni:
NOTA: I passaggi sono stati eseguiti in una cappa di biosicurezza di classe 2.- Preparare 2 tubi da 2 ml, ciascuno riempito con 1,5 mL di paraformaldeide al 4% (PFA).
- Tagliare l'estremità di una punta della pipetta P1000 (per renderla a foro largo) e trasferire delicatamente ogni organoide in uno dei tubi da 2 ml preparati sopra (un organoide per tubo).
NOTA: per evitare l'eccessiva miscelazione di fluidi DM con il PFA, lasciare che l'organoide affondi verso l'apertura della punta della pipetta P1000 e appoggiare la punta appena sopra la parte superiore del PFA nel tubo prima di pipettare l'organoide. Ciò consentirà al ricercatore di trasferire solo l'organoide e mezzi molto minimi. - Lasciare che il processo di fissazione avvenga a 4°C per 1 ora.
- Utilizzando una punta della pipetta P1000 non tagliata, aspirare accuratamente il PFA in eccesso e aggiungere 1,5 ml di PBS 1x freddo.
- Trasferire i tubi su uno shaker orbitale impostato a 70 giri / min per 10 minuti a RT.
- Ripetere il processo di lavaggio con 1x PBS freddo tre volte per assicurarsi che tutti i PFA siano stati accuratamente rimossi.
NOTA: Non smaltire il PFA nei normali contenitori per rifiuti; invece, preparare uno specifico contenitore per lo smaltimento dei rifiuti chimici per questo, poiché il PFA è un pericolo. - Immergere gli organoidi in 1x PBS contenente il 30% di saccarosio e incubare a 4 °C fino a quando tutti gli organoidi non sono affondati sul fondo del tubo.
NOTA: Il tempo necessario per consentire l'affondamento degli organoidi dipende dalle dimensioni/età degli organoidi. Gli organoidi di 3 mesi possono richiedere fino a 5 ore. - Utilizzando una punta della pipetta P1000 tagliata a foro largo, trasferire delicatamente da tre a cinque organoidi in uno stampo di montaggio contenente una soluzione di montaggio composta al 30% da saccarosio e da un mezzo a temperatura di taglio ottimale (OCT) al 100%, con un rapporto di 3:2.
- Utilizzare una punta di pipetta da 10 μL con l'aiuto di uno stereomicroscopio per orientare e posizionare gli organoidi in uno schema a griglia.
- Posizionare lo stampo su ghiaccio secco per solidificare la soluzione OCT di saccarosio prima di procedere con il criosezionamento (16-20 μm) utilizzando un criostato.
NOTA: Per la beta-galattosidasi associata alla senescenza, tutti i tessuti devono essere trattati per il sezionamento una volta affondati. Per l'immunofluorescenza, i tessuti possono essere elaborati per il sezionamento il giorno successivo. Tutti i vetrini contenenti sezioni devono essere conservati a -20 °C prima della successiva immunofluorescenza o beta-galattosidasi, se non macchiati immediatamente.
- Processo per l'analisi della senescenza negli organoidi cerebrali corticali:
NOTA: i seguenti passaggi possono essere eseguiti su un normale banco di laboratorio.- Trasferire i vetrini in un contenitore di colorazione per vetrini per microscopio con un coperchio e lavare il tessuto organoide sezionato tre volte con 1x PBS per 10 minuti a RT per rimuovere qualsiasi soluzione di montaggio in eccesso.
- Successivamente, incubare il tessuto lavato con una soluzione colorante di beta-galattosidasi appena fatta durante la notte a 37 °C.
NOTA: La soluzione colorante beta-galattosidasi è costituita da tampone fosfato (per 10 mL di tampone fosfato: 8,15 mL di 1 M NaH2PO4, 1,85 mL di 1M Na2HPO4) pH aggiustato = 6, 100 mM di esacianoferrato di potassio (III), 100 mM di esacianoferrato di potassio (II) triidrato, 5 M di NaCl, 1 M di MgCl2, 20 mg/mL di X-Gal. Evitare l'uso di un incubatore di colture cellulari standard contenente CO2 poiché la CO2 altererà il pH della soluzione di colorazione della beta-galattosidasi. - Lavare i tessuti macchiati con 1x PBS tre volte per 10 minuti ciascuno a RT per rimuovere la soluzione di beta-galattosidasi.
- Montare i tessuti lavati con un montante antideflagrante in vetro e lasciare che la soluzione di montaggio si solidifichi per 30 minuti a RT prima di visualizzare al microscopio.
Qui abbiamo descritto un protocollo robusto che consente ai ricercatori di generare organoidi cerebrali corticali omogenei derivati da hPSC che imitano la regione del cervello corticale umano in vivo entro 1-3 mesi dalla coltura. Le colonie di hPSC vengono prima coltivate in mezzi di differenziazione per generare colonie neuroectodermiche, che possono quindi essere utilizzate per formare sferoidi neurali. Questi sferoidi vengono successivamente incorporati in una matrice di membrana basale e mantenuti per periodi di tempo prolungati per produrre organoidi che possono essere utilizzati per modellare l'invecchiamento neuronale (vedere la Figura 1 per uno schema del protocollo). Vale la pena notare che la coltivazione di questi organoidi in piastre ultra-non rivestite a 24 pozzetti provoca stress cellulare e promuove fenotipi associati alla senescenza per 13 settimane di coltura in vitro . Gli organoidi derivati da questo protocollo possono anche essere mantenuti in bioreattori agitati per una crescita e una differenziazione ottimali delle cellule neurali della piastra corticale o all'interfaccia aria-liquido.
Per iniziare, le colonie di hPSC vengono coltivate per 1 giorno prima della differenziazione neuroectodermica. È fondamentale che queste colonie di hPSC siano coltivate con una confluenza solo del 20%-30% e siano della massima qualità possibile: un monostrato piatto stretto senza cellule differenziate che contaminano le colonie (Figura 2A, B). La pluripotenza delle colonie di hPSC deve essere confermata dall'espressione di marcatori come NANOG (Figura 2C). Le colonie di hPSC convalidate vengono quindi esposte al mezzo di differenziazione neuroectodermica N2 con SB-431542 e LDN 193189. Dopo 3 giorni di mantenimento in questo mezzo, le colonie di hPSC dovrebbero essersi differenziate in colonie neuroectodermiche e non mostrare più la stessa morfologia monostrato piatta delle hPSC (Figura 2B), ma piuttosto, diventeranno cellule a forma di colonna più lunghe (Figura 2B). Queste cellule saranno anche negative per i marcatori di pluripotenza come NANOG (Figura 2C).
È in questa fase che le colonie neuroectodermiche sono enzimaticamente staccate con dispasi, e ogni colonia sana e distaccata con successo è autorizzata ad auto-organizzarsi e formare un giovane sferoide neurale (Figura 2D, Video supplementare 1). Solo le colonie neuroectodermiche sane e pulite si staccano nel periodo di tempo specificato per l'attività di dispasi; tutte le altre colonie dovrebbero essere ignorate in quanto si tradurranno in una qualità inferiore dello sferoide. Con l'esposizione giornaliera a FGF2 nel mezzo N2, le cellule staminali neurali (SOX2+) in questi sferoidi (Figura 2D, giorno 1) proliferano e formano un numero significativo di rosette neurali (Figura 2D, giorno 4). Queste rosette esprimeranno la giunzione stretta e il marcatore epiteliale ZO1 nelle cellule situate all'interno del centro delle rosette e lungo il bordo esterno dello sferoide, dimostrando la polarità apicale-basale dello sferoide (Figura 2D, giorno 4). Il metodo per l'imaging 3D a montaggio intero degli sferoidi è stato descritto prima del13. L'ispezione quotidiana degli sferoidi dovrebbe chiarire la formazione di un bordo esterno stretto e scuro e di una periferia luminosa degli sferoidi, essendo questo lo strato neuroepiteliale. Questo strato dovrebbe essere sufficientemente formato dopo 3-4 giorni con un diametro approssimativo di 500 μm, momento in cui gli sferoidi possono essere incorporati nella matrice del seminterrato. Se questo strato non è presente o è solo debolmente formato, gli sferoidi non sono sufficientemente sviluppati per andare avanti. Si consiglia di attendere un altro giorno per osservare qualsiasi cambiamento, ma se questo non viene osservato, ignorare questi sferoidi.
Un'immagine rappresentativa in campo luminoso degli sferoidi dopo 3 giorni di coltura può essere vista nella Figura 2D. Gli sferoidi con uno stretto strato neuroepiteliale che non si sono fusi con altri sferoidi vicini, hanno tessuto semitrasparente e dimostrano la formazione di rosette neurali, sono scelti per essere incorporati nella matrice del seminterrato. Una volta incorporato, lo sferoide prolifererà rapidamente e inizierà a germogliare: appariranno nodi di tessuto compatto, espandendosi verso l'esterno dal corpo principale dello sferoide. Ciò è evidente tra 1-3 settimane nella matrice del seminterrato e può essere osservato su più linee cellulari (Figura 3A). L'analisi quantitativa degli sferoidi incorporati conferma la presenza di cellule epiteliali fino al 100% degli sferoidi su tre diverse linee cellulari, affermando l'omogeneità e la riproducibilità attese da questo protocollo (Figura 3B). La quantificazione del diametro degli organoidi durante la differenziazione in vitro conferma ulteriormente la riproducibilità su diverse linee di hPSC (Figura 3C). Se il germogliamento non si verifica, gli sferoidi non si sviluppano in modo appropriato e devono essere scartati. Una volta che gli sferoidi sono stati incorporati in una matrice, il loro sviluppo progredisce e gli sferoidi sono ora indicati come organoidi. La colorazione a immunofluorescenza conferma anche la presenza di cellule progenitrici neurali (PAX6) e di marcatori dello strato corticale colorati con CTIP2 e SATB2 negli organoidi con stratificazione chiara (Figura 3D,E). Questa stratificazione è osservabile in diversi punti temporali di manutenzione degli organoidi (Figura 3D,E). Il metodo di immunoistochimica dei tessuti è stato descritto prima dei14 anni.
Una possibile applicazione di questi organoidi è studiare come i processi legati all'invecchiamento neuronale influenzano il cervello. Per indagare su questo, gli organoidi generati con successo vengono raccolti da più punti temporali diversi per il sezionamento e la colorazione per i biomarcatori molecolari standard della senescenza come la beta-galattosidasi associata alla senescenza e la p21. La Figura 4A mostra un'immagine rappresentativa della colorazione beta-galattosidasi associata alla senescenza degli organoidi 4 e 13 settimane dopo l'incorporazione nella matrice della membrana basale. Tra le settimane 4 e 13, c'è un marcato aumento della presenza di beta-galattosidasi associata alla senescenza, suggerendo che la senescenza cellulare, un driver riconosciuto dell'invecchiamento dell'organismo, si è verificata in questo periodo in coltura. La colorazione di immunofluorescenza degli organoidi alla settimana 13 ha confermato la presenza di un altro marcatore di senescenza, p21, co-marcato con il marcatore neuronale corticale maturo (CTIP2) e può essere visto nella Figura 4B. Va tuttavia notato che la presenza di p21 è un marker di arresto del ciclo cellulare e di per sé non è un marker definitivo di senescenza, e il rilevamento di altri marcatori di senescenza come p16 e SASP (senescence-associated secretory phenotype) fattori sono raccomandati per identificare definitivamente le cellule come senescenti.
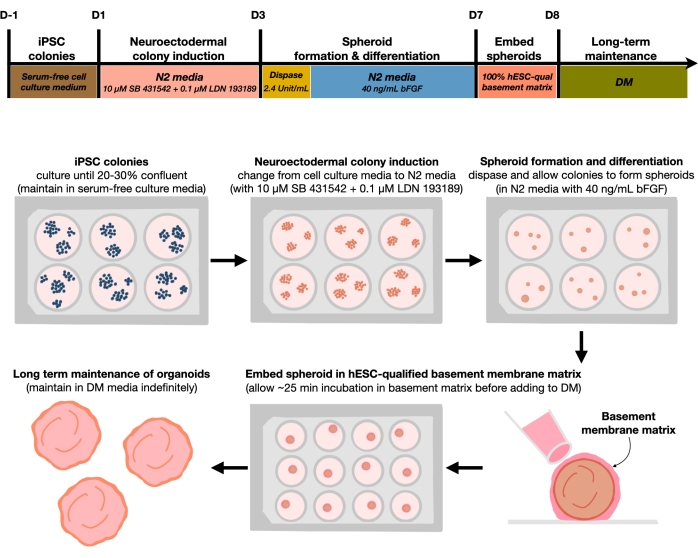
Figura 1: Diagramma schematico per la generazione di organoidi cerebrali corticali riproducibili. Flusso di lavoro schematico della procedura sperimentale per la generazione di organoidi cerebrali corticali da hPSC mantenuti nel mezzo privo di alimentatore. Il flusso di lavoro fornisce una panoramica di sei passaggi coinvolti per differenziare le hPSC 2D in tessuti umani a piastre corticali modellate in 3D in organoidi. Fare clic qui per visualizzare una versione più grande di questa figura.
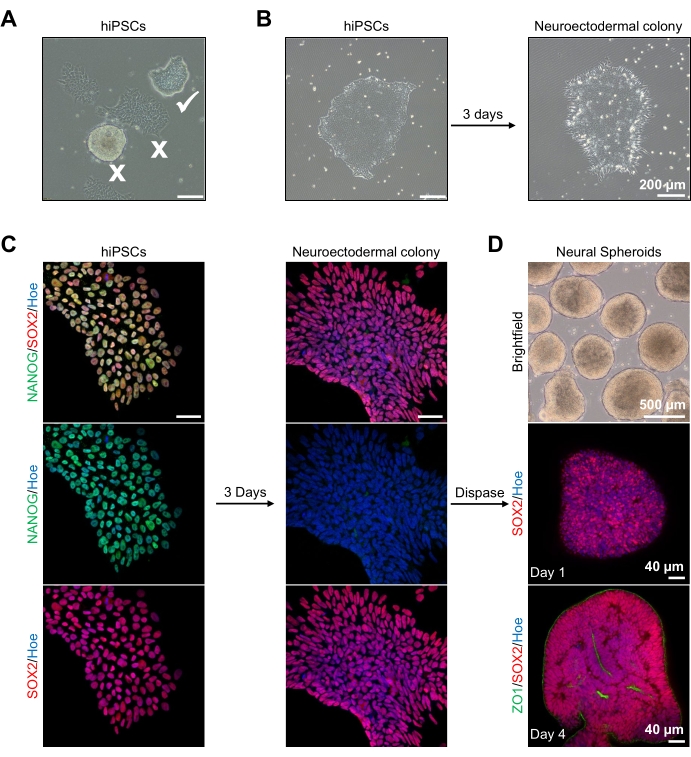
Figura 2: Generazione di sferoidi neurali derivati da colonie neuroectodermiche-hPSC. (A) Immagini rappresentative di PSC umano che mostrano colonie ottimali (zecca bianca) e differenziate (croce bianca). Barra di scala: 200 μm, ingrandimento 4x. (B) Immagine rappresentativa della colonia neuroectodermica derivata dalle hPSC dopo 3 giorni di doppio trattamento con inibitori SMAD. Barra di scala: 200 μm, ingrandimento 4x. (C) Le colonie umane di PSC sono state differenziate verso colonie neuroectodermiche. Le immagini rappresentano la colorazione di PSC (al giorno 1) e le colonie neuroectodermiche (al giorno 3) con SOX2 (rosso), NANOG (verde), tutti i nuclei sono stati controcolorati con Hoechst 33342 (blu). Barra di scala: 40 μm, ingrandimento 100x. (D) Immagini che mostrano le fasi di sviluppo degli sferoidi cerebrali corticali nel tempo in coltura in vitro sotto campo luminoso, e immunostained a tutto monte con SOX2 (rosso) al giorno 1, e doppia immunocolorato con SOX2 (rosso) e ZO1 (verde) al giorno 4. La barra di scala dell'immagine in campo luminoso è 500 μm, ingrandimento 4x, le barre di scala delle immagini inferiori sono 40 μm, ingrandimento 20x. Fare clic qui per visualizzare una versione più grande di questa figura.
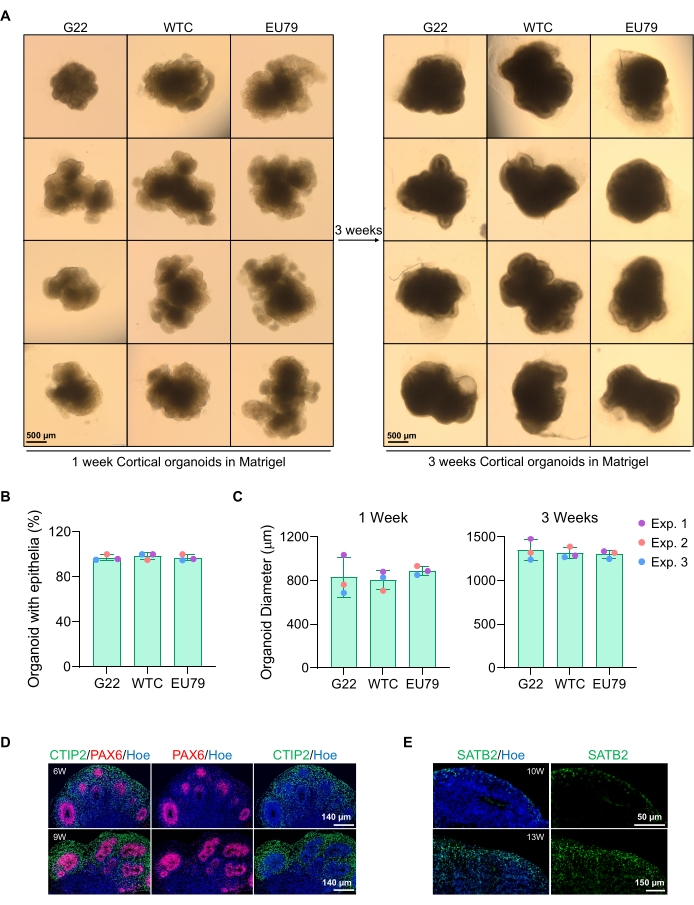
Figura 3: Caratterizzazione di organoidi cerebrali corticali derivati da diverse linee hPSC. (A) Immagini rappresentative di organoidi cerebrali corticali derivati da linee iPSC umane G22, WTC ed EU79 coltivate nell'arco di 3 settimane in vitro. La barra di scala di tutte le immagini è 500 μm, ingrandimento 2x. (B) Percentuali della generazione riuscita di organoidi cerebrali corticali a 3 settimane di differenziazione in vitro in diverse linee hPSC (G22, WTC e EU79). N = 3. I dati sono presentati come deviazione media ± standard. (C) Grafici a barre che mostrano la crescita di organoidi cerebrali corticali (basati sul diametro medio) alle settimane 1 e 3 di differenziazione in vitro in diverse linee di linee di cellule staminali pluripotenti umane (G22, WTC e EU79). N = 3. I dati sono presentati come deviazione media ± standard. (D) Immagini rappresentative di sezioni di organoidi cerebrali corticali di 6 settimane e 9 settimane derivati da HPSC G22, immunostituiti per pax6 della zona ventricolare (rosso) e piastra corticale CTIP2 (verde). Tutte le sezioni sono state controbilanciate con Hoechst 33342 (blu). Barra di scala = 140 μm, ingrandimento 20x. W è settimana. (E) Immagini rappresentative di sezioni di organoidi cerebrali corticali di 10 e 13 settimane derivati da hPSC del WTC, immunocolorati per lo strato corticale IV SATB2 (verde). Tutte le sezioni sono state controbilanciate con Hoechst 33342 (blu). 10 settimane immagine Barra della scala = 50 μm, ingrandimento 40x. Barra della scala dell'immagine di 13 settimane = 150 μm, ingrandimento 40x. W è settimana. Fare clic qui per visualizzare una versione più grande di questa figura.
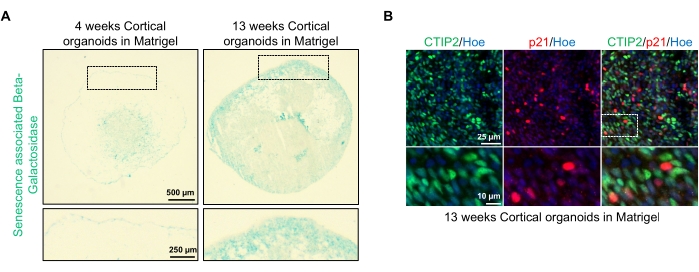
Figura 4: Caratterizzazione della senescenza in organoidi cerebrali corticali derivati da hPSC. (A) Immagini rappresentative di sezioni di organoidi cerebrali corticali umani derivati da hPSC WTC coltivati per 4 e 13 settimane in vitro e colorati con SA-β-gal. Barra di scala = 500 μm, barra di scala di immagini ingrandite = 250 μm, ingrandimento 4x. La casella tratteggiata indica un'immagine ingrandita. (B) Immagini rappresentative di sezioni di organoidi cerebrali corticali di 13 settimane derivati da hPSC eu79 umane, immunocolorati per i neuroni corticali CTIP2 (verde) e p21 (rosso). Tutte le sezioni sono state controbilanciate con Hoechst 33342 (blu). Barra di scala = 25 μm, barra di scala di immagini ingrandite = 10 μm, ingrandimento 40x. Fare clic qui per visualizzare una versione più grande di questa figura.
| Componenti multimediali | Concentrazione |
| DMEM Miscela di nutrienti F12 10x 500 mL (DMEM/F-12) | |
| Supplemento N2 5 mL (100x) | Supplmentato all'1% |
| B 27 Supplemento 10 ml | Supplemento al 2% |
| Soluzione di aminoacidi non essenziali MEM (100x) | Integrato all'1% |
| Penicillina-Streptomicina (10.000 U/mL) | Integrato all'1% |
| 2-Mercaptoetanolo 50 mL (1000x) | Integrato allo 0,1% |
Tabella 1: N2 Medio. La tabella elenca i reagenti necessari per preparare il mezzo N2.
| Componenti multimediali | Concentrazione |
| DMEM Miscela di nutrienti F12 10x 500 mL (DMEM/F-12) | I supporti DM sono realizzati con un rapporto 1:1 di DMEM/F12 e supporti neurobasali |
| Mezzo Neurobasale | |
| Supplemento N2 5 mL (100x) | Supplmentato allo 0,5% |
| B 27 Supplemento 10 ml | Integrato all'1% |
| Soluzione di aminoacidi non essenziali MEM (100x) | Integrato all'1% |
| GlutaMAX Supplemento 100x | Integrato all'1% |
| Penicillina-Streptomicina (10.000 U/mL) | Integrato all'1% |
| Soluzione insulinica Ricombinante umano | 12,5 μL per 50 mL di fluido |
| 2-Mercaptoetanolo 50 mL (1000x ) | 17,5 μL per 50 mL di fluido |
Tabella 2: Mezzo di differenziazione (DM). La tabella elenca i reagenti necessari per preparare il mezzo di differenziazione.
Video supplementare 1. Imaging dal vivo della conversione indotta di fogli/colonie hNEct 2D in 3D sotto trattamento di bFGF. Le colonie indotte di hNEct sono state delicatamente staccate dal piatto con dispase come descritto sopra e trasferite in una piastra di coltura a 6 pozzetti a attacco basso. Le colonie 2D hNEct sono state convertite in sferoidi 3D hNEct entro 12 ore. Le immagini seriali sono state catturate ogni 5 minuti. Barra della scala = 100 μm. Fare clic qui per scaricare questo video.
Per consentire l'uso di organoidi cerebrali derivati da hPSC nello screening farmacologico e nella modellazione delle malattie, è fondamentale realizzare organoidi seguendo un protocolloreplicabile e affidabile 15. Gli organoidi cerebrali sono comunemente generati da corpi embrioidi derivati da hPSC, che vengono poi incorporati in una matrice extracellulare che promuove l'espansione dei tessuti e la differenziazione neurale. Se confrontati con protocolli come 1,16,17 di Lancaster e Velasco 18, che partono da corpi embrioidi e consentono di seguire un percorso di differenziazione predefinito da parte degli organoidi in via di sviluppo, abbiamo scoperto che iniziare la creazione di organoidi cerebrali corticali con cellule NEct umane piuttosto che con corpi embrioidi migliora la coerenza della formazione di organoidi cerebrali corticali. Ciò consente di conseguenza anche il ridimensionamento richiesto per lo screening farmacologico e fenotipico. Poiché le cellule NEct umane non solo possono essere espanse in quantità considerevoli, ma possono anche essere facilmente crioconservate, questo approccio migliora anche la replicabilità tra gli esperimenti. Va inoltre notato che, rispetto ad altri protocolli che hanno adottato l'uso di Bioreattori e tecnologie simili, non sono necessarie attrezzature specializzate per questo protocollo, rendendolo adatto a qualsiasi laboratorio6. Infine, il tempo necessario per generare organoidi maturi positivi per i marcatori dello strato corticale come SATB2 è ridotto rispetto ai protocolli Lancaster1 e bioreattore 6,19 rendendolo più adatto per studiare la traiettoria di sviluppo dello sviluppo corticale umano in salute e malattie 1,6,16.
Inoltre, dato l'impatto sanitario globale in continua crescita delle malattie legate all'invecchiamento come la demenza, che sono associate ad un aumento dei tipi di cellule senescenti nel cervello che contribuisce alla patogenesi, la capacità di identificare e testare composti che possono migliorare l'invecchiamento cerebrale sono di enorme interesse. Nonostante le hPSC siano note per essere epigeneticamente ringiovanite durante il processo di riprogrammazione20, troviamo robusti aumenti delle cellule senescenti negli organoidi cerebrali corticali coltivati per periodi di tempo prolungati. Questo è uno sviluppo promettente che ora consente lo screening di farmaci che eliminano tali cellule senescenti dal cervello (senolitici) o che rallentano questo processo (senostatici)21. Poiché gli organoidi cerebrali corticali derivati da NEct umani sono di origine umana, questo approccio probabilmente accorcerà il percorso tradizionale per commercializzare tali nuove terapie.
Ci sono due passaggi critici in questo protocollo. Il primo è il corretto livello di confluenza delle colonie di hPSC al momento della differenziazione. Le colonie di hPSC devono essere al massimo confluenti al 30% per garantire che le colonie di NEct generate non si fondano con le colonie vicine e che i singoli organoidi siano guidati clonalmente. Il secondo passo critico prevede l'uso corretto della dispasi per sollevare le colonie di NEct e produrre gli sferoidi neurali. La tempistica dell'incubazione con dispasi è fondamentale per l'eventuale qualità degli sferoidi neurali generati. Questo perché la sovraesposizione delle colonie con dispasi è tossica per le cellule22 e alla fine influisce sulla qualità degli organoidi generati. Il limite di questo protocollo è che è difficile controllare la dimensione degli sferoidi neurali perché dipende dalle dimensioni delle colonie iniziali che vengono sollevate con la dispasi. Tuttavia, questo problema può essere superato selezionando sferoidi neurali di dimensioni simili quando si procede alla fase di incorporamento.
Infine, le applicazioni future potrebbero estendersi all'uso di questi organoidi corticali riproducibili nell'analisi robotica e negli approcci di screening biofarmaceutico tipicamente utilizzati in quel settore. Ciò è supportato da dati preliminari del nostro laboratorio che indicano che la generazione di organoidi cerebrali corticali da cellule NEct umane può essere facilmente automatizzata, rendendola compatibile con questi approcci.
Gli autori non hanno nulla da rivelare.
Questo lavoro è supportato dal Medical Research Future Fund-Accelerated Research, Leukodystrophy flagship Massimo's Mission (EPCD000034), Medical Research Future Fund-Stem Cell Mission (APP2007653). Gli autori desiderano ringraziare il Dr. Ju-Hyun Lee (Korea University) per aver generato dati nel video supplementare 1.
| Name | Company | Catalog Number | Comments |
| 16% Formaldehyde (W/V) Methanol-free | Thermo Fisher Scientific | 28908 | 4% of PFA are diluted in 1x PBS |
| 2-Mercaptoethanol 50 mL(1000x) | Life Technologies Australia (TFS) | 21985023 | Used in NM and DM media |
| B 27 Supplement 10 mL | Life Technologies Australia (TFS) | 17504044 | Used in NM and DM media |
| CKX53 microscope with SC50 camera | Olympus | ||
| Corning Costar 6 well cell culture plates | Sigma Aldrich Pty Ltd | CLS3516-50EA | |
| Dispase II powder | Thermo Fisher Scientific | 17105041 | Powder is dissolve in HBSS, filtered through 0.22 µm filter, aliquote at 10 mL and store at -20 °C |
| DMEM Nutrient Mix F12 10x 500 mL (DMEM/F-12) | Thermofisher | 11320082 | Used in NM and DM media |
| DMSO Dimethyl Sulfoxide | Sigma Aldrich Pty Ltd | D2650-100ML | |
| Dulbecco's Phosphate Buffered Saline | Sigma Aldrich Pty Ltd | D1408-500ML | |
| Falcon Matrigel hESC-qualified Matrix | In Vitro Technologies Pty Ltd | FAL354277 | Make aliquotes of 100 µL and stored at -20 °C |
| GlutaMAX Supplement 100x | Thermo Fisher Scientific | 35050061 | Used in NM and DM media |
| Hanks Balanced Salt Solution | Sigma Aldrich Pty Ltd | H8264 | |
| Human induced pluripotent stem cells (EU79) | In-house reporogrammed from skin fibroblast | ||
| Human induced pluripotent stem cells (G22) | Genea Biocells | Obtained from Genea Biocells (San Diego, United States) | |
| Human induced pluripotent stem cells (WTC) | Gift from Professor Bruce Conklin | ||
| InSolution TGF-Β RI Kinase Inhibitor VI, SB431542 | Merck | US1616464-5MG | |
| Insulin Solution Human Recombinant | Sigma Aldrich Pty Ltd | I9278 | Used in NM and DM media |
| LDN193189 Dihydrochloride | Sigma Aldrich Pty Ltd | SML0559-5MG | Used during differentiation |
| MEM Non-Essential Amino Acids Solution (100x) | Thermo Fisher Scientific | 11140050 | Used in NM and DM media |
| mTeSR Plus | STEMCELL TECHNOLOGIES | 100-0276 | Used to maintain hiPSC colonies prior to differentiation with NM media |
| N2 Supplement 5 mL (100x) | Life Technologies Australia Pty Ltd | 17502048 | Used in NM and DM media |
| Neurobasal Medium | Thermo Fisher Scientific | 21103049 | Used in DM media |
| OCT Embedding Compound Sakura Clear (118 mL/Bottle) | Tissue Tek | 4583 | |
| Parafilm M Roll Size 4 in. x 125 Ft | Sigma Aldrich Pty Ltd | P7793 | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 | Used in NM and DM media |
| Potassium Hexacyanoferrate (II) Trihydrate | Sigma Aldrich Pty Ltd | CP1087 | |
| Potassium hexacyanoferrate(III) | Sigma Aldrich Pty Ltd | 455946 | |
| Prolong Glass Antifade Mountant | Life Technologies Australia (TFS) | P36980 | |
| Recombinant Human FGF basic | R&D Systems | 233-FB-01M | Aliquotes are made at 20 µg/mL and stored at -20 °C |
| SB431542 | Tocris | 1614 | Used during differentiation |
| Sucrose | Sigma Aldrich Pty Ltd | PHR1001-1G | 30% of sucrose are diluted in 1x PBS |
| Ultra-Low attachment multiwell plates , 24 well plate, polystyrene | Sigma Aldrich Pty Ltd | CLS3473-24EA | |
| X-GAL EA | Life Technologies Australia (TFS) | R0404 | Make aliquotes of 20 mg/mL and storde at -80 °C |
- Lancaster, M. A., Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nature Protocols. 9 (10), 2329-2340 (2014).
- Mansour, A. A., et al. An in vivo model of functional and vascularized human brain organoids. Nature Biotechnology. 36 (5), 432-441 (2018).
- Xiang, Y., et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell. 21 (3), 383-398 (2017).
- Bagley, J. A., Reumann, D., Bian, S., Lévi-Strauss, J., Knoblich, J. A. Fused cerebral organoids model interactions between brain regions. Nature Methods. 14 (7), 743 (2017).
- Kwak, T. H., et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson's disease modeling. Stem Cells. 38 (6), 727-740 (2020).
- Qian, X., et al. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nature Protocols. 13 (3), 565-580 (2018).
- Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Reports. 10 (4), 537-550 (2015).
- Shaker, M. R., Cooper-White, J., Wolvetang, E. J. Self-organizing 3D human choroid plexus-ventricle-cortical organoids. BioRxiv. , (2020).
- Shaker, M. R., Aguado, J., Chaggar, H. K., Wolvetang, E. J. Klotho inhibits neuronal senescence in human brain organoids. npj Aging and Mechanisms of Disease. 7 (1), 1-12 (2021).
- Shaker, M. R., et al. Anteroposterior Wnt-RA gradient defines adhesion and migration properties of neural progenitors in developing spinal cord. Stem Cell Reports. 15 (4), 898-911 (2020).
- Chambers, S. M., et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology. 27 (3), 275-280 (2009).
- Shaker, M. R., et al. Rapid and efficient generation of myelinating human oligodendrocytes in organoids. Frontiers in Cellular Neuroscience. 15, 631548 (2021).
- Lee, J. -. H., Shaker, M. R., Lee, E., Lee, B., Sun, W. NeuroCore formation during differentiation of neurospheres of mouse embryonic neural stem cells. Stem Cell Research. 34, 101691 (2020).
- Shaker, M. R., et al. Spatiotemporal contribution of neuromesodermal progenitor-derived neural cells in the elongation of developing mouse spinal cord. Life Sciences. 282, 119393 (2021).
- Shaker, M. R., et al. Neural epidermal growth factor-like like protein 2 Is expressed in human oligodendroglial cell types. Frontiers in Cell and Developmental Biology. 10, 803061 (2022).
- Lancaster, M. A., et al. Cerebral organoids model human brain development and microcephaly. Nature. 501 (7467), 373-379 (2013).
- Giandomenico, S. L., Sutcliffe, M., Lancaster, M. A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nature Protocols. 16 (2), 579-602 (2021).
- Velasco, S., et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 570 (7762), 523-527 (2019).
- Qian, X., et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 165 (5), 1238-1254 (2016).
- Hunter, Z. L., Leeson, H. C., Shaker, M. R., Wolvetang, E. J., Vadlamudi, L. Human induced pluripotent stem cells generated from epilepsy patients for use as in vitro models for drug screening. Stem Cell Research. 60, 102673 (2022).
- Kaur, A., Macip, S., Stover, C. M. An appraisal on the value of using nutraceutical based senolytics and senostatics in aging. Frontiers in Cell and Developmental Biology. 8, 218 (2020).
- Wang, F., et al. Safety and efficacy of dispase and plasmin in pharmacologic vitreolysis. Investigative Ophthalmology & Visual Science. 45 (9), 3286-3290 (2004).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved