
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Neuroscience
Geração robusta e altamente reprodutível de organoides cerebrais corticais para modelar senescência neuronal cerebral in vitro
Neste estudo, fornecemos uma técnica detalhada para um sistema de cultura organoide cortical simples, mas robusto, usando culturas hPSC padrão sem alimentador. Este é um protocolo rápido, eficiente e reprodutível para gerar organoides que modelam aspectos da senescência cerebral in vitro.
Organoides cerebrais são modelos tridimensionais do cérebro humano em desenvolvimento e fornecem uma plataforma convincente e de ponta para modelagem de doenças e rastreamento genômico e medicamentos em larga escala. Devido à natureza auto-organizada das células em organoides cerebrais e à crescente gama de protocolos disponíveis para sua geração, foram identificados problemas com heterogeneidade e variabilidade entre organoides. Neste documento de protocolo, descrevemos um protocolo robusto e replicável que supera em grande parte essas questões e gera organoides corticais de progenitores neuroectodérmicos dentro de 1 mês, e que pode ser mantido por mais de 1 ano. Este protocolo altamente reprodutível pode ser facilmente realizado em uma sala padrão de cultura tecidual e resulta em organoides com uma rica diversidade de tipos celulares tipicamente encontrados no córtex humano em desenvolvimento. Apesar de sua maquiagem de desenvolvimento precoce, neurônios e outros tipos de células cerebrais humanas começarão a exibir os sinais típicos de senescência em células neuronais após uma cultura in vitro prolongada, tornando-as uma plataforma valiosa e útil para estudar processos neuronais relacionados ao envelhecimento. Este protocolo também descreve um método para detectar tais células senescentes em organoides cerebrais corticais usando coloração beta-galactosidase associada à senescência.
Nosso conhecimento atual do cérebro humano tem sido em grande parte baseado em modelos animais e espécimes cerebrais pós-morte. A biologia das células-tronco é um campo que avança rapidamente que fornece novas percepções sobre a biologia básica do desenvolvimento cerebral humano e os condutores patológicos das doenças cerebrais humanas. As células-tronco pluripotentes humanas (hPSCs) são uma ferramenta inestimável para modelar o cérebro humano através da geração de organoides, tecido tridimensional (3D) semelhante a órgãos que tipicamente recapitula as trajetórias de desenvolvimento, maquiagem celular e arquitetura do cérebro humano em desenvolvimento. Os organoides cerebrais são auto-montados e compostos de células-tronco neurais, progenitores neurais especificados, neurônios maduros e tipos de células gliais. Os organoides, portanto, proporcionam uma oportunidade única para estudar o cérebro humano primitivo, que muitas vezes é inacessível para experimentação direta, mas também tem limitações intrínsecas, como a ausência de vasculatura e um sistema imunológico.
Metodologias para gerar organoides cerebrais têm sido perseguidas de duas maneiras diferentes: diferenciação não guiada e guiada. Métodos organoides cerebrais não guiados dependem das capacidades espontâneas de diferenciação intrínseca das células-tronco que impulsionam a morfogênesetecidual 1,2 e permitem o surgimento de uma variedade de identidades de linhagem celular que vão desde cérebro, cérebro médio e cérebro traseiro, até plexo choroide, retina e mesoderme. Em contraste, métodos organoides cerebrais guiados requerem o uso substancial de fatores externos para conduzir os HPSCs em direção à padronização desejada de linhagens neuronais representando um tipo de região cerebral, como eminência gangliônica medial3, cérebro4, cérebro médio5, hipotálamo6, cerebelo7 e plexo coralid8. Essa capacidade de gerar diferentes regiões cerebrais com diferentes linhagens celulares, e o potencial de fundi-las à vontade, torna os organoides cerebrais um excelente modelo para investigar o desenvolvimento cerebral humano e decifrar os mecanismos subjacentes de doenças relacionadas ao cérebro. Embora esses métodos de geração de organoides cerebrais ofereçam um avanço na modelagem de regiões cerebrais humanas, a variabilidade e a heterogeneidade entre organoides continuam sendo uma limitação significativa para estudos sistemáticos e quantitativos, como o rastreamento de drogas.
O protocolo atual baseia-se em um método desenvolvido em nosso recente artigo9 e envolve a diferenciação seletiva das colônias hPSC em direção à identidade neuroectoderm (NEct) com inibidores duplos de SMAD (SB-431542 e LDN 193189), que então têm a capacidade de se auto-organizar dentro de 4 dias em spheroids neuroepithelium 3D sob a influência da sinalização FGF2. Esses esferoides neuroepithelium geram de forma confiável organoides cortical homogêneos com uma composição celular in vivo dentro de 4 semanas de diferenciação. O protocolo aqui descrito é baseado em nossos achados anteriores mostrando que a inibição da sinalização dual SMAD (Supressor de Mães Contra Decapentaplégica) promove a diferenciação de hPSCs em relação às células-tronco neurais rostrais derivadas de progenitores neuroectodérmicos10 por, entre outros, inibir o destino celular endrodérmico, mesodérmico e trophectoderm . Além disso, a incorporação dos esferoides neuroepithelium na matriz de membrana de porão qualificada pelo HESC desencadeia um brotamento significativo da neuroepiteelia, formando ventrículos com polaridade apicobasal. A cultura em larga escala mostrou reprodutibilidade e homogeneidade de organoides cortical independentes de linhas celulares, clones ou lotes, e, portanto, representa um sistema de células-tronco confiável e estável para imitar o desenvolvimento cortical humano precoce em saúde e doenças in vitro. Delineamos ainda um protocolo para detectar marcadores de células neuronais senescentes em organoides cerebrais cortical derivados de hPSCs que foram cultivados por longos períodos de tempo.
Depois de emplacar hPSCs a uma densidade de semeadura de 20%-30%, as células são tratadas com inibidores duplos de SMAD por 3 dias para diferenciar colônias hPSC em direção a colônias neuroectodérmicas. Estas colônias são então suavemente levantadas com despase e semeadas em placas de 6 poços de fixação ultra-baixas complementadas com FGF2. As colônias 2D flutuantes se auto-organizam em esferoides neuroectodérmicos 3D durante a noite e são mantidas por 4 dias em N2 médio suplementado diariamente com FGF2. Uma vez que os esferoides tenham estabelecido a camada neuroepitelial, eles podem ser incorporados na matriz de membrana do porão. Adicionando rotineiramente um novo meio de diferenciação terminal, os pesquisadores observarão a expansão progressiva e o brotamento da neuroepiteelia em organoides corticais. Os pesquisadores podem querer dissociar esses organoides para realizar perfis transcricionais e proteômicos. Além disso, a imagem de campo brilhante é recomendada para monitorar a qualidade dos organoides cortical. A análise pode ser realizada por fixação, crioseção e imunostensão. Descrições e métodos para essas técnicas foram descritos anteriormente12. Em última análise, este protocolo permite que os pesquisadores gerem de forma rápida e robusta organoides cerebrais cortical homogêneos para modelar o cérebro cortical humano em desenvolvimento, com baixo custo e equipamentos limitados, e para estudar aspectos da senescência neuronal celular, conforme descrito neste artigo.
1. Geração de organoides cerebrais cortical
NOTA: Todas as etapas desta seção do protocolo ocorrerão em um capô de biossegurança classe 2, a menos que seja indicado o contrário.
- Indução de colônias neuroectodérmicas 2D da cultura 2D hPSC (Dias -1 a 3)
- Antes da indução, emplaque as colônias hPSC em uma matriz de membrana de porão qualificada hESC em uma placa de 6 poços com densidade de 20%-30%. Alcance essa densidade passando colônias hPSC de um poço de uma placa de 6 poços a 60% de confluência em três poços de uma placa de 6 poços.
- Para revestimento da matriz da membrana do porão, diluir a matriz da membrana do porão a uma proporção de 1:50 em um meio basal simples. Deposite uniformemente 1 mL/poço de uma placa de 6 poços, incubar por 1h à temperatura ambiente (RT) e, em seguida, aspirar.
- Mantenha colônias hPSC por 1 dia em 2 mL de um meio de cultura celular livre de soro antes da diferenciação.
- No dia da diferenciação do HPSC, inspecione as colônias hPSC usando microscopia de campo brilhante em 4x a 10x de ampliação para garantir colônias saudáveis sem diferenciação detectável.
NOTA: HPSCs saudáveis formarão colônias de bordas apertadas com células que possuem um núcleo grande, citoplasma muito pequeno e nucleoli proeminente. Colônias diferenciadas de iPSC apresentarão diferenças morfológicas claras em relação às das colônias hPSC descritas acima, particularmente ao redor das bordas externas das colônias ou no centro. - Adicione reagentes listados na Tabela 1 para compor o meio N2 necessário para diferenciação. Leve este meio para RT antes de usar.
- Uma vez na RT, aspire o meio de cultura celular livre de soro de cada poço da placa de 6 poços e substitua por 2 mL de média N2 suavemente adicionada com uma pipeta sorológica de 5 mL.
- Adicione os inibidores duplos SMAD, SB-431542 (10 μM) e LDN 193189 (100 nM).
NOTA: Os inibidores SMAD podem ser adicionados ao meio N2 após o meio ter sido colocado em cada poço, ou à quantidade necessária de meio N2 antes de substituir o meio de cultura celular livre de soro. Os inibidores podem ser incorporados uniformemente ao meio girando suavemente a placa ou invertendo o tubo contendo o meio e os inibidores 3-4 vezes. - Adicione mídia N2 fresca suplementada com SB-431542 (10 μM) e LDN 193189 (100 nM) diariamente para cada poço pelos próximos 2 dias.
NOTA: O meio N2 fresco é adicionado para reduzir a exposição prolongada de células ao DMSO que é usado para dissolver compostos SB-431542 e LDN 193189 e prevenir a citotoxicidade.
- Geração de esferoides neuroectodérmicos 3D de colônias neuroectodérmicas induzidas (Dias 3 a 7)
- Levante as colônias neuroectodérmicas induzidas usando dispase seguindo as etapas 1.2.2-1.2.8.
- Primeiro, remova 2 mL de meio N2 da placa de 6 poços e lave 1x com HBSS para garantir que todo o meio N2 seja removido.
NOTA: O meio N2 pode interferir na atividade enzimática da dispase, impedindo o descolamento adequado das colônias neuroectodérmicas do poço. - Adicione 1 mL de 2,4 unidade/mL para dispase a cada poço contendo colônia.
- Incubar o poço por 20-25 min (máximo 30 min) a 37 °C. Verifique se há desprendimento da colônia regularmente.
NOTA: Pequenas colônias podem se desprender dentro de 20 minutos. Todas as colônias que permanecerem presas após 30 minutos devem ser ignoradas. - Após a incubação, adicione 1 mL de N2 médio ao poço para parar a atividade da enzima despase e transfira as colônias para um tubo de 15 mL usando uma ponta de pipeta P1000 larga ou uma ponta de pipeta P1000 modificada cortada com tesoura estéril (tornando-a uma ponta P1000 larga).
- Permita que os aglomerados da colônia afundem no fundo do tubo com gravidade.
NOTA: Este processo levará aproximadamente 1 min. - Uma vez que os aglomerados tenham afundado, remova cuidadosamente o supernatante com uma ponta de pipeta P1000 padrão e substitua-o por 1 mL de meio N2 fresco. Repita esta etapa de lavagem três vezes para garantir a remoção completa da despase.
NOTA: Qualquer despase restante impedirá uma formação uniforme de esferoides neuroectodérmicos e induzirá a morte celular. - Após a lavagem, resuspenque as células em 3 mL de média N2 e transfira para um poço de uma placa de 6 poços e adicione 40 ng/mL de bFGF.
NOTA: Se um alto número de colônias neuroectodérmicas foram separadas, essas colônias poderiam ser banhadas em dois ou mais poços de uma placa de 6 poços para evitar a fusão de esferoides. 24 h após o desprendimento das colônias, verifique se os esferoides se formaram. - Mantenha os esferoides na mesma mídia pelos próximos 3-4 dias, mas adicione bFGF fresco (40 ng/mL) a cada poço diariamente para promover a proliferação neuroectodérmica celular, auto-organização e induzir e expandir a neuroepiteelia.
NOTA: Se os spheróides tiverem sido banhados a uma densidade maior, é provável que a mídia se transforme em amarelo e exija a substituição a cada 2 dias por bFGF fresco (40 ng/mL). No entanto, isso não é recomendado e, em vez disso, um número menor de esferoides deve ser mantido em cada poço para evitar esse problema. Esferoides podem ser incorporados na matriz de membrana do porão após 3 dias se a neuroepiteelia for evidente. Se a neuroepithelia não for evidente ou não parecer forte, então mantenha os esferoides por mais um dia e verifique novamente.
- Diferenciação e manutenção organoides do cérebro cortical (Dia 8)
- Prepare a mídia de diferenciação de terminal (DM) utilizando os reagentes listados na Tabela 2. Traga essa mídia para rt.
- Descongele matriz de membrana de porão 100% hESC no gelo.
- Prepare uma folha de parafilm com covinhas esterilizadas com 70% de etanol em uma placa de Petri de 10 cm e coloque-a em um estereoscópio sob um capô.
NOTA: Uma bandeja vazia de 200 pontas de pipeta μL pode ser usada para gerar uma grade de covinhas. - Corte a ponta de uma ponta de pipeta de 100 μL (para torná-la larga). Isso será usado para incorporar esferoides neurais na matriz do porão sem quebrá-los.
- Usando um estereómico, selecione esferoides neurais de tamanho semelhante (500 μm) da placa de 6 poços e transfira-os para as covinhas do parafilme usando a ponta de pipeta de 100 μL de furo largo, colocando uma única esceréquia neural.
- Remova suavemente qualquer excesso de mídia, deixando apenas o suficiente para cobrir os esferoides.
NOTA: Isto é para manter a qualidade dos esferoides antes de adicionar a matriz do porão e garantir que eles não sequem. - Adicione suavemente 18 μL de matriz de porão sobre o esferoide, posicionando o esferoide dentro do centro da queda da matriz. Use a ponta de uma pipeta de 10 μL para centralizar os esferoides na matriz.
NOTA: Tente adicionar a matriz do porão o mais rápido possível para evitar que o excesso de mídia ao redor dos esferoides seque. - Cubra a placa de Petri de 10 cm e transfira para a incubadora e incubar a placa de parafilm com esferoides embutidos em matriz de porão a 37 °C por 25 min.
- Após a incubação, enxágue os esferoides incorporados em uma placa de 24 poços de baixa fixação usando uma ponta P1000 com 0,5 mL da mídia DM, certificando-se de que os esferoides incorporados individuais sejam colocados em um poço cada.
NOTA: Se mais de um esferoide incorporado cair em um poço, use uma ponta P1000 larga para transferir o outro esferoide para um novo poço. - Mantenha os organoides cerebrais corticais diferenciados na mídia DM por períodos prolongados de tempo, com mudanças na mídia ocorrendo a cada 2 dias quando os organoides se tornam maiores e mais velhos.
NOTA: Durante a primeira semana de diferenciação, o meio pode ser trocado a cada 3 dias.
2. Caracterização do envelhecimento neuronal em organoides cortical
- Processe organoides cortical para criosecções:
NOTA: As etapas foram realizadas em um capô biossegurança classe 2.- Prepare tubos de 2 mL, cada um preenchido com 1,5 mL de 4% de paraformaldeído (PFA).
- Corte a extremidade de uma ponta de pipeta P1000 (para torná-la um furo largo) e transfira suavemente cada organoide para um dos tubos de 2 mL preparados acima (um organoide por tubo).
NOTA: Para evitar que o excesso de mídia DM se misture com o PFA, permita que o organoide afunde em direção à abertura da ponta de pipeta P1000 e descanse a ponta logo acima da parte superior do PFA no tubo antes de encanar o organoide para fora. Isso permitirá ao pesquisador transferir apenas o organoide e a mídia muito mínima. - Permita que o processo de fixação ocorra a 4°C durante 1h.
- Usando uma ponta de pipeta P1000 sem cortes, aspire cuidadosamente o excesso de PFA e adicione 1,5 mL de PBS frio de 1x.
- Transfira os tubos para um agitador orbital a 70 rpm por 10 min na RT.
- Repita o processo de lavagem com PBS frio de 1x três vezes para garantir que todo o PFA tenha sido completamente removido.
NOTA: Não descarte a PFA em recipientes de resíduos regulares; em vez disso, prepare um recipiente específico de descarte de resíduos químicos para isso, pois a PFA é um perigo. - Mergulhe os organoides em 1x PBS contendo 30% de sacarose e incubar a 4 °C até que todos os organoides tenham afundado na parte inferior do tubo.
NOTA: O tempo necessário para permitir que os organoides afundem depende do tamanho/idade dos organoides. Organoides de 3 meses de idade podem levar até 5h. - Usando uma ponta de pipeta P1000 cortada e larga, transfira suavemente de três a cinco organoides em um molde de montagem contendo uma solução de montagem feita de 30% de sacarose e 100% de temperatura de corte (OCT) ótima, em uma proporção de 3:2.
- Use uma ponta de pipeta de 10 μL com o auxílio de um estereoscópio para orientar e posicionar organoides em um padrão semelhante à grade.
- Coloque o molde em gelo seco para solidificar a solução oct de sacarose antes de prosseguir com crio-seção (16-20 μm) usando um criostat.
NOTA: Para a beta-galactosidase associada à senescência, todos os tecidos devem ser processados para secção depois de afundados. Para a imunofluorescência, os tecidos podem ser processados para secção no dia seguinte. Todos os slides contendo seções devem ser armazenados a -20 °C antes da imunofluorescência subsequente ou beta-galactosidase, se não forem manchados imediatamente.
- Processo para análise da senescência nos organoides cerebrais corticais:
NOTA: As seguintes etapas podem ser realizadas em um banco de laboratório regular.- Transfira os slides para um recipiente de coloração de slides de microscópio com uma tampa e lave o tecido organoide seccionado três vezes com 1x PBS por 10 minutos em RT para remover qualquer solução de montagem em excesso.
- Na sequência, incubar o tecido lavado com solução de coloração beta-galactosidase recém-feita durante a noite a 37 °C.
NOTA: A solução de coloração beta-galactosidase é feita de tampão fosfato (para 10 mL de tampão fosfato: 8,15 mL de 1 M NaH2PO4, 1,85 mL de 1M Na2HPO4) pH ajustado = 6,100 mM de hexacianoferato de potássio (III), 100 mM de hexacianoferato de potássio (II) trihidrato, 5 M de NaCl, 1 M de MgCl2, 20 mg/mL de X-Gal. Evite usar uma incubadora de cultura celular padrão contendo CO2 , pois o CO2 alterará o pH da solução de coloração beta-galactosidase. - Lave os tecidos manchados com 1x PBS três vezes por 10 minutos cada no RT para remover a solução beta-galactosidase.
- Monte os tecidos lavados com um montador de antifato de vidro e permita que a solução de montagem se solidifique por 30 minutos no RT antes de visualizar sob o microscópio.
Aqui descrevemos um protocolo robusto que permite aos pesquisadores gerar organoides cerebrais cortical derivados do hPSC homogêneos que imitam a região do cérebro cortical humano in vivo dentro de 1-3 meses de cultura. colônias hPSC são primeiramente cultivadas em mídia de diferenciação para gerar colônias neuroectodérmicas, que podem então ser usadas para formar esferoides neurais. Estes esferoides são posteriormente incorporados em uma matriz de membrana de porão e mantidos por longos períodos de tempo para produzir organoides que podem ser usados para modelar o envelhecimento neuronal (ver Figura 1 para um esboço do protocolo). Vale ressaltar que cultivar esses organoides em placas ultra-não revestidas de 24 poços causa estresse celular e promove fenótipos associados à senescência ao longo de 13 semanas de cultura in vitro . Organoides derivados deste protocolo também podem ser mantidos em bioreatores agitados para o crescimento ideal e diferenciação de células neurais de placa cortical ou na interface ar-líquido.
Para começar, as colônias hPSC são cultivadas por 1 dia antes da diferenciação neuroectodérmica. É fundamental que essas colônias hPSC sejam cultivadas com apenas 20%-30% de confluência e sejam da mais alta qualidade possível: uma monocamada plana apertada sem células diferenciadas contaminando as colônias (Figura 2A,B). A pluripotência das colônias hPSC deve ser confirmada pela expressão de marcadores como NANOG (Figura 2C). As colônias hPSC validadas são então expostas aos meios de diferenciação neuroectodérmica N2 com sb-431542 e LDN 193189. Após 3 dias de manutenção nesta mídia, as colônias hPSC deveriam ter se diferenciado em colônias neuroectodérmicas e não mais mostrar a mesma morfologia monocamada plana apertada dos hPSCs (Figura 2B), mas sim, elas se tornarão células mais longas em forma de coluna (Figura 2B). Essas células também serão negativas para marcadores de pluripotência, como NANOG (Figura 2C).
É nesta fase que as colônias neuroectodérmicas são enzimáticamente separadas com dispase, e cada colônia saudável e com sucesso separada é permitida a auto-organização e formação de um jovem esferoide neural (Figura 2D, Vídeo Suplementar 1). Somente colônias neuroectodérmicas saudáveis e limpas se desprenderão no prazo especificado para atividade despase; todas as outras colônias devem ser ignoradas, pois resultarão em uma pior qualidade de esferoide. Com a exposição diária ao FGF2 na mídia N2, as células-tronco neurais (SOX2+) nestes esferoides (Figura 2D, dia 1) proliferarão e formarão um número significativo de rosetas neurais (Figura 2D, dia 4). Estas rosetas expressarão a junção apertada e o marcador epitelial ZO1 em células localizadas dentro do centro das rosetas e ao longo da borda externa do esferoide, demonstrando a polaridade apical-basal da esferoide (Figura 2D, dia 4). O método para imagens 3D de spheroids foi descrito antes das13. A inspeção diária dos esferoides deve elucidar a formação de uma borda externa apertada e escura e periferia brilhante dos esferoides, sendo esta a camada neuroepiteelial. Esta camada deve ser suficientemente formada após 3-4 dias com um diâmetro aproximado de 500 μm, momento em que os esferoides podem ser incorporados na matriz do porão. Se esta camada não estiver presente ou apenas estiver fracamente formada, os esferoides não são suficientemente desenvolvidos o suficiente para avançar. Recomenda-se esperar mais um dia para observar qualquer mudança, mas se isso não for observado, desconsidere esses esferoides.
Uma imagem representativa dos esferoides após 3 dias de cultura pode ser vista na Figura 2D. Esferoides com uma camada neuroefitelial apertada que não se fundiram com outros esferoides vizinhos, têm tecido semi-transparente e demonstram formação de rosetas neurais, são escolhidos para serem incorporados na matriz do porão. Uma vez incorporado, o esferoide se proliferará rapidamente e começará a brotar: acenos de tecido compacto aparecerão, expandindo-se para fora do corpo principal do esferoide. Isso é evidente entre 1-3 semanas na matriz do porão e pode ser observado em múltiplas linhas celulares (Figura 3A). A análise quantitativa dos esferoides incorporados confirma a presença de células epiteliais em até 100% dos esferoides em três diferentes linhas celulares, afirmando a homogeneidade e reprodutibilidade esperadas a partir deste protocolo (Figura 3B). A quantificação do diâmetro dos organoides durante a diferenciação in vitro confirma ainda mais a reprodutibilidade em diferentes linhas de hPSC (Figura 3C). Se o brotamento não ocorrer, os esferoides não estão se desenvolvendo adequadamente e devem ser descartados. Uma vez que os esferoides foram incorporados em uma matriz, seu desenvolvimento progride, e os esferoides são agora referidos como organoides. A coloração da imunofluorescência também confirma a presença de células progenitoras neurais (PAX6), bem como marcadores de camada cortical manchados com CTIP2 e SATB2 nos organoides com camadas claras (Figura 3D,E). Esta camada é observável em diferentes pontos de tempo de manutenção organoide (Figura 3D,E). O método de imunohistoquímica dos tecidos foi descrito antes dos14 anos.
Uma possível aplicação desses organoides é estudar como processos relacionados ao envelhecimento neuronal afetam o cérebro. Para investigar isso, organoides gerados com sucesso são colhidos a partir de vários pontos de tempo diferentes para seção e coloração para biomarcadores moleculares padrão de senescência, como beta-galactosidase e p21 associados à senescência. A Figura 4A mostra uma imagem representativa da mancha beta-galactosidase associada à senescência dos organoides 4 e 13 semanas após a incorporação na matriz de membrana do porão. Entre as semanas 4 e 13, há um aumento acentuado na presença de beta-galactosidase associada à senescência, sugerindo que a senescência celular, um reconhecido condutor do envelhecimento organismo, ocorreu ao longo deste tempo na cultura. A coloração de imunofluorescência de organoides na semana 13 confirmou a presença de outro marcador de senescência, p21, co-rotulado com o marcador neuronal cortical maduro (CTIP2) e pode ser visto na Figura 4B. Deve-se notar, no entanto, que a presença do p21 é um marcador de parada do ciclo celular e por si só não é um marcador definitivo de senescência, e a detecção de outros marcadores de senescência como p16 e SASP (fenótipo secreto associado à senescência) são recomendados para identificar definitivamente as células como senescentes.
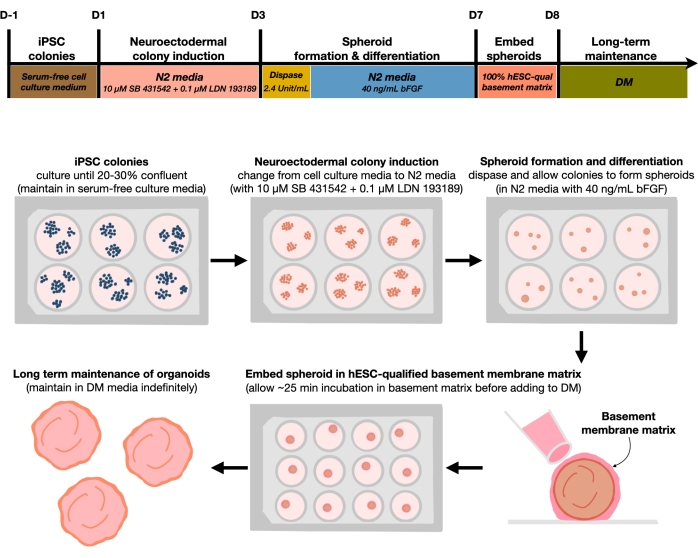
Figura 1: Diagrama esquemático para geração de organoides cerebrais cortical reprodutíveis. Fluxo de trabalho esquemático do procedimento experimental para a geração de organoides cerebrais cortical a partir de hPSCs mantidos no meio sem alimentação. O fluxo de trabalho fornece uma visão geral de seis etapas envolvidas para diferenciar os hPSCs 2D em tecidos humanos de placa cortical padronizados em 3D em organoides. Clique aqui para ver uma versão maior desta figura.
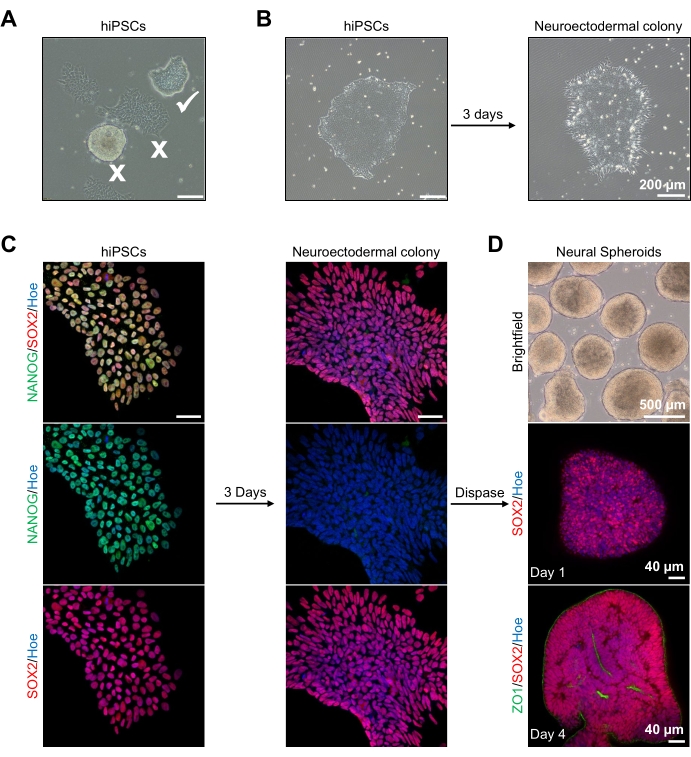
Figura 2: Geração de esferoides neurais derivados de colônias neuroectodérmicas-hPSCs. (A) Imagens representativas do PSC humano exibindo colônias ótimas (carrapato branco) e diferenciadas (cruz branca). Barra de escala: 200 μm, ampliação 4x. (B) Imagem representativa da colônia neuroectodérmica derivada de hPSCs após 3 dias de tratamentos inibidores duplos de SMAD. Barra de escala: 200 μm, ampliação 4x. (C) As colônias humanas de PSC foram diferenciadas em relação às colônias neuroectodérmicas. As imagens representam a coloração de PSC (no dia 1) e colônias neuroectodérmicas (no dia 3) com sox2 (Vermelho), NANOG (Verde), todos os núcleos foram neutralizados com Hoechst 33342 (azul). Barra de escala: 40 μm, ampliação de 100x. (D) Imagens que mostram os estágios de desenvolvimento de esferoides cerebrais corticais ao longo do tempo na cultura in vitro sob campo brilhante, e todo o valor imunossuperudo com SOX2 (Vermelho) no dia 1, e duplamente imunossuadas com SOX2 (vermelho) e ZO1 (Verde) no dia 4. A barra de escala da imagem brightfield é de 500 μm, ampliação de 4x, barras de escala das imagens inferiores são 40 μm, ampliação de 20x. Clique aqui para ver uma versão maior desta figura.
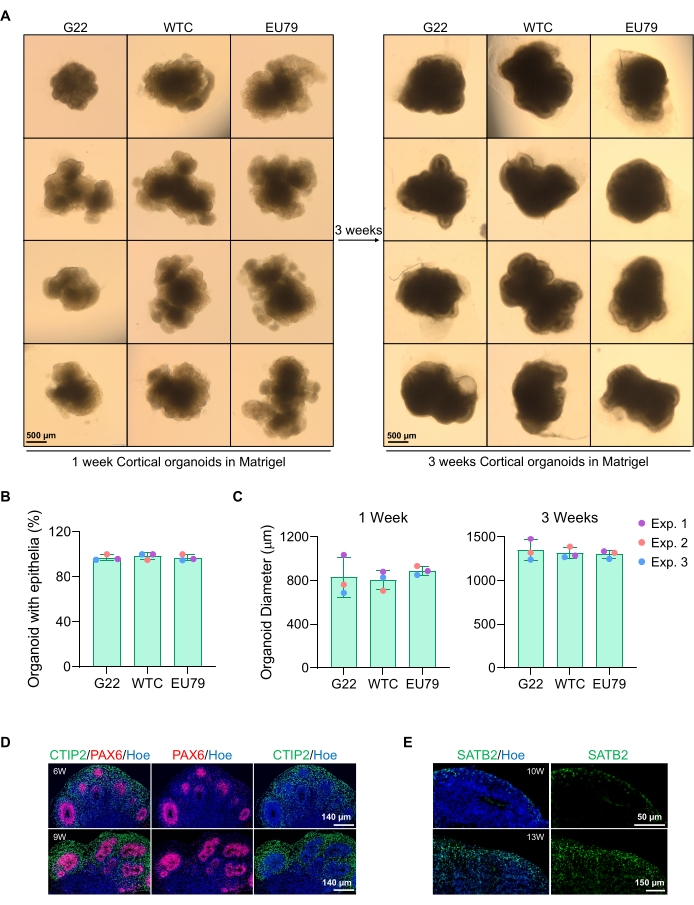
Figura 3: Caracterização de organoides cerebrais cortical derivados de diferentes linhas hPSC. (A) Imagens representativas de organoides cerebrais corticais derivados de linhas de G22, WTC e IPSC humanos cultivadas ao longo de 3 semanas in vitro. A barra de escala de todas as imagens é de 500 μm, ampliação de 2x. (B) Percentuais da geração bem sucedida de organoides cerebrais corticais a 3 semanas de diferenciação in vitro em diferentes linhas hPSC (G22, WTC e EU79). N = 3. Os dados são apresentados como ± desvio padrão. (C) Gráficos de barras mostrando o crescimento de organoides cerebrais corticais (baseados no diâmetro médio) nas semanas 1 e 3 de diferenciação in vitro em diferentes linhas de linhas de células-tronco pluripotentes humanas (G22, WTC e EU79). N = 3. Os dados são apresentados como ± desvio padrão. (D) Imagens representativas de seções de organoides cerebrais cortical de 6 semanas e 9 semanas de idade derivadas de G22 hPSCs, imunoss detidas para a zona ventrícula PAX6 (vermelha) e placa cortical CTIP2 (verde). Todas as seções foram contra-manchadas com Hoechst 33342 (azul). Barra de escala = 140 μm, ampliação de 20x. W é semana. (E) Imagens representativas de seções de organoides cerebrais cortical de 10 semanas e 13 semanas de idade derivadas de HPSCs WTC, imunoss detidas para camada cortical IV SATB2 (verde). Todas as seções foram contra-manchadas com Hoechst 33342 (azul). 10 semanas de imagem Barra de escala = 50 μm, ampliação de 40x. 13 semanas imagem Barra de escala = 150 μm, ampliação de 40x. W é semana. Clique aqui para ver uma versão maior desta figura.
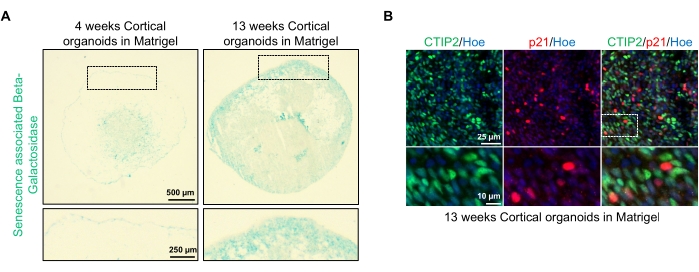
Figura 4: Caracterização da senescência em organoides cerebrais corticais derivados de hPSCs. (A) Imagens representativas de seções de organoides cerebrais cortical humanos derivados de HPSCs WTC cultivados por 4 e 13 semanas in vitro e manchados com SA-β-gal. Barra de escala = 500 μm, barra de escala de imagens ampliadas = 250 μm, ampliação de 4x. A caixa pontilhada indica uma imagem ampliada. (B) Imagens representativas de seções de organoides cerebrais cortical de 13 semanas de idade derivadas de eu79 hPSCs humanos, imunossu detidas para neurônios corticais CTIP2 (verde) e p21 (vermelho). Todas as seções foram contra-manchadas com Hoechst 33342 (azul). Barra de escala = 25 μm, barra de escala de imagens ampliadas = 10 μm, ampliação de 40x. Clique aqui para ver uma versão maior desta figura.
| Componentes de mídia | Concentração |
| Mistura de nutrientes DMEM F12 10x 500 mL (DMEM/F-12) | |
| Suplemento N2 5 mL (100x) | Supplmentado em 1% |
| B 27 Suplemento 10 mL | Suplementado em 2% |
| Solução de aminoácidos não essenciais MEM (100x) | Suplementado em 1% |
| Penicilina-Streptomicina (10.000 U/mL) | Suplementado em 1% |
| 2-Mercaptoethanol 50 mL(1000x ) | Suplementado em 0,1% |
Tabela 1: N2 Medium. A tabela lista os reagentes necessários para preparar o meio N2.
| Componentes de mídia | Concentração |
| Mistura de nutrientes DMEM F12 10x 500 mL (DMEM/F-12) | A mídia DM é feita com proporção 1:1 de mídia DMEM/F12 e Neurobásal |
| Meio Neurobásal | |
| Suplemento N2 5 mL (100x) | Supplmentado em 0,5% |
| B 27 Suplemento 10 mL | Suplementado em 1% |
| Solução de aminoácidos não essenciais MEM (100x) | Suplementado em 1% |
| Suplemento GlutaMAX 100x | Suplementado em 1% |
| Penicilina-Streptomicina (10.000 U/mL) | Suplementado em 1% |
| Solução de insulina Recombinante humano | 12,5 μL para 50 mL de mídia |
| 2-Mercaptoetanol 50 mL (1000x ) | 17,5 μL para 50 mL de mídia |
Tabela 2: Meio de diferenciação (DM). A tabela lista os reagentes necessários para preparar o meio de diferenciação.
Vídeo suplementar 1. Imagem viva da conversão de folha/colônia 2D induzida de hNEct para 3D sob o tratamento de bFGF. Colônias induzidas de hNEct foram suavemente separadas do prato com despase como descrito acima e transferidas para uma placa de cultura de baixo acessório de 6 poços. As colônias 2D hNEct foram convertidas em spheróides 3D hNEct dentro de 12 h. Imagens em série foram capturadas a cada 5 minutos. Barra de escala = 100 μm. Clique aqui para baixar este Vídeo.
Para permitir o uso de organoides cerebrais derivados do hPSC na triagem de medicamentos e modelagem de doenças, é crucial fazer organoides seguindo um protocolo replicável e confiável15. Organoides cerebrais são comumente gerados a partir de corpos embrionários derivados de hPSCs, que são então incorporados em uma matriz extracelular que promove a expansão do tecido e a diferenciação neural. Quando comparados a protocolos comoos 1,16,17 e Velasco18 de Lancaster, que começam a partir de corpos embrionários e permitem que um caminho de diferenciação padrão seja seguido pelos organoides em desenvolvimento, descobrimos que iniciar a criação de organoides cerebrais corticais com células de NEct humanas em vez de corpos embrionários melhora a consistência da formação de órgãos cerebrais corticais. Isso, consequentemente, também permite o dimensionamento necessário para a triagem de drogas e fenotípicas. Uma vez que as células de NEct humanos não só podem ser expandidas em quantidades consideráveis, mas também podem ser prontamente criopreservadas, essa abordagem também melhora a replicabilidade entre experimentos. Deve-se notar também que, em comparação com outros protocolos que adotaram o uso de Bioreatores e tecnologias similares, não é necessário equipamento especializado para este protocolo, tornando-o adequado para qualquer laboratório6. Finalmente, o tempo necessário para gerar organoides maduros que são positivos para marcadores de camada cortical como o SATB2 é reduzido em comparação com os protocolos Lancaster1 e bioreator 6,19 tornando-o mais adequado para estudar a trajetória de desenvolvimento do desenvolvimento cortical humano em saúde e doenças 1,6,16.
Além disso, dado o crescente impacto global nos cuidados de saúde de doenças relacionadas ao envelhecimento, como a demência, que estão associadas a um aumento dos tipos de células senescentes no cérebro que contribuem para a patogênese, a capacidade de identificar e testar compostos que podem amenizar o envelhecimento cerebral são de enorme interesse. Apesar de os HPSCs serem conhecidos por serem epigeneticamente rejuvenescidos durante o processo de reprogramação20, encontramos aumentos robustos em células senescentes em organoides cerebrais corticais cultivados por períodos prolongados de tempo. Este é um desenvolvimento promissor que agora permite a triagem de drogas que eliminam tais células senescentes do cérebro (senolíticos) ou que retardam esse processo (senostáticas)21. Uma vez que os organoides cerebrais cortical derivados do NECT humano são de origem humana, essa abordagem provavelmente encurtará o caminho tradicional para comercializar tais novas terapêuticas.
Há dois passos críticos neste protocolo. O primeiro é o nível correto de confluência das colônias hPSC no momento da diferenciação. as colônias hPSC devem ser no máximo 30% confluentes para garantir que as colônias de NEct geradas não se fundam com colônias vizinhas e que os organoides individuais sejam clonalmente conduzidos. O segundo passo crítico envolve o uso correto de dispase para levantar as colônias de NEct e produzir os esferoides neurais. O tempo de incubação com dispase é fundamental para a eventual qualidade dos esferoides neurais gerados. Isso porque a exposição excessiva de colônias com despase é tóxica para as células22 e eventualmente afeta a qualidade dos organoides gerados. A limitação deste protocolo é que é difícil controlar o tamanho dos esferoides neurais porque depende do tamanho das colônias iniciais que são levantadas com despase. No entanto, essa questão pode ser superada selecionando esferoides neurais de tamanho semelhante ao prosseguir para o estágio de incorporação.
Finalmente, aplicações futuras poderiam se estender ao uso desses organoides cortical reprodutíveis em análise robótica e abordagens de triagem biofarmacêutica tipicamente utilizadas nessa indústria. Isso é apoiado por dados preliminares de nosso laboratório indicando que a geração de organoides cerebrais corticais a partir de células NEct humanas pode ser facilmente automatizada, tornando-o compatível com essas abordagens.
Os autores não têm nada a revelar.
Este trabalho é apoiado pela Pesquisa Acelerada do Fundo futuro de Pesquisa Médica, Leucodystrophy flagship Massimo's Mission (EPCD000034), Medical Research Future Fund-Stem Cell Mission (APP2007653). Os autores gostariam de agradecer ao Dr. Ju-Hyun Lee (Korea University) por gerar dados no Vídeo Suplementar 1.
| Name | Company | Catalog Number | Comments |
| 16% Formaldehyde (W/V) Methanol-free | Thermo Fisher Scientific | 28908 | 4% of PFA are diluted in 1x PBS |
| 2-Mercaptoethanol 50 mL(1000x) | Life Technologies Australia (TFS) | 21985023 | Used in NM and DM media |
| B 27 Supplement 10 mL | Life Technologies Australia (TFS) | 17504044 | Used in NM and DM media |
| CKX53 microscope with SC50 camera | Olympus | ||
| Corning Costar 6 well cell culture plates | Sigma Aldrich Pty Ltd | CLS3516-50EA | |
| Dispase II powder | Thermo Fisher Scientific | 17105041 | Powder is dissolve in HBSS, filtered through 0.22 µm filter, aliquote at 10 mL and store at -20 °C |
| DMEM Nutrient Mix F12 10x 500 mL (DMEM/F-12) | Thermofisher | 11320082 | Used in NM and DM media |
| DMSO Dimethyl Sulfoxide | Sigma Aldrich Pty Ltd | D2650-100ML | |
| Dulbecco's Phosphate Buffered Saline | Sigma Aldrich Pty Ltd | D1408-500ML | |
| Falcon Matrigel hESC-qualified Matrix | In Vitro Technologies Pty Ltd | FAL354277 | Make aliquotes of 100 µL and stored at -20 °C |
| GlutaMAX Supplement 100x | Thermo Fisher Scientific | 35050061 | Used in NM and DM media |
| Hanks Balanced Salt Solution | Sigma Aldrich Pty Ltd | H8264 | |
| Human induced pluripotent stem cells (EU79) | In-house reporogrammed from skin fibroblast | ||
| Human induced pluripotent stem cells (G22) | Genea Biocells | Obtained from Genea Biocells (San Diego, United States) | |
| Human induced pluripotent stem cells (WTC) | Gift from Professor Bruce Conklin | ||
| InSolution TGF-Β RI Kinase Inhibitor VI, SB431542 | Merck | US1616464-5MG | |
| Insulin Solution Human Recombinant | Sigma Aldrich Pty Ltd | I9278 | Used in NM and DM media |
| LDN193189 Dihydrochloride | Sigma Aldrich Pty Ltd | SML0559-5MG | Used during differentiation |
| MEM Non-Essential Amino Acids Solution (100x) | Thermo Fisher Scientific | 11140050 | Used in NM and DM media |
| mTeSR Plus | STEMCELL TECHNOLOGIES | 100-0276 | Used to maintain hiPSC colonies prior to differentiation with NM media |
| N2 Supplement 5 mL (100x) | Life Technologies Australia Pty Ltd | 17502048 | Used in NM and DM media |
| Neurobasal Medium | Thermo Fisher Scientific | 21103049 | Used in DM media |
| OCT Embedding Compound Sakura Clear (118 mL/Bottle) | Tissue Tek | 4583 | |
| Parafilm M Roll Size 4 in. x 125 Ft | Sigma Aldrich Pty Ltd | P7793 | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 | Used in NM and DM media |
| Potassium Hexacyanoferrate (II) Trihydrate | Sigma Aldrich Pty Ltd | CP1087 | |
| Potassium hexacyanoferrate(III) | Sigma Aldrich Pty Ltd | 455946 | |
| Prolong Glass Antifade Mountant | Life Technologies Australia (TFS) | P36980 | |
| Recombinant Human FGF basic | R&D Systems | 233-FB-01M | Aliquotes are made at 20 µg/mL and stored at -20 °C |
| SB431542 | Tocris | 1614 | Used during differentiation |
| Sucrose | Sigma Aldrich Pty Ltd | PHR1001-1G | 30% of sucrose are diluted in 1x PBS |
| Ultra-Low attachment multiwell plates , 24 well plate, polystyrene | Sigma Aldrich Pty Ltd | CLS3473-24EA | |
| X-GAL EA | Life Technologies Australia (TFS) | R0404 | Make aliquotes of 20 mg/mL and storde at -80 °C |
- Lancaster, M. A., Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nature Protocols. 9 (10), 2329-2340 (2014).
- Mansour, A. A., et al. An in vivo model of functional and vascularized human brain organoids. Nature Biotechnology. 36 (5), 432-441 (2018).
- Xiang, Y., et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell. 21 (3), 383-398 (2017).
- Bagley, J. A., Reumann, D., Bian, S., Lévi-Strauss, J., Knoblich, J. A. Fused cerebral organoids model interactions between brain regions. Nature Methods. 14 (7), 743 (2017).
- Kwak, T. H., et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson's disease modeling. Stem Cells. 38 (6), 727-740 (2020).
- Qian, X., et al. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nature Protocols. 13 (3), 565-580 (2018).
- Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Reports. 10 (4), 537-550 (2015).
- Shaker, M. R., Cooper-White, J., Wolvetang, E. J. Self-organizing 3D human choroid plexus-ventricle-cortical organoids. BioRxiv. , (2020).
- Shaker, M. R., Aguado, J., Chaggar, H. K., Wolvetang, E. J. Klotho inhibits neuronal senescence in human brain organoids. npj Aging and Mechanisms of Disease. 7 (1), 1-12 (2021).
- Shaker, M. R., et al. Anteroposterior Wnt-RA gradient defines adhesion and migration properties of neural progenitors in developing spinal cord. Stem Cell Reports. 15 (4), 898-911 (2020).
- Chambers, S. M., et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology. 27 (3), 275-280 (2009).
- Shaker, M. R., et al. Rapid and efficient generation of myelinating human oligodendrocytes in organoids. Frontiers in Cellular Neuroscience. 15, 631548 (2021).
- Lee, J. -. H., Shaker, M. R., Lee, E., Lee, B., Sun, W. NeuroCore formation during differentiation of neurospheres of mouse embryonic neural stem cells. Stem Cell Research. 34, 101691 (2020).
- Shaker, M. R., et al. Spatiotemporal contribution of neuromesodermal progenitor-derived neural cells in the elongation of developing mouse spinal cord. Life Sciences. 282, 119393 (2021).
- Shaker, M. R., et al. Neural epidermal growth factor-like like protein 2 Is expressed in human oligodendroglial cell types. Frontiers in Cell and Developmental Biology. 10, 803061 (2022).
- Lancaster, M. A., et al. Cerebral organoids model human brain development and microcephaly. Nature. 501 (7467), 373-379 (2013).
- Giandomenico, S. L., Sutcliffe, M., Lancaster, M. A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nature Protocols. 16 (2), 579-602 (2021).
- Velasco, S., et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 570 (7762), 523-527 (2019).
- Qian, X., et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 165 (5), 1238-1254 (2016).
- Hunter, Z. L., Leeson, H. C., Shaker, M. R., Wolvetang, E. J., Vadlamudi, L. Human induced pluripotent stem cells generated from epilepsy patients for use as in vitro models for drug screening. Stem Cell Research. 60, 102673 (2022).
- Kaur, A., Macip, S., Stover, C. M. An appraisal on the value of using nutraceutical based senolytics and senostatics in aging. Frontiers in Cell and Developmental Biology. 8, 218 (2020).
- Wang, F., et al. Safety and efficacy of dispase and plasmin in pharmacologic vitreolysis. Investigative Ophthalmology & Visual Science. 45 (9), 3286-3290 (2004).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved