
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
微管触觉的自组装
本文介绍了一种使用基于植物的微管交联剂MAP65和PEG作为填充剂形成触头形状的微管组件的方案。
细胞骨架负责细胞内的主要内部组织和重组,所有这些都没有经理来指导变化。在有丝分裂或减数分裂期间尤其如此,其中微管在细胞分裂期间形成纺锤体。纺锤体是用于在细胞分裂过程中分离遗传物质的机器。为了 在体外创建自组织纺锤体,我们最近开发了一种技术,将微管重组为纺锤体样组件,其中包含一组最小的微管相关蛋白质和拥挤剂。具体而言,使用了MAP65,它是来自植物的抗平行微管交联剂,来自酵母的Ase1和来自哺乳动物的PRC1的同源物。该交联剂将微管自组织成长而薄的纺锤状微管自组织组件。这些组件也类似于液晶触头,微管可以用作中尺度介系原。这里提出了用于创建这些微管触觉的方案,以及使用荧光显微镜表征组件的形状以及使用光漂白后的荧光恢复来表征成分的迁移率的方案。
通过有丝分裂的细胞分裂是维持生命的最重要的生物过程之一。由微管蛋白二聚体组成的微管细丝是该过程的基本结构元件。当染色体在细胞中心对齐时,中期产生的瞬态机制由于其形状而被称为有丝分裂纺锤体,其形状就像被螺纹覆盖的织机的纺锤体(图1A)。在许多生物体中,微管在中期使用微管将凝聚的染色体推拉到细胞的中心,将它们对齐并将它们连接到微管上,微管将在间期将它们拉开(图1B,C)。纺锤体在减数分裂(图1B)和有丝分裂(图1C)中形成,由许多重叠的微管形成,这些微管不像螺纹一样缠绕在中心轴上,而是平行于界面。创建这些基于微管的结构需要交联的相关蛋白质和相关的酶,这些酶可以作为马达来帮助推动和拉动1号染色体。
减数分裂纺锤体的研究表明,微管在交联阵列2,3,4,5,6中短,动态且重叠(图1Di)。由于这些短微管的物理组织,减数分裂纺锤体类似于液晶触头(图1E)。事实上,纺锤体已被证明可以合并和合并,正如人们对液晶触头5所期望的那样。
许多可追溯到20世纪60年代的研究都使用固定,连续切片和电子显微镜来确定有丝分裂纺锤体内有两种类型的微管7,8,9,10。第一种类型称为运动微管,它将纺锤形极连接到运动细胞。第二种类型称为插值或极性微管,它们生长在染色体之外并在中区重叠(图1Dii)8,9,10。第三种类型称为星体微管,它位于纺锤体外部,并将极点连接到细胞边缘;这些微管组织不在当前讨论的范围之内。最近有关于 Augmin6 和 γ-微管蛋白环复合物之间相互作用的研究,这些相互作用会影响微管的成核中心,从而导致有丝分裂纺锤体具有较短的微管,如图 1D 所示。
由于微管长于宽,具有高纵横比和高刚度,因此它们就像液晶分子的放大版本。在软物质物理学中,原子和分子已经使用最小的相互作用来推断相变的物理机制,包括成核和晶体的熔化11。类似地,微管是中尺度物体,是液晶分子的放大版本,可以深入了解液晶动力学的物理原理,包括各向同性向列相的成核和生长。此外,如上所述,减数分裂纺锤体显示出类似于液晶触头的性质,这是一种向列态,其成核并从液晶分子的各向同性状态3,4,5生长。对于类触子,成核和生长与其他晶体类似(即,需要相对高浓度的介质[形成液晶的分子])。触头的独特“纺锤体”形状来自与向列相对齐的液晶介质的局部对准(图1E)。它们不能形成圆形晶体,因为分子高度不对称。鉴于微管的性质,由高局部浓度的微管制成的有丝分裂纺锤体机械也具有相同的形状,无论是称为触头还是纺锤体,这也许并不奇怪。触觉可以是双极性的,在锥形末端有极点(图1Ei),也可以是均匀的,极点在无穷远处有效(图1Eii)。
鉴于纺锤体形成的重要性,通过展示微管通过离子物质12,13,产生消耗相互作用的拥挤剂和特定的微管交联蛋白13,16,17,18,19,在体外自组织纺锤体形成的努力正在进行中,21.令人惊讶的是,尽管这些药物都有助于增加微管的局部浓度,但它们通常会导致长微管束,但不会导致触头。这些束很长的一个原因可能是组成它们的微管也很长。最近使用较短的微管的工作也报告了较长的束,这些束在15末端没有锥形;在这种情况下,束与运动蛋白结合在一起,导致束的延伸,从而使它们更长。如图所示,锥形纺锤状组件需要具有非伸缩交联剂的短微管。
最近,我们开发了一种技术,可以在有核稳定的微管22存在下,使用反平行交联剂MAP65创建微管触觉。微管需要短,但很少有已知的微管长度调节器可以限制微管免受动态不稳定或端到端退火的影响。相反,GMPCPP用于在生长后成核和稳定细丝。这允许产生高密度的短微管,可以自我组织成触觉。这些触觉在双折射下观察时是均匀的。除了短的微管外,还使用了特定的反平行交联剂MAP65来形成触头(图2)。MAP65是有丝分裂交联剂PRC1/Ase1家族23中的一种植物微管相关蛋白。MAP65以二聚体的形式存在,具有很强的亲和力与自身以及微管24结合。与用肌动蛋白丝25,26,27观察到的减数分裂纺锤体和触头不同,它们是双极性的,具有液晶的液体状性质,微管触头被观察到是固体状的22,28。
这里提出了用于创建微管触觉并使用基于荧光的技术表征组件的形状和成分的迁移率的方案。
注意:除非另有说明,否则实验的某些部分可以在实验室工作台上进行,同时穿着适当的防护设备(手套)。
1. 盖玻片硅烷化
注意:盖玻片必须经过硅烷化处理,才能与这些实验中使用的聚合物刷涂层一起使用。这是一种疏水性硅烷化处理,允许具有疏水性中心嵌段的嵌段共聚物结合并形成聚合物刷。应在通风橱中执行以下步骤,以防止戴手套时接触有毒蒸气。二甲基二氯硅烷具有剧毒,必须非常小心地处理。
- 按顺序用ddH2O,70%乙醇和ddH2O冲洗盖玻片。每次冲洗之间用不起毛的实验室湿巾擦干它们。这在处理前从表面去除灰尘和水溶性或有机颗粒。
- 将盖玻片放入金属盖玻片夹持架中,并将盖玻片转移到紫外线臭氧(UVO)机器上。用UVO照射盖玻片20分钟以除去任何背景荧光。可以使用等离子体室代替 UVO。
- 使用镊子,将盖玻片从用于UVO处理的金属架转移到用于硅烷化的其他金属架上。不要对两者使用相同的机架,因为当应用UVO时,它会导致高水平的氧化。用水和乙醇对机架进行预清洁,以免残留先前使用的化学品。
- 将装有盖玻片的架子浸入装有100%丙酮的容器中1小时。用自来水冲洗容器3x,然后用ddH2O冲洗3x以除去所有丙酮。
- 用盖玻片将架子浸入100%乙醇中10分钟。用自来水冲洗容器3x,然后用ddH2O冲洗3x以除去所有乙醇。
- 用盖玻片将机架浸入ddH 2 O中3x,每次5分钟。
- 将装有盖玻片的培养架浸入 0.1 M KOH(50 mL 1 M KOH,450 mL ddH2O)中 15 分钟。用自来水冲洗容器3倍,然后用ddH2O冲洗3倍以除去所有KOH。
- 用盖玻片将机架浸入ddH 2 O中3x,每次5分钟。
- 用盖玻片将机架在通风橱或层流通风橱中风干过夜。
- 完全干燥架子和盖玻片后,将它们浸入2%二甲基二氯硅(DDS)中5分钟,该容器取于专门用于硅烷的不同容器中。不要让任何不干燥的东西与硅烷接触。
- 将架子和盖玻片浸入装有100%乙醇的容器中2x5分钟。用自来水冲洗容器3倍,然后用ddH2O冲洗3倍。
- 将机架和盖玻片浸入ddH2O中3x,每次5分钟。
- 用盖玻片将机架在通风橱或层流通风橱中风干过夜。
- 在最后的干燥步骤之后,使用镊子将盖玻片移回盖玻片盒中。这些盖玻片可以在未来1-2个月内使用。旧的盖玻片将开始失去其涂层,应丢弃。
2. 微管蛋白制剂
注意:购买的微管蛋白是冻干粉末,未标记或标有荧光基团。冻干的微管蛋白储存在−80°C冰箱中。以下程序用于将未标记的微管蛋白与标记的微管蛋白以有利于可视化的比例混合。
- 从−80°C冰箱中取出一等分试样的含有1mg冻干微管蛋白粉的未标记微管蛋白,并将其保存在冰上。向试管中加入200μL冷PEM-80,使微管蛋白浓度达到5mg / mL。将其保存在冰上10分钟以溶解所有冻干物。
- 从−80°C冰箱中取出含有20μg冻干微管蛋白粉末的罗丹明标记的微管蛋白等分试样,并将其保存在冰上。向试管中加入4μL冷PEM-80,使微管蛋白浓度达到5mg / mL。将其保存在冰上10分钟以完全溶解冻干物。
- 溶解后,将100μL未标记的重悬微管蛋白溶液加入到罗丹明标记的微管蛋白溶液中。移液器6x-7x非常缓慢地混合。如果聚集体可见,则将溶解的微管蛋白以90,000× g 离心10分钟,通过丢弃沉淀并保留上清液来除去聚集体。这种微管蛋白混合物将产生~4%标记的微管蛋白。
- 将剩余的100μL未标记的微管蛋白滴冻到液氮(LN2)中,并将其储存在-80°C以用于额外的微管蛋白混合物。
- 取标记的微管蛋白混合物并等分到七个管中,每个管15μL。每个等分试样可用于单个实验室。滴冻剩余的等分试样并将其储存在-80°C以备将来实验。
3. MAP65净化
注意:MAP65不是商业上可获得的,因此需要为这项工作进行净化。该协议之前已在若干出版物23,29中详细阐述。
- 将 MAP65 质粒和 GFP-MAP65 质粒转化为 BL21 细菌菌株以进行蛋白表达。
- 在600nm处将BL21细菌培养成0.6-1的光密度。使用lac操作器诱导蛋白质产生,并在一夜之间生长细菌。
- 沉淀培养物并裂解细菌。
- 离心后收集裂解物,并用具有可用于结合6x-组氨酸标签的镍离子的磁珠孵育。
- 使用咪唑洗脱蛋白质并脱盐。
- 用液氮滴冻蛋白质并将其储存在−80°C以在1年内使用。
4. 流室的组装
注意:实验在由载玻片和硅烷化盖玻片制成的流动室中进行(图3)。
- 取载玻片,按顺序使用ddH2O,乙醇和ddH2O清洁。每次冲洗之间用不起毛的实验室湿巾擦干。
- 使用一段双面胶带创建流路径。使用戴手套的手,将胶带切成~25-30毫米的长度。纵向拆分磁带以创建两条较细的条带。将两条胶带放在载玻片上,它们之间约5-8毫米。
注意:由于胶带的厚度标准化为约80-100μm,因此胶带片之间的路径宽度将决定腔室中的体积。 - 将硅烷化盖玻片放在流道的顶部。用笔背面轻轻按压胶带区域,将载玻片和盖玻片密封到双面胶带上。确保整个区域保持良好的密封;当密封性良好时,胶带应从半透明变为透明。
- 取下边缘上多余的胶带,用剃须刀片切割胶带,从流室入口仅留下1毫米。
- 根据需要,用实验参数的信息标记腔室。
5. 触觉实验
注意:一旦产生所有试剂和耗材,它们就可用于在流动室中对微管触觉进行成核和聚合。
- 收集所有要使用的试剂。将它们解冻在冰上,并在工作时将它们储存在冰上。为实验制作几个流动室。
- 每个实验使用一个流室。通过在溶解在PEM-80中的20μL5%非离子嵌段共聚物表面活性剂(材料表)中流动,用聚合物刷涂覆流动室表面,并在腔室两端滴小滴以防止内部形成气泡。将其保存在潮湿的腔室中(即,用湿的无绒实验室湿巾培养皿)直到准备使用(至少5-7分钟)。
- 在无菌管中,混合以下物质以产生微管蛋白- MAP混合物:9.5μLPEM-80;4微升10毫升转基因酚酯;4 μL 5%普隆尼克-F127;1 μL 1M 数码热脱硫;1μL葡萄糖;2微升聚乙二醇;从步骤2.5开始,12μL5mg / mL微管蛋白混合物(13.6μM终浓度);和 5.5 μL MAP65 工作储备液,其中 10% 为 GFP-MAP65,用于可视化。混合时保持在冰上。
注意:建议使用容积式移液器处理粘性PEG溶液。切尖后可以使用常规移液器,使开口更大;但是,这种方法不太准确。 - 通过移液混合5x-6x。
- 在加入腔室之前,将1μL葡萄糖氧化酶(0.5mg / mL)和过氧化氢酶(0.15mg / mL)(脱氧)的预混合溶液加入微管蛋白- MAP混合物中并混合7x-8x。将溶液的总体积(40μL)分成两部分,在单独的腔室中使用。
- 将微管蛋白-MAP混合物流入腔室。由于腔室中已经含有非离子嵌段共聚物表面活性剂,因此如果不去除旧液体,就无法添加更多液体。为此,请在腔室的另一端使用一张滤纸或无绒的实验室湿巾,通过毛细管作用除去液体。
- 一旦样品完全在腔室内,使用5分钟环氧树脂密封腔室的两端,并将其保持在37°C下约30分钟以成核并生长微管触觉。
6. 荧光显微镜
- 使用荧光显微镜对触觉进行成像。
注意:全内反射荧光显微镜或旋转盘共聚焦显微镜可以很好地去除游离微管蛋白的背景荧光,但是一旦形成,通过常规的落射荧光甚至透射光显微镜也可以看到触觉,这使得该过程无需专用设备即可进入。 - 使用1.2 NA或更高的数值孔径(NA)物镜,放大倍率为60倍或更高,以收集足够的荧光光。这些物镜通常需要浸入ddH2O或油中。
- 使用立体图像感应器或 CCD 相机记录图像。在相机上使用 108 nm 的有效像素大小。
注:像素大小取决于相机和所使用的放大倍率,在本例中为 60 倍或 100 倍,具有高数值孔径(1.2 或 1.49 NA)。可以在相机之前使用额外的图像扩展器来实现所需的像素尺寸。 - 使用设置为该温度的环境室将样品保持在37°C。或者,使用其他阶段加热器,包括热空气级加热器和带循环温水的客观温控环。
- 使用对所需荧光正确的激发源。对于罗丹明微管蛋白,在样品上使用功率至少为1 mW的561nm激光器,对于GFP-MAP65,使用488nm激光器,在样品处使用至少1 mW功率的488nm激光器。
注意:如果使用宽视场落射荧光显微镜,请使用激发:540 ± 12.5 nm,二向色性:545 nm ± 12.5 nm 截止时间和发射:575 nm 长通量的罗丹明滤光片立方体,其激发:480 ± 15 nm,二向色性:505 nm ± 15 nm 截止,发射:515 nm 长通。 - 拍摄至少 10 张不同区域的图像,以拍摄 100 多个触觉。在红色和绿色通道中拍摄图像,并将其另存为 16 位 tiff 图像以供分析。确保照明功率和曝光时间使照相机的强度刻度不饱和。
7. 光漂白后的荧光恢复 (FRAP)
注意:为了研究触头内部成分的迁移率,使用了FRAP。FRAP的工作原理是光漂白罗丹明标记的微管蛋白和GFP标记的MAP65触头的选定部分,然后观察该区域中荧光随时间的恢复。回收率取决于被光漂白物种的周转率。这种周转率可能取决于扩散和结合反应。对于 MAP65 与触头的结合,可以估计绑定汇率。FRAP使用额外的405 nm激光系统执行,该系统可以扫描任何形状的激光。执行FRAP有许多可能性,包括使用透射灯和光孔对局部区域14进行光漂白。
- 在腔室中选择一个孤立的触觉,以创建一个覆盖部分触觉和周围溶液的感兴趣区域(ROI)。
- 使用带有额外405nm激光的显微镜进行FRAP,以同时光漂白微管蛋白和MAP65。或者,可以通过光圈14的场停止使用明亮的灯。根据经验调整光漂白系统的比强度,以避免在漂白过程中损坏蛋白质。
- 在光漂白之前,将触觉记录为时间序列电影30-60秒,以获取有关漂白前强度的信息。记录红色和绿色通道。
- 通过根据需要用激光或灯暴露ROI来光漂白触头,以便在不损坏触头的情况下进行光漂白。根据经验确定强度和时间。
- 在光漂白后,继续在两个彩色通道中录制短片5-10分钟,或直到恢复似乎达到平衡。
- 目视检查 GFP-MAP65 通道的恢复情况。
8. 数据分析
注意:对触觉图像进行了定量分析,以了解通过不同的拥挤剂,离子条件和其他因素的添加施加的环境变化的影响。
- 触头形状表征
- 从用共聚焦显微镜拍摄的红色和绿色图像中量化触头的长度和宽度。
- 使用斐济/图像J打开图像。
- 如果原始数据以 16 位为单位,请根据需要调整亮度和对比度。选择 “图像>调整>亮度和对比度 ”以调整图像,以便能够清楚地看到触觉。在不应用设置的情况下调整亮度和对比度,以免意外更改强度数据。
- 一旦触头清晰可见,选择要测量的良好触头(图4Bi)。确保触头清晰可见,不与任何其他触头或聚集体重叠,并且没有弯曲或弯曲,以便能够使用直线测量工具。
- 接下来,检查是否为图像设置了正确的像素大小。显微镜图像带有有关像素大小的元数据。当使用没有元数据的其他相机或可以更改预期有效像素大小的外部图像扩展系统时,请手动调整像素大小。在斐济/ImageJ 中,转到 分析>设置比例 以设置正确的像素转换。
- 使用斐济/ImageJ工具栏中的 直线 工具,单击触觉的一端,然后将光标拖动到触头的另一端(图4Bii)。选择线 ROI 后,选择 分析>测量 以测量长度。如果默认情况下未测量长度,请确保在“ 分析>设置 测量值”对话框中将测量值设置为包括长度。
注意:通常,使用 “直线” 工具进行测量时,它会给出所绘制线条的长度和角度。例如, 图4Bii 显示了一条在触头的侧面绘制的直线,以使后者可见,但直接在触头上进行测量。 - 进行测量后,使用工具栏中的 “文本” 工具标记触觉。创建文本框,添加数字标签,然后选择 “编辑>绘制 ”以将标签固定到图像中。将图像另存为单独的 ROI 文件。
注意:标记并保存此文件可让调查人员从原始数据中知道哪个测量值对应于哪个触觉。确保测量每个触觉一次。 - 测量整个图像的触觉后,将“ 结果 ”窗口中的数据保存到逗号或制表符分隔的文本文件中(使用 “文件>另存为”),然后在电子表格程序中打开数据以将数据解析为数字。将所有数据(原始图像数据、ROI 图像和结果文本文件)一起收集到具有适当命名约定的文件夹中,以保持所有内容井井有条。
注意:虽然触头长度测量是用手进行的,但鉴于触头宽度较窄,最好采用不同的方法来测量触头宽度(见下文)以减少测量误差。 - 使用图像 J/斐济,使用直线工具绘制 直线 区域。将线绘制为与触状长轴垂直的平分扇区(图4Bii)。
- 选择分析>绘图剖面图以创建线性双扇区的强度剖面图(图 4Biii)。将出现一个绘图。要从绘图中检索并保存数据,请选择左下角的“列表”按钮;这将沿绘制的线的长度生成强度数据的文本文件列表。将文本文件另存为.csv或.txt文件。
- 在合适的程序中打开文本文件,如 MatLab、蟒蛇 (sciPy) 或其他程序。用高斯函数的形式拟合强度数据:
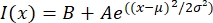 ,其中 I(x) 是沿长度 x 的灰度值;B是背景级别;A是高斯振幅;μ是高斯的均值或中心;σ是高斯的标准差。
,其中 I(x) 是沿长度 x 的灰度值;B是背景级别;A是高斯振幅;μ是高斯的均值或中心;σ是高斯的标准差。 - 报告 2σ 为触头的宽度。通过计算高斯(不包括背景)下的面积来估计触头中微管的强度。
注意:如果图像在相机的线性强度范围内,并且以相同的曝光时间和激发强度拍摄,则可以比较积分强度以估计触头中微管的相对数量。
- 熔断分析
注意:测试微管迁移率的实验和MAP65使用FRAM来记录由于分子运动引起的特定光漂白和强度恢复(图5A)。数据是使用ImageJ/斐济从图像时间序列数据中量化的。- 使用图像J/斐济打开电影数据。
- 随着时间的推移注册堆栈(时间序列数据)以消除漂移。使用 堆栈注册 插件以及辅助的 涡轮重置 插件;有关使用插件的说明,请参阅 材料表中给出的网络链接。选择平移以移动帧的位置,从而注册图像。
- 注册图像以消除漂移后,通过选择“图像” >“变换”>“旋转”,旋转图像,使触觉在帧中垂直或水平。选择要旋转的角度,然后使用 “预览” 确定触觉是否旋转得足够快。当预览显示触觉是垂直或水平时,选择“ 确定” 以旋转影片中的所有图像。
- 使用工具栏中的矩形选择工具在触觉的光漂白区域上创建矩形截面。使用图像>堆栈>测量堆栈记录每帧的 ROI 区域的集成强度。使用“分析”将测量类型设置为“集成密度>设置测量值”。通过选择“文件”(>另存为)将“结果”窗口中显示的分析强度数据保存为.csv或.txt格式的文本文件。
注意: 图5B 显示了在漂白区域中测量的微管和GFP-MAP65通道的原始16位强度数据的示例。 - 由于成像引起的光漂白,图像的整体强度将随着时间的推移而全局褪色,因此必须纠正这种全局光漂白。为此,请使用相同的 ROI 大小(步骤 8.2.4.),并将其移动到图像背景中不可见微管或 MAP65 的区域。按照步骤 8.2.4 中所述测量堆栈的集成强度。将结果另存为第二个文本文件。
注: 图5B 显示了在背景区域中为微管和GFP-MAP65通道测量的原始16位强度数据的示例。 - 要校正背景褪色,请将触头上的信号强度除以同一时间点的背景强度。将 我校正的 (t) 计算为:
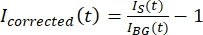 ,其中 IS (t)(信号)是在漂白区域进行的测量, IBG (t)(噪声)是在背景区域进行的测量(图5C)。这将计算每帧的信噪比,并减去噪声。
,其中 IS (t)(信号)是在漂白区域进行的测量, IBG (t)(噪声)是在背景区域进行的测量(图5C)。这将计算每帧的信噪比,并减去噪声。 - 然后,使用
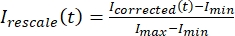 将数据重新缩放到 0 和 1 之间的范围,其中 Imin 和 Imax 分别表示 I校正 数据在整个时间内的全局最小值和最大值(图 5C)。
将数据重新缩放到 0 和 1 之间的范围,其中 Imin 和 Imax 分别表示 I校正 数据在整个时间内的全局最小值和最大值(图 5C)。 - 将此数据拟合到以下形式的衰减指数:
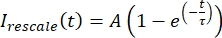 ,其中 A 是恢复的振幅,τ 是恢复的时间刻度(图 5C)。
,其中 A 是恢复的振幅,τ 是恢复的时间刻度(图 5C)。
仅使用少量组分,微管蛋白二聚体和微管交联剂,即可形成微管触点(图2A)。虽然该方案描述了在培养箱中成核和生长微管的孵育,但成核和生长可以直接在显微镜下观察(在30分钟内完成)(图2B)。微管蛋白的浓度保持在13.6μm,MAP65-MT结合在10%。
图 4 表示成功的数据。微管蛋白通道中的561nm激光器和MAP65通道中的488nm激光器都应该可见,它们彼此完全重叠(图4A)。该系统的一个谜团是,在各种实验变化下,触觉的宽度似乎没有变化,包括改变微管长度,MAP65浓度和拥挤剂(图4B)22,28。长度变化更大,取决于微管长度和MAP65浓度(图4B)22,28。
执行 FRAP 时,已观察到 MAP65 信号恢复,但微管信号不恢复(图 5)。FRAP中的恢复是由于标记和光漂白物体的移动性和移动性。在MAP65的情况下,变暗的分子解离并远离微管,新的分子进入该区域(图5)。MAP65结合处于平衡状态,因此结合和解绑的速率相等(以每秒分子数测量)。对于微管,没有看到恢复,这意味着微管不能离开触头(图5A,Bi,C)。此外,没有看到变暗区域的扩散,这表明微管局部不动,而不是触头形状内的流体。
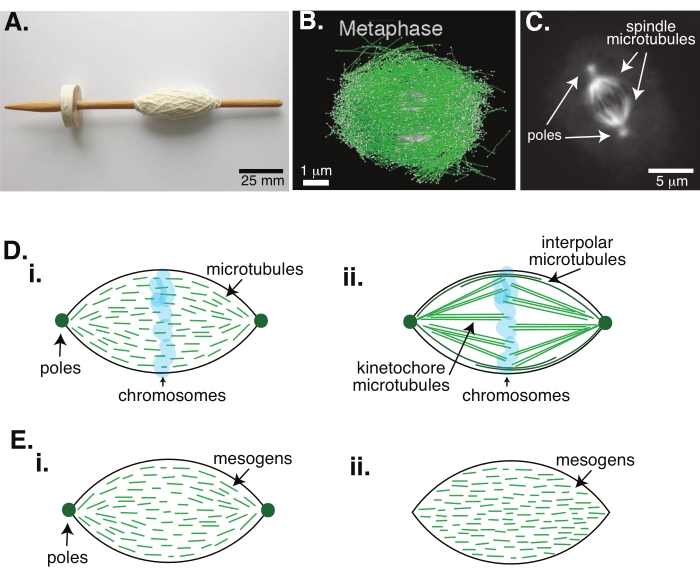
图 1:不同型号的主轴形成。 有丝分裂纺锤体是由微管及其相关蛋白质和酶制成的机器,在细胞分裂过程中将染色体对齐并分离成两个新的子细胞。(A)中世纪早期用荷兰细纱线制作的锭子复制品的图像。此图已从维基媒体图像中修改,由彼得·范德斯鲁伊斯30。(B)野生型减数分裂II不同阶段微管的三维重建。微管以绿色显示,染色体以灰色显示。比例尺 = 1 μm。这个数字是从兰奇等人31修改而来的。(C)分裂的Sf9细胞的有丝分裂纺锤体中微管的显微镜图像。纺锤体极和纺锤体的微管用绿色荧光蛋白标记。比例尺 = 5 μm。这个数字是从阿德瓦尼等人32修改而来的。(D)有丝分裂和减数分裂纺锤体微管如何组织的不同模型。(i)以前观察到由非洲爪蟾卵提取物产生的减数分裂纺锤体,微管(绿色)被推断为在整个纺锤体中短而动态。这类似于液晶内部的双极触状组织。(ii)有丝分裂纺锤体内微管组织的规范模型有两种类型的微管:在染色体周围的中带交联的插管或极性微管(深绿色)和运动微管(浅绿色),它们捆绑并从极点延伸到运动管以推动和拉动染色体。在所有图像中,染色体以透明蓝色显示,纺锤体极以深绿色表示。(E)液晶触头中介质(绿线)的示意图,用于(i)双极和(ii)均质触头。双极触觉在触觉的末端有两个极点,介质重新定向以指向这些极点。均匀的触觉在无穷远处有极点,并且介系原不会沿着触头的长度改变方向。 请点击此处查看此图的大图。
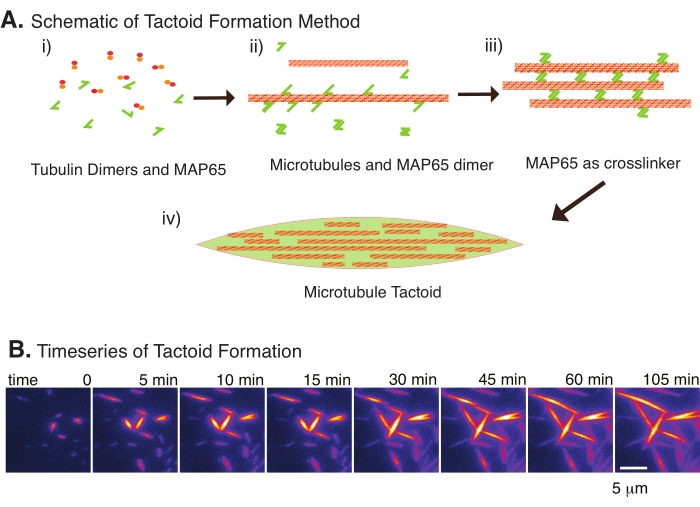
(A)微管可以通过多种方法捆绑和交联,包括离子物质,拥挤剂引起的消耗力以及特定的微管交联剂,如MAP65。(i)将微管蛋白二聚体和MAP65蛋白混合成核并生长微管。(ii)微管成核并从微管蛋白中生长,MAP65立即与微管,另一种MAP65单体或两者结合并导致捆绑。(iii)交联束中的微管成核并生长。(iv)最终配置是类似于纺锤体的微管触觉。(B)微管触觉成核并在105分钟内生长的时间序列。比例尺 = 5 μm。图改编自埃多齐等人22。请点击此处查看此图的大图。
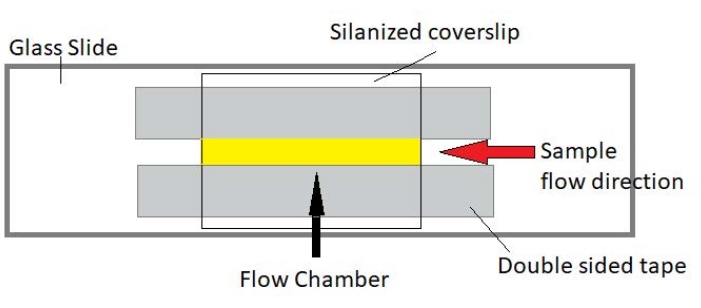
图 3:流室组件。 流动室使用载玻片、硅烷化盖板玻璃和永久双面胶带制成。黄色突出显示的区域是样品流动和观察的流动路径。流动室的体积约为20μL,环氧树脂用于密封腔室的末端,以防止样品在数小时内长期成像期间蒸发。 请点击此处查看此图的大图。
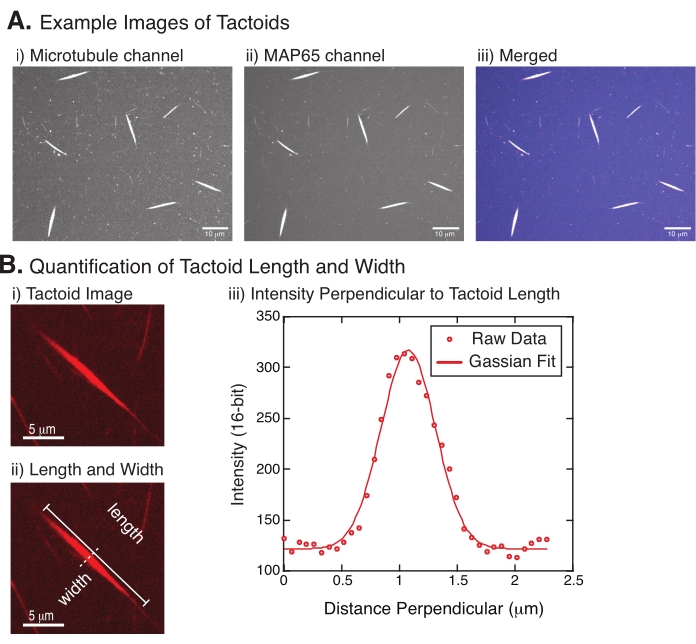
图 4:触觉图像以及长度和宽度分析。 (A)使用旋转盘共聚焦形成和使用旋转盘共聚焦成像的触觉类化合物的示例数据显示(i)使用561nm激光对罗丹明标记的微管蛋白进行微管蛋白成像,(ii)使用488nm激光对GFP进行GFP的GFP成像,以及(iii)来自微管通道(洋红色)和GFP-MAP65通道(青色)的合并叠加图像。重叠区域显示为白色,并表明微管和 MAP65 完全共定位。比例尺 = 10 μm,适用于 (A) 中的所有图像。(B)触觉长度和宽度的量化。(i) 在没有标签的情况下进行分析的触觉图像。比例尺 = 5 μm. (ii) 与 (i) 中的图像相同,其中表示长度(带线帽的实线)和宽度(虚线)测量值。(iii)宽度是通过在(ii)中表示的垂直双扇区(虚线)处的触头上测量宽度的。强度分布适合高斯函数,以显示触觉的振幅和宽度。 请点击此处查看此图的大图。
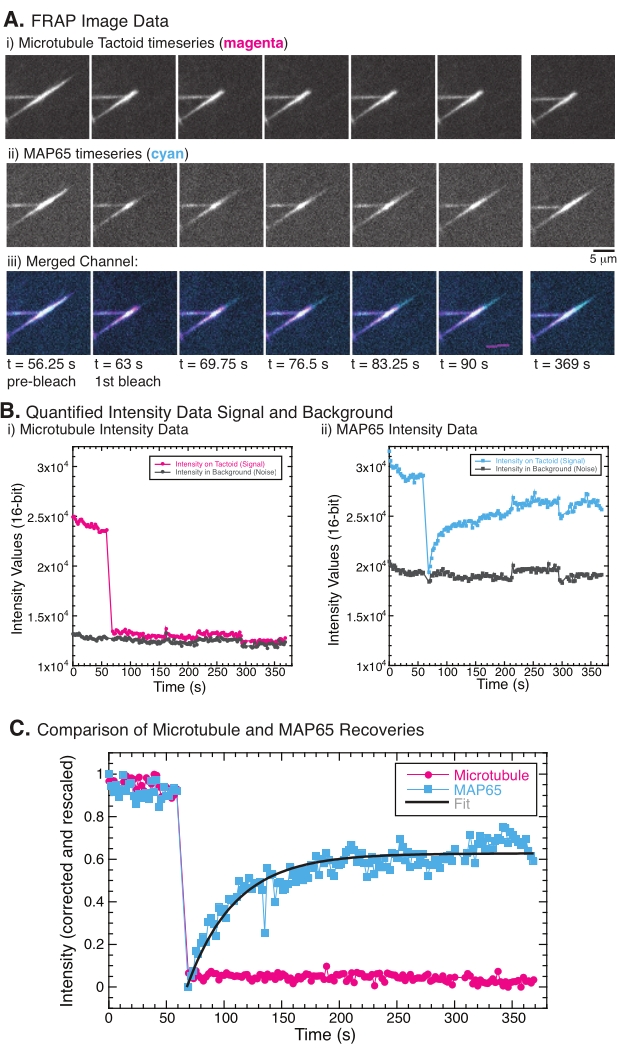
(A)(i)微管触头和(ii)GFP-MAP65的显微镜时间序列数据,以及(iii)两个通道的叠加图像,在洋红色中与GFP-MAP65中的青色中,在63秒时光漂白并额外观察5分钟(B)(i)微管通道在漂白区域(洋红色圆圈)和背景(深灰色圆圈)和(ii)GFP-MAP65通道在漂白区域(青色方块)和背景(深灰色方块)。(C)对数据进行背景噪声校正,并对微管通道(洋红色圆圈)和GFP-MAP65通道(青色方块)进行重新缩放。微管不会恢复,但GFP-MAP65确实可以并且可能适合(深灰色线)到上升的指数衰减中,以找到恢复的振幅和时间尺度。请点击此处查看此图的大图。
这里描述的方法已在几篇论文中用于制造微管触觉(图2)22,28。这些实验在生物学上是相关的,有助于揭示控制大多数细胞类型中有丝分裂或减数分裂纺锤体的形状和稳定性的组织原理。此外,微管是模型液晶介素,可以帮助更多地了解液晶如何成核并从各向同性相中生长向列相。
这里概述的程序对于探索微管自组织有几个优点。首先,它是高度可重复的,许多学生(包括高中生)在实验室中进行了实验,在开始进入实验室之前几乎没有预知或培训。触点是双折射22,除了荧光显微镜之外,还可以在透射光下观察它们,这使得这种方法可以被许多实验室使用,并且除了高端研究之外,该实验程序还适用于教育目的。最后,这个过程开辟了继续以精简的还原主义方法理解和探测生物系统的途径,使人们能够了解每个附加条件,蛋白质或添加剂如何改变触觉的自组织,也许最终改变纺锤体。更好的仿生学目标包括活性、流动性和灯丝极性分选。
可能有几个因素会影响实验,从而产生意想不到的结果。例如,如果触头未形成(图2),但观察到扇形图案,则MAP65可能不存在或不与微管22,28结合。这在MAP65荧光通道中也应该很明显,因为GFP-MAP65不会与微管结合。
如果触头未形成,并且背景在玻璃上显示为斑点,则可能是由于表面涂层。一旦进行,硅烷化仅在盖玻片上持续1个月。当它磨损时,微管蛋白将能够非特异性地结合到暴露的表面。此绑定将以奇怪的模式发生。
如果触觉不形成,并且在各种形状和大小的聚集体中观察到微管蛋白,这可能是由于质量差的微管蛋白。微管蛋白可以离心以除去初始聚集体,这些聚集体可以驱动这种旁路聚集而不是微管聚合。如果表面与微管蛋白结合,它也会耗尽溶液中的微管蛋白。低浓度的微管蛋白,低于聚合微管的临界浓度,可导致聚集体。
在FRAP实验中,如果MAP65通道没有显示出任何恢复(图5),则可能是光漂白对微管进行了光损伤。光损伤导致细丝的局部破坏。这可以通过在传输信道中进行检查来检查。微管触状体通过与周围水的高折射率不匹配在透射通道中可见。光致光损伤将在受光漂白的ROI位置的透射光成像中表现为烧伤标记或对比度损失。如果发生这种情况,必须降低激光或光功率以抑制蛋白质的光损伤。
在这一程序和方法中遇到了一些挑战。一个问题是,长度测量目前是通过单击图像手动执行的。这种方法虽然简单明了,但可能导致高不确定性。使用横截面并拟合高斯的宽度测量是量化尺寸的更好方法。对于长度,可以采用类似的方法。第二个问题是,有时,触觉,因为它们又长又薄,可以弯曲。这使得量化长度变得更加困难。可以使用分割线量化等高线长度,但每次添加线段时都会增加不确定性。
从科学的角度来看,这种方法在用作液晶或纺锤体的模型方面还有其他一些挑战。第一个挑战是微管产生的触觉的长而薄的形状(图3和图4)。如先前的出版物22所述,微管触觉是均质的触头,而不是双极性。这意味着构成形状的微管不会重新定向以指向结构的尖端。相反,所有的微管都平行于长轴,“极点”位于无穷远处。这与分子液晶甚至肌动蛋白或DNA观察到的触觉非常不同,肌动蛋白或DNA也可以充当液晶中间源。在这些其他系统中,触点是双极的,当在交叉偏振片中观察时,它们显示出杆重新定向的迹象。
该系统的第二个主要挑战是微管在触头内是不动的。从FRAP实验和分析中可以清楚地看出这一点,因为微管的回收率非常低。它们的固体性质使得微管触觉不如大规模液晶类似物有价值。液晶的向列相应同时具有液体(流体)和晶体(有组织)性质。虽然形状似乎适合主轴,但固定性使系统不像有丝分裂纺锤体那样令人兴奋。另一方面,这个问题提供了机会来研究如何修改实验以在系统中创造更多的流动性。
这些科学挑战提供了令人兴奋的机会,将允许对该系统有新的了解。为了使微管触觉更具有双极性,可以使用较短的微管。然而,还有一个额外的挑战,因为微管没有许多表征良好的封端蛋白来控制肌动蛋白的长度。成核和生长的使用需要使用非常高浓度的微管蛋白和GMPCPP来制造短的微管。高微管蛋白浓度导致系统中的细丝数量较多,这使得将触头彼此分离变得更加困难。添加新的微管封盖机,如DARPin33,可能有助于解决这种情况。微管不动的第二个问题可以通过添加运动蛋白来缓解,例如驱动蛋白-534,它们是用于有丝分裂的运动的四聚体。或者,可以使用二聚体驱动蛋白-1的人造二聚体15。
增加流动性的另一种方法是允许微管执行其动态不稳定性,即微管的生长和收缩。目前,用稳定的GMPCPP细丝播种然后经历动态不稳定的微管远远长于形成纺锤体或触头的预期时间,这将导致非常长的组织,如风扇或束。因此,需要小心地添加微管动态不稳定性以保持触觉形状。添加可以控制长度的相关蛋白质和酶可能会缓解此问题。例如,可能需要解聚激肽,如激肽-1335,或切断酶,如片宁36。这些实验是复杂而困难的,尽管无论结果如何,它们都会非常有见地。无论未来的实验采取哪个方向,这里开发的用于创建微管触觉的平台都可以在微管组织的物理基础上暴露新的信息。
作者声明他们没有竞争的经济利益。
作者要感谢所有2021年夏季罗斯实验室的成员,特别是K.爱丽丝林赛,他们的帮助。这项工作得到了NSF BIO-2134215的资助,该赠款支持了S.萨胡,N.古德比,H.B.李和J.L.罗斯。凯克基金会(雷·安德森,美元,首席PI)的赠款部分支持了R.布兰奇和P.肖汉
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
Tags
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved