
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
Selvmontering af mikrotubulus tactoider
Denne artikel præsenterer en protokol til dannelse af mikrotubulussamlinger i form af taktoider ved hjælp af MAP65, en plantebaseret mikrotubuluskrydsbinding og PEG som trængselsmiddel.
Cytoskelettet er ansvarlig for større intern organisation og omorganisering i cellen, alt sammen uden en leder til at lede ændringerne. Dette er især tilfældet under mitose eller meiose, hvor mikrotubuli danner spindlen under celledeling. Spindlen er det maskineri, der bruges til at adskille genetisk materiale under celledeling. Mod at skabe selvorganiserede spindler in vitro udviklede vi for nylig en teknik til at rekonstituere mikrotubuli i spindellignende samlinger med et minimalt sæt mikrotubulusassocierede proteiner og trængselsmidler. Specifikt blev MAP65 anvendt, som er en antiparallel mikrotubulus tværbinding fra planter, en homolog af Ase1 fra gær og PRC1 fra pattedyrorganismer. Denne tværbinding organiserer selv mikrotubuli i lange, tynde, spindellignende mikrotubulus selvorganiserede samlinger. Disse samlinger ligner også flydende krystal tactoider, og mikrotubuli kan anvendes som mesoskala mesogener. Her præsenteres protokoller til oprettelse af disse mikrotubulus tactoider samt til karakterisering af samlingernes form ved hjælp af fluorescensmikroskopi og mobiliteten af bestanddelene ved hjælp af fluorescensgendannelse efter fotobleaching.
Celledeling via mitose er en af de vigtigste biologiske processer for at opretholde livet. Mikrotubulusfilamenterne, der består af tubulindimere, er væsentlige strukturelle elementer i denne proces. Det forbigående maskineri, der skabes i metafase, når kromosomerne justeres i cellecentret, kaldes den mitotiske spindel på grund af dens form, som er som en spindel af en væv dækket af tråde (figur 1A). Det er veletableret på tværs af mange organismer, at mikrotubuli bruges i metafase til at skubbe og trække kondenserede kromosomer ind i midten af cellen, justere dem og tilslutte dem til mikrotubuli, der vil trække dem fra hinanden i anafase (figur 1B, C). Spindlen dannes i både meiose (figur 1B) og mitose (figur 1C), skabt af mange overlappende mikrotubuli, der ikke er viklet rundt om den centrale akse som tråd, men løber parallelt med grænsefladen. Oprettelse af disse mikrotubulusbaserede strukturer kræver tilknyttede proteiner, der tværbinding og tilhørende enzymer, der kan fungere som motorer til at hjælpe med at skubbe og trække kromosomerne1.
Undersøgelser af meiotiske spindler har vist, at mikrotubuli er korte, dynamiske og overlappende i tværbundne arrays 2,3,4,5,6 (figur 1Di). På grund af den fysiske organisering af disse korte mikrotubuli ligner den meiotiske spindel en flydende krystal taktoid (figur 1E). Faktisk har spindler vist sig at samle sig og fusionere, som man ville forvente af flydende krystal tactoider5.
Mange undersøgelser, der går tilbage til 1960'erne, har brugt fiksering, serielle sektioner og elektronmikroskopi for at bestemme, at der er to typer mikrotubuli inde i den mitotiske spindel 7,8,9,10. Den første type kaldes kinetochore mikrotubuli, som forbinder spindelstangen til kinetochore. Den anden type kaldes de interpolære eller polære mikrotubuli, som vokser forbi kromosomerne og overlapper ved midtzonen (figur 1Dii)8,9,10. En tredje type kaldes de astrale mikrotubuli, som er uden for spindlen og forbinder polerne til cellekanten; disse mikrotubuliorganisationer er uden for rammerne af den aktuelle diskussion. Der har været nylige undersøgelser af interaktionen mellem augmin6 og gamma-tubulin ringkompleks, der påvirker kimdannelsescentre for mikrotubuli, hvilket resulterer i en mitotisk spindel med kortere mikrotubuli som i figur 1D.
Da mikrotubuli er længere, end de er brede, med et højt billedformat og høj stivhed, er de som opskalerede versioner af flydende krystalmolekyler. I blødstoffysik er atomer og molekyler blevet tilnærmet ved hjælp af minimale interaktioner for at udlede de fysiske mekanismer for faseovergange, herunder kimdannelse og smeltning af krystaller11. På samme måde er mikrotubuli mesoskalaobjekter, der er opskalerede versioner af flydende krystalmolekyler, hvilket giver indsigt i fysikken i flydende krystaldynamik, herunder kimdannelse og vækst af de nematiske faser fra isotropiske. Som diskuteret ovenfor viser den meiotiske spindel egenskaber som dem af en flydende krystal taktoid, en nematisk tilstand, der nukleerer og vokser fra den isotropiske tilstand af flydende krystalmolekyler 3,4,5. For taktoider er kimdannelse og vækst som for andre krystaller (dvs. kræver en relativt høj koncentration af mesogener [molekylerne, der danner flydende krystaller]). Den unikke "spindel" form af tactoid kommer fra den lokale justering af de flydende krystal mesogener, der justerer ind i den nematiske fase (figur 1E). De kan ikke danne en afrundet krystal, fordi molekylerne er meget asymmetriske. I betragtning af mikrotubuliens art er det måske ikke overraskende, at det mitotiske spindelmaskineri fremstillet af en høj lokal koncentration af mikrotubuli også har samme form, hvad enten det kaldes en taktoid eller spindel. Tactoider kan være bipolære med poler i de koniske ender (figur 1Ei) eller homogene, med poler effektivt i uendelig (figur 1Eii).
I betragtning af betydningen af spindeldannelse har der været bestræbelser i gang mod selvorganiseret spindeldannelse in vitro ved at demonstrere mikrotubuluskondensation i bundter via ioniske arter12,13, trængselsmidler, der skaber udtømningsinteraktioner 14,15, og specifikke mikrotubulus tværbindende proteiner 13,16,17,18,19, 21. Overraskende nok, selvom disse midler alle arbejder for at øge den lokale koncentration af mikrotubuli, resulterer de ofte i lange mikrotubulusbundter, men ikke taktoider. En af grundene til, at disse bundter er lange, kan være, at mikrotubuli, der omfatter dem, også er lange. Nyligt arbejde ved hjælp af kortere mikrotubuli rapporterede også længere bundter, der ikke er tilspidset i slutningenaf 15; i dette tilfælde holdes bundterne sammen med motorproteiner, der forårsager forlængelse af bundterne og derved gør dem længere. Korte mikrotubuli med ikke-ekstensile tværbindinger er nødvendige til koniske, spindellignende samlinger, som beskrevet her.
For nylig har vi udviklet en teknik til at muliggøre dannelsen af mikrotubulus tactoider ved hjælp af antiparallel tværbinding, MAP65, i nærværelse af nucleating stabile mikrotubuli22. Mikrotubuli skulle være korte, men få kendte regulatorer af mikrotubuluslængde kan dække mikrotubuli mod dynamisk ustabilitet eller ende-til-ende-udglødning. I stedet blev GMPCPP brugt til at nukleere og stabilisere filamenterne efter vækst. Dette gjorde det muligt at skabe en høj tæthed af korte mikrotubuli, der kunne selvorganisere sig i taktarter. Disse taktoider var homogene, når de blev set under birefringence. Ud over korte mikrotubuli blev en specifik antiparallel tværbinding, MAP65, anvendt til at danne taktoiderne (figur 2). MAP65 er et plantemikrotubulus-associeret protein i PRC1/Ase1-familien af mitotiske tværbindinger23. MAP65 eksisterer som en dimer, med en stærk affinitet til at binde sig til sig selv såvel som mikrotubuli24. I modsætning til den meiotiske spindel og taktoider, der observeres med actinfilamenter 25,26,27, som er bipolære og har flydende egenskaber som flydende krystaller, er mikrotubulus tactoider blevet observeret at være fastlignende 22,28.
Her præsenteres protokoller til oprettelse af mikrotubulus tactoider og karakterisering af samlingernes form og bestanddelenes mobilitet ved hjælp af fluorescensbaserede teknikker.
BEMÆRK: Medmindre andet er angivet, kan dele af eksperimentet udføres på en laboratoriebænk, mens der bæres passende beskyttelsesudstyr (handsker).
1. Silanisering af coverslip
BEMÆRK: Dækslips skal silaniseres for at blive brugt med den polymerbørstebelægning, der anvendes i disse forsøg. Dette er en hydrofob silaniseringsbehandling, der tillader en blokcopolymer med en hydrofob central blok at binde og skabe en polymerbørste. Følgende trin skal udføres i en røghætte for at forhindre udsættelse for giftige dampe, mens du bærer handsker. Dimethyldichlorolsilan er meget giftigt og skal håndteres med største forsigtighed.
- Skyl dækslerne med ddH2O, 70% ethanol og ddH2O i rækkefølge. Tør dem med fnugfri laboratorieservietter mellem hver skylning. Dette fjerner støv og vandopløselige eller organiske partikler fra overfladen før behandling.
- Anbring dækslerne i et metaldækslerstativ, og overfør stativet til en UV-Ozon (UVO) maskine. Bestråle dækslerne med UVO i 20 minutter for at fjerne eventuel baggrundsfluorescens. Et plasmakammer kan bruges i stedet for UVO.
- Brug pincet til at overføre dækslerne fra metalstativet, der bruges til UVO-behandling, til et andet metalstativ, der bruges til silanisering. Brug ikke de samme stativer til begge, da det vil medføre høje niveauer af oxidation, når UVO påføres. Forrens stativerne med vand og ethanol, så der ikke er rester af kemikalier tilbage fra tidligere anvendelser.
- Dyp stativet med dækslerne i en beholder med 100% acetone i 1 time. Skyl beholderen 3x med ledningsvand og derefter 3x med ddH2O for at fjerne al acetone.
- Dyp stativet med dækslerne i 100% ethanol i 10 min. Skyl beholderen 3x med ledningsvand og derefter 3x med ddH2O for at fjerne al ethanol.
- Dyp stativet med dækslerne 3x i ddH2O i 5 min hver.
- Dyp stativet med dækslerne i 0,1 M KOH (50 ml 1 M KOH i 450 ml ddH2O) i 15 min. Skyl beholderen 3x med ledningsvand og derefter 3x med ddH2O for at fjerne al KOH.
- Dyp stativet med dækslips 3x i ddH2O i 5 min hver.
- Lufttør stativet med dækslerne natten over i en røghætte eller laminær flowhætte.
- Efter fuldstændig tørring af stativet og dækslerne nedsænkes dem i 5 minutter i 2% dimethyldichlorsilan (DDS) taget i en anden beholder, der anvendes specifikt til silan. Lad ikke noget, der ikke er tørt, komme i kontakt med silanen.
- Dyp stativet og dækslerne 2x i en beholder med 100% ethanol i 5 min. Skyl beholderen 3x med ledningsvand og derefter 3x med ddH2O.
- Dyp stativet og dækslerne 3x i ddH2O i 5 min hver.
- Lufttør stativet med dækslerne natten over i en røghætte eller laminær flowhætte.
- Efter dette sidste tørretrin skal du overføre dækslipsene tilbage til dækslipbokse ved hjælp af pincet. Disse coverslips kan bruges i de næste 1-2 måneder. Gamle dækslips begynder at miste deres belægning og skal kasseres.
2. Forberedelse af tubulin
BEMÆRK: Det købte tubulin kommer som et frysetørret pulver, der enten er umærket eller mærket med fluoroforer. Frysetørret tubulin opbevares i en fryser på −80 °C. Følgende procedure bruges til at blande umærket tubulin med mærket tubulin i et forhold, der er godt til visualisering.
- Fra -80 °C fryseren fjernes en alikvote umærket tubulin indeholdende 1 mg fryseboks, og det opbevares på is. Der tilsættes 200 μL kold PEM-80 til røret for at bringe tubulinkoncentrationen op på 5 mg/ml. Opbevar det på is i 10 minutter for at opløse alt lyofilatet.
- Fra fryseren på −80 °C fjernes en alikvote rhodaminmærket tubulin indeholdende 20 μg frysebokset tubulinpulver, og det opbevares på is. Der tilsættes 4 μL kold PEM-80 til røret for at bringe tubulinkoncentrationen op på 5 mg/ml. Opbevar det på is i 10 minutter for helt at opløse frysepunktet.
- Når det er opløst, tilsættes 100 μL umærket resuspenderet tubulinopløsning til 4 μL opløsningen af rhodaminmærket tubulin. Pipetter 6x-7x meget langsomt for at blande. Hvis aggregater er synlige, centrifugeres det opløselige tubulin i 10 minutter ved 90.000 x g for at fjerne aggregater ved at kassere pelleten og beholde supernatanten. Denne tubulinblanding vil resultere i ~ 4% mærket tubulin.
- De resterende 100 μL umærket tubulin nedfryses til flydende nitrogen (LN2) og opbevares ved −80 °C, der skal anvendes til yderligere tubulinblandinger.
- Tag den mærkede tubulinblanding og aliquot i syv rør med 15 μL hver. Hver aliquot kan bruges til et enkelt eksperimentelt kammer. Drop-frys de resterende alikvoter og opbevar dem ved -80 ° C til fremtidige forsøg.
3. MAP65 rensning
BEMÆRK: MAP65 er ikke kommercielt tilgængelig og skal derfor renses for dette arbejde. Protokollen er tidligere blevet uddybet i flere publikationer23,29.
- Map65-plasmidet og GFP-MAP65-plasmidet omdannes til BL21-bakteriestammen til proteinekspression.
- Dyrk BL21 bakterier til en optisk tæthed på 0,6-1 ved 600 nm. Inducer proteinproduktion ved hjælp af lac-operatøren og dyrk bakterierne natten over.
- Pellets kulturerne og lyse bakterierne.
- Saml lysatet efter centrifugering og inkuber det med perler, der har en nikkelion til rådighed til at binde 6x-histidinmærket.
- Eluter proteinet ved hjælp af imidazol og afsalt det.
- Drop-frys proteinet med flydende nitrogen og opbevar det ved -80 ° C til brug inden for 1 år.
4. Samling af flowkamre
BEMÆRK: Eksperimenter udføres i flowkamre lavet af et glasglas og silaniseret dækglas (figur 3).
- Tag et glasglas og rengør det med ddH2O, ethanol og ddH2O i rækkefølge. Tør med en fnugfri laboratorieserstat mellem hver skylning.
- Brug et stykke dobbeltklæbende tape til at oprette en flowkurve. Brug behandskede hænder til at skære båndet til ~ 25-30 mm i længden. Del båndet i længderetningen for at skabe to tyndere strimler. Placer de to båndstrimler på diaset med ca. 5-8 mm mellem dem.
BEMÆRK: Da båndets tykkelse er standardiseret til ca. 80-100 μm, bestemmer bredden af stien mellem båndstykkerne volumenet i kammeret. - Placer de silaniserede dækslips oven på strømningsstien. Forsegl glide- og dækslippen til de dobbeltsidede tapestrimler ved forsigtigt at trykke på båndområdet med bagsiden af en pen. Sørg for at få en god forsegling over hele området; båndet skal dreje fra gennemskinneligt til klart, når tætningen er lavet godt.
- Fjern det ekstra bånd på kanterne, og efterlad kun 1 mm fra indgangen til flowkammeret ved at skære båndet med et barberblad.
- Mærk kammeret med oplysninger om de eksperimentelle parametre efter behov.
5. Tactoid eksperimenter
BEMÆRK: Når alle reagenser og forsyninger er genereret, kan de bruges til at nukleere og polymerisere mikrotubulus tactoider i flowkammeret.
- Saml alle de reagenser, der skal bruges. Tø dem op på is og opbevar dem på is, mens du arbejder. Lav flere flowkamre til eksperimenterne.
- Brug et flowkammer til hvert eksperiment. Overtræk flowkammeroverfladerne med en polymerbørste ved at flyde i 20 μL 5% ikke-ionisk blok co-polymer overfladeaktivt stof (Material Table of Materials) opløst i PEM-80, med små dråber i begge ender af kammeret for at forhindre dannelse af luftbobler indeni. Opbevar dette i et fugtigt kammer (dvs. petriskål med en våd fnugfri laboratorieseriet), indtil den er klar til brug (mindst 5-7 min).
- I et sterilt rør blandes følgende for at skabe Tubulin-MAP-blandingen: 9,5 μL PEM-80; 4 μL 10 mM GMPCPP; 4 μL af 5% Pluronic-F127; 1 μL af 1M DTT; 1 μL glucose; 2 μL polyethylenglycol (PEG); 12 μL 5 mg/ml tubulinblanding (13,6 μM slutkoncentration) fra trin 2.5 og 5,5 μL arbejdslager af MAP65, hvor 10% er GFP-MAP65 til visualisering. Opbevares på is, mens du blander.
BEMÆRK: Det anbefales at bruge en positiv forskydningspipette til håndtering af den viskøse PEG-opløsning. Regelmæssige pipetter kan bruges efter skæring af spidsen for at gøre åbningen større; denne metode er dog mindre nøjagtig. - Bland 5x-6x ved pipettering.
- Lige før tilsætning til kammeret tilsættes 1 μL af en forblandet opløsning af glucoseoxidase (0,5 mg / ml) og katalase (0,15 mg / ml) (Deoxy) i Tubulin-MAP-blandingen og blandes 7x-8x. Opdel det samlede volumen af opløsningen (40 μL) i to portioner, der skal anvendes i separate kamre.
- Flow Tubulin-MAP-blandingen i kamre. Da kamrene allerede har ikke-ionisk blok-co-polymer overfladeaktivt stof i dem, kan der ikke tilsættes mere væske uden at fjerne den gamle væske. For at gøre dette skal du bruge et stykke filterpapir eller en fnugfri laboratorierietiet i den anden ende af kammeret for at fjerne væske via kapillærvirkning.
- Når prøven er helt inde i kammeret, forsegles de to ender af kammeret ved hjælp af 5-minutters epoxy og opbevares ved 37 ° C i ~ 30 minutter for at nukleere og dyrke mikrotubulus tactoider.
6. Fluorescensmikroskopi
- Brug et fluorescensmikroskop til billeddannelse af taktoiderne.
BEMÆRK: Total intern refleksion fluorescensmikroskopi eller roterende diskkonfokal mikroskopi er gode til at fjerne baggrundsfluorescens fra frit tubulin, men taktoider er også synlige ved regelmæssig epifluorescens og endda transmitteret lysmikroskopi, når de er dannet, hvilket gør denne procedure tilgængelig uden specialudstyr. - Brug et numerisk blændemål (NA) på 1,2 NA eller højere med en forstørrelse på 60x eller højere for at opsamle nok lys i fluorescens. Disse mål kræver ofte nedsænkning i enten ddH2O eller olie.
- Optag billeder med CMOS- eller CCD-kamera. Brug en effektiv pixelstørrelse på kameraet på 108 nm.
BEMÆRK: Pixelstørrelsen afhænger af kameraet og den anvendte forstørrelse, som i dette tilfælde var 60x eller 100x med en høj numerisk blænde (1,2 eller 1,49 NA). Yderligere billedudvidelser kan bruges før kameraet for at opnå den nødvendige pixelstørrelse. - Prøven opbevares ved 37 °C ved hjælp af et miljøkammer indstillet til denne temperatur. Alternativt kan du anvende andre scenevarmere, herunder varmluftstrinvarmere og objektive temperaturstyrede kraver med cirkulerende varmt vand.
- Brug exciteringskilder, der er korrekte for den nødvendige fluorescens. For rhodamin tubulin skal der anvendes en 561 nm laser med mindst 1 mW effekt ved prøven, og for GFP-MAP65 anvendes en 488 nm laser med mindst 1 mW effekt ved prøven.
BEMÆRK: Hvis du bruger wide-field epi-fluorescensmikroskopi, skal du bruge en rhodaminfilterterning med excitation: 540 ± 12,5 nm, dikroisk: 545 nm ± 12,5 nm cut-off og emission: 575 nm lang pass og en GFP filterterning med excitation: 480 ± 15 nm, dikroisk: 505 nm ± 15 nm cut-off og emission: 515 nm lang pass. - Tag mindst 10 billeder af forskellige områder for at afbilde over 100 takter. Tag billeder i både de røde og grønne kanaler, og gem dem som 16-bit tiff-billeder til analyse. Sørg for, at belysningseffekten og eksponeringstiderne er sådan, at kameraets intensitetsskala ikke er mættet.
7. Fluorescensgendannelse efter fotobleaching (FRAP)
BEMÆRK: For at studere mobiliteten af de interne bestanddele af tactoider blev FRAP anvendt. FRAP fungerer ved at fotobleaching en udvalgt del af rhodamin-mærket tubulin og GFP-mærket MAP65 tactoid og derefter observere genopretningen af fluorescensen med tiden i denne region. Genopretningshastigheden afhænger af omsætningen af de arter, der fotobleges. Denne omsætningshastighed kan afhænge af diffusions- og bindingsreaktioner. For MAP65, der binder til tactoider, kan bindende valutakurser estimeres. FRAP udføres ved hjælp af et ekstra 405 nm lasersystem, der kan scanne laseren i enhver form. Der er mange muligheder for at udføre FRAP, herunder brug af den transmitterede lampe og blænde til at fotoblege et lokalområde14.
- Vælg en isoleret takt i kammeret for at skabe et interesseområde (ROI), der dækker dele af taktoiderne og den omgivende opløsning.
- Brug et mikroskop med en ekstra 405 nm laser til FRAP til at fotoblechere både tubulin og MAP65 samtidigt. Alternativt kan en lys lampe bruges gennem iris14's feltstop. Juster den specifikke intensitet af fotoblegningssystemerne empirisk for at undgå at beskadige proteinerne under blegning.
- Optag tactoid som en tidsseriefilm i 30-60 s før fotobleaching for at få information om intensiteten før blegemidlet. Optag både de røde og grønne kanaler.
- Fotobleach takten ved at udsætte ROI med enten lasere eller lampen så længe som nødvendigt for at fotobleach uden at beskadige taktoidet. Bestem intensiteten og tiden empirisk.
- Fortsæt med at optage filmen i begge farvekanaler i 5-10 minutter efter fotobleaching, eller indtil gendannelsen ser ud til at have nået ligevægt.
- Undersøg visuelt GFP-MAP65-kanalen for gendannelse.
8. Analyse af data
BEMÆRK: Kvantitativ analyse af billederne af tactoider blev udført for at lære om virkningerne af miljøændringer pålagt via forskellige trængselsmidler, ioniske forhold og tilføjelse af andre faktorer.
- Karakterisering af taktoid form
- Kvantificer længden og bredden af taktoiderne fra de røde og grønne billeder taget med konfokal mikroskopi.
- Åbn billederne ved hjælp af FIJI/ImageJ.
- Hvis rådata tages i 16-bit, skal du justere lysstyrken og kontrasten om nødvendigt. Vælg Billede > Juster > lysstyrke og kontrast for at justere billedet, så det kan se taktoidet tydeligt. Juster lysstyrken og kontrasten uden at anvende indstillingen for ikke at ændre intensitetsdataene ved et uheld.
- Når taktoiderne er tydeligt synlige, skal du vælge gode taktoider, der skal måles (figur 4Bi). Sørg for, at taktoiderne er tydeligt synlige uden overlapning med andre taktoider eller aggregater og ikke er buede eller bøjede for at kunne bruge lineære måleværktøjer.
- Kontroller derefter, at den korrekte pixelstørrelse er indstillet til billederne. Mikroskopbillederne leveres med metadata om pixelstørrelsen. Når du bruger et andet kamera, der ikke har metadata, eller eksterne billedudvidelsessystemer, der kan ændre den forventede effektive pixelstørrelse, skal du justere pixelstørrelsen manuelt. I FIJI/ImageJ skal du gå til Analysér > Indstil skala for at indstille den korrekte pixelkonvertering.
- Brug værktøjet Lige linje fra værktøjslinjen i FIJI/ImageJ til at klikke på den ene ende af taktoidet og trække markøren til den anden ende af taktbladet (figur 4Bii). Når linjen ROI er valgt, skal du vælge Analyser > Måling for at måle længden. Hvis længden ikke måles som standard, skal du sørge for at angive målingen til at medtage længde i dialogboksen Analysér > Angiv målinger .
BEMÆRK: Når du måler ved hjælp af værktøjet Lige linje , vil det typisk give længden og vinklen på den tegnede linje. Som et eksempel viser figur 4Bii en lige linje trukket til siden af taktoidet for at gøre sidstnævnte synlig, men foretage målingen direkte på taktoidet. - Når du har foretaget målingen, skal du bruge tekstværktøjet på værktøjslinjen til at mærke taktoidet. Opret et tekstfelt, tilføj en taletiket, og vælg Rediger > Tegn for at rette etiketten i billedet. Gem billedet som en separat ROI-fil.
BEMÆRK: Mærkning og lagring af denne fil giver efterforskeren mulighed for at vide, hvilken måling der svarer til hvilken taktoid fra de rå data. Sørg for at måle hver taktoid en gang. - Når taktoiderne for hele billedet er målt, skal du gemme dataene i vinduet Resultater i en kommasepareret eller tabulatorsepareret tekstfil (ved hjælp af File > Save As) og åbne dataene i et regnearksprogram for at analysere dataene i tal. Saml alle data sammen (rå billeddata, ROI-billede og tekstfil med resultater) i en mappe med en passende navngivningskonvention for at holde alt organiseret.
BEMÆRK: Selvom taktoidlængdemålinger udføres manuelt, da taktbredderne er smalle, er det bedre at anvende en anden metode til måling af taktformet bredde (se nedenfor) for at reducere målefejl. - Brug ImageJ/FIJI til at tegne et linjeområde ved hjælp af værktøjet Lige linje . Tegn linjen som en vinkelret bisektor til den taktformede lange akse (figur 4Bii).
- Vælg Analyser > plotprofil for at oprette intensitetsprofilen for den lineære bisektor (Figur 4Biii). Et plot vises. For at hente og gemme dataene fra plottet skal du vælge knappen Liste nederst til venstre; dette genererer tekstfillisten over intensitetsdataene langs længden af den tegnede linje. Gem tekstfilen som en .csv- eller .txt fil.
- Åbn tekstfilen i et passende program som MatLab, Python (sciPy) eller andre programmer. Tilpas intensitetsdataene med en gaussisk funktion af formularen:
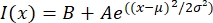 , hvor I(x) er gråtoneværdien langs længden, x; B er baggrundsniveauet; A er amplituden af gaussian; μ er middelværdien eller centrum af gaussianeren; og σ er gaussernes standardafvigelse.
, hvor I(x) er gråtoneværdien langs længden, x; B er baggrundsniveauet; A er amplituden af gaussian; μ er middelværdien eller centrum af gaussianeren; og σ er gaussernes standardafvigelse. - Rapport 2σ som bredden af taktoidet. Estimer intensiteten af mikrotubuli i taktoidet ved at beregne arealet under Gaussian (ikke inklusive baggrunden).
BEMÆRK: Hvis billederne er inden for kameraets lineære intensitetsområde og er taget med samme eksponeringstid og excitationsintensitet, kan integrerede intensiteter sammenlignes for at estimere det relative antal mikrotubuli i taktoidet.
- FRAP-analyse
BEMÆRK: Eksperimenter til test af mikrotubulis mobilitet og MAP65 brugte FRAP til at registrere den specifikke fotobleaching og genopretning af intensitet på grund af molekylær bevægelse (figur 5A). Dataene blev kvantificeret ud fra billedtidsseriedataene ved hjælp af ImageJ/FIJI.- Brug ImageJ/FIJI til at åbne filmdataene.
- Registrer stakke (tidsseriedata) over tid for at fjerne drift. Brug StackReg-pluginet sammen med det supplerende TurboReg-plugin ; for instruktioner om brug af plugins henvises til weblinks, der er angivet i materialetabellen. Vælg oversættelse for at flytte placeringen af rammerne og dermed registrere billederne.
- Når billederne er registreret for at fjerne drift, skal du rotere billedet, så taktoidet enten er lodret eller vandret i rammen ved at vælge Image > Transform > Rotate. Vælg den vinkel, der skal roteres, og brug Eksempel til at afgøre, om taktoidet roteres nok. Når eksemplet viser, at taktbladet enten er lodret eller vandret, skal du vælge OK for at rotere alle billederne i filmen.
- Brug markeringsværktøjet Rektangel på værktøjslinjen til at oprette en rektangulær sektion over det fotobleachede område af taktoidet. Registrer den integrerede intensitet af ROI-området for hver ramme ved hjælp af Image > Stacks > Measure Stack. Indstil måletypen til Integreret densitet ved hjælp af Analysér > Indstil målinger. Gem de analyserede intensitetsdata, der vises i vinduet Resultater , som en tekstfil i .csv- eller .txt format ved at vælge Filer > Gem som.
BEMÆRK: Figur 5B viser et eksempel på de rå 16-bit intensitetsdata målt for mikrotubulus- og GFP-MAP65-kanalerne i blegemiddelområdet. - Da den samlede intensitet af billederne vil falme over tid globalt på grund af fotobleaching forårsaget af billeddannelse, skal denne globale fotobleaching korrigeres. Det gør du ved at bruge den samme ROI-størrelse (trin 8.2.4.) og flytte den til et område i baggrunden af billedet, hvor ingen mikrotubuli eller MAP65 er synlige. Mål stakkens integrerede intensitet som beskrevet i trin 8.2.4. Gem resultaterne som en anden tekstfil.
BEMÆRK: Figur 5B viser et eksempel på de rå 16-bit intensitetsdata, der er målt for mikrotubulus- og GFP-MAP65-kanalerne i baggrundsområdet. - For at korrigere baggrundens fading skal du dividere signalintensiteten på taktoidet med baggrundsintensiteten for det samme tidspunkt. Beregn Ikorrigeret (t) som:
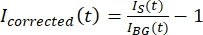 , hvor IS (t) (signalet) er målingen foretaget på det blegede område og IBG (t) (støjen) er målingen foretaget på baggrundsområdet (figur 5C). Dette beregner signal/støj-forholdet for hver ramme og trækker også støjen fra.
, hvor IS (t) (signalet) er målingen foretaget på det blegede område og IBG (t) (støjen) er målingen foretaget på baggrundsområdet (figur 5C). Dette beregner signal/støj-forholdet for hver ramme og trækker også støjen fra. - Derefter skaleres dataene igen til at ligge mellem nul og en ved hjælp af
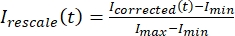 , hvor Imin og Imax angiver henholdsvis det globale minimum og maksimum for deI-korrigerede data over hele tiden (figur 5C).
, hvor Imin og Imax angiver henholdsvis det globale minimum og maksimum for deI-korrigerede data over hele tiden (figur 5C). - Tilpas disse data til en henfaldende eksponentiel af formen:
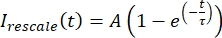 , hvor A er amplituden af opsvinget og τ er tidsskalaen for genoprettelsen (figur 5C).
, hvor A er amplituden af opsvinget og τ er tidsskalaen for genoprettelsen (figur 5C).
Med kun et lille antal komponenter, tubulindimere og mikrotubuluskrydslinkere kan der dannes mikrotubulus tactoider (figur 2A). Selvom denne protokol beskriver inkubation for at nukleere og dyrke mikrotubuli i en inkubator, kan kimdannelsen og væksten observeres direkte under mikroskopet (som er afsluttet inden for 30 minutter) (figur 2B). Koncentrationen af tubulin holdes konstant ved 13,6 μm og MAP65-MT binding ved 10%.
Figur 4 repræsenterer vellykkede data. Taktoiderne skal være synlige med både en 561 nm laser i tubulinkanalen og 488 nm i MAP65-kanalen, som overlapper hinanden perfekt (figur 4A). Et mysterium ved systemet har været, at bredden af taktoiderne ikke synes at variere under en række eksperimentelle ændringer, herunder ændring af mikrotubuluslængderne, MAP65-koncentrationen og trængselsmidlerne (figur 4B)22,28. Længden er meget mere variabel og afhænger af både mikrotubuluslængderne og MAP65-koncentrationen (figur 4B)22,28.
Ved udførelse af FRAP er det blevet observeret, at MAP65-signalet gendannes, men mikrotubulussignalet gendannes ikke (figur 5). Gendannelsen i FRAP skyldes mobiliteten og bevægelsen af de mærkede og fotobleachede objekter. I tilfælde af MAP65 dissocieres de mørke molekyler og bevæger sig væk fra mikrotubulusen, og nye bevæger sig ind i regionen (figur 5). MAP65-bindingen er i ligevægt, så bindingshastigheden og afbindingen er ens (målt i molekyler pr. Sekund). For mikrotubuli blev der ikke set nogen genopretning, hvilket tyder på, at mikrotubuli ikke er i stand til at forlade taktoidet (figur 5A, Bi, C). Endvidere blev der ikke set nogen spredning af det mørke område, hvilket tyder på, at mikrotubuli er lokalt immobile og ikke en væske inden for taktformen.
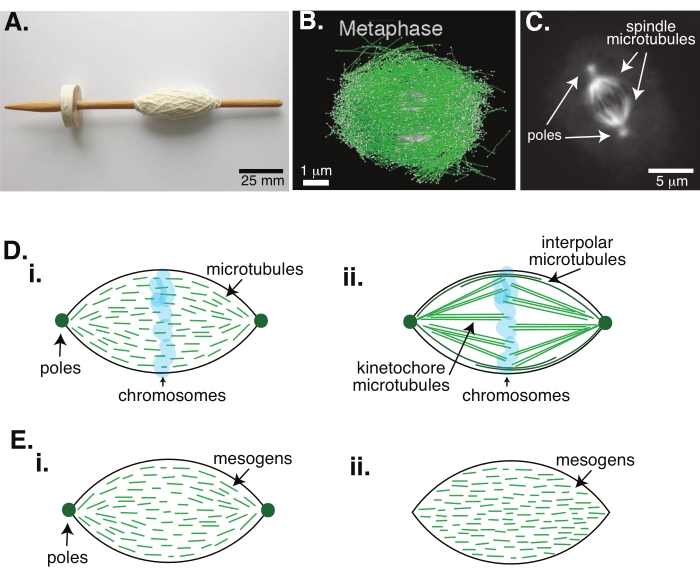
Figur 1: Forskellige modeller af spindeldannelse. En mitotisk spindel er en maskine lavet af mikrotubuli og deres tilknyttede proteiner og enzymer, der justerer og adskiller kromosomerne i de to nye datterceller under celledeling. (A) Billede af en tidlig middelalder dråbe spindel replika med fint garn fra Holland. Denne figur er blevet ændret fra et Wikimedia-billede af Peter van der Sluijs30. (B) Tredimensionel rekonstruktion af mikrotubuli på forskellige stadier af vildtypemeiose II. Mikrotubuli er vist i grønt, og kromosomer er vist i gråt. Skala bar = 1 μm. Dette tal er ændret fra Lantzsch et al.31. (C) Mikroskopibillede af mikrotubuli i en mitotisk spindel af en delende Sf9-celle. Spindelstængerne og spindlens mikrotubuli er mærket med et grønt fluorescerende protein. Skala bar = 5 μm. Dette tal er blevet ændret fra Advani et al.32. (D) Forskellige modeller af, hvordan mitotiske og meiotiske spindelmikrotubuli er organiseret. (i) Mikrotubuli (grøn), der tidligere blev observeret for meiotiske spindler fremstillet af Xenopus-ægekstrakter, blev udledt til at være korte og dynamiske i hele spindlen. Dette ligner en bipolar taktoid organisation inde i en flydende krystal. (ii) Den kanoniske model for mikrotubulusorganisation inde i en mitotisk spindel har to typer mikrotubuli: interpolære eller polære mikrotubuli (mørkegrøn), der tværbinder i midtzonen omkring kromosomerne og kinetochore mikrotubuli (lysegrøn), der er bundtet og strakt fra polen til kinetochore for at skubbe og trække kromosomerne. På alle billeder vises kromosomer i gennemsigtig blå, og spindelstænger er repræsenteret i mørkegrøn. (E) Skemaer af mesogener (grønne linjer) i en flydende krystal tactoid for (i) bipolære og (ii) homogene taktoider. Bipolære taktoider har to poler i slutningen af taktoidet, og mesogenerne omorienterer sig for at pege på disse poler. Homogene taktoider har poler i uendelighed, og mesogenerne ændrer ikke orientering langs længden af taktoidet. Klik her for at se en større version af denne figur.
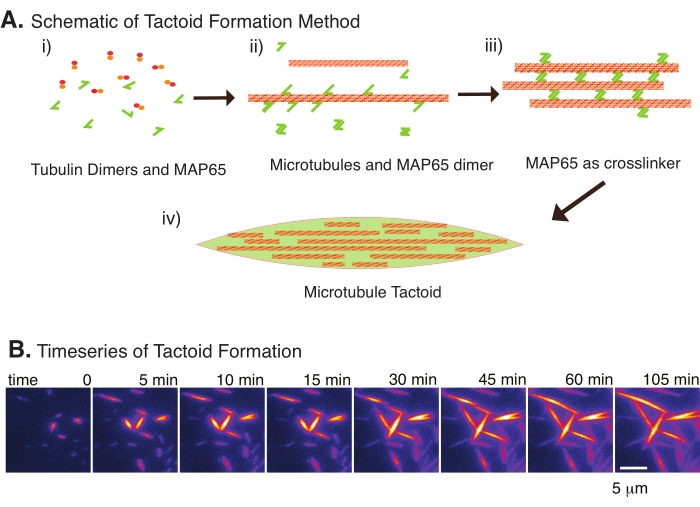
Figur 2: Mikrotubuluskondensation. (A) Mikrotubuli kan bundtes og tværbindes ved hjælp af en række forskellige metoder, herunder ioniske arter, udtømningskræfter forårsaget af trængselsmidler og specifikke mikrotubuluskrydsbindinger, såsom MAP65. i) Tubulindimere og MAP65-proteiner blandes for at nukleere og dyrke mikrotubuli. (ii) Mikrotubuli nukleerer og vokser fra tubulinet, og MAP65 binder sig straks til mikrotubuli, en anden MAP65-monomer eller begge dele og forårsager bundtning. iii) Mikrotubuli i de tværbundne bundter nukleerer og vokser. iv) Den endelige konfiguration er en mikrotubulus tactoid svarende til en spindel. (B) Tidsserier af mikrotubulus tactoider, der nukles og vokser over 105 min. Skala bar = 5 μm. Figur tilpasset fra Edozie et al.22. Klik her for at se en større version af denne figur.
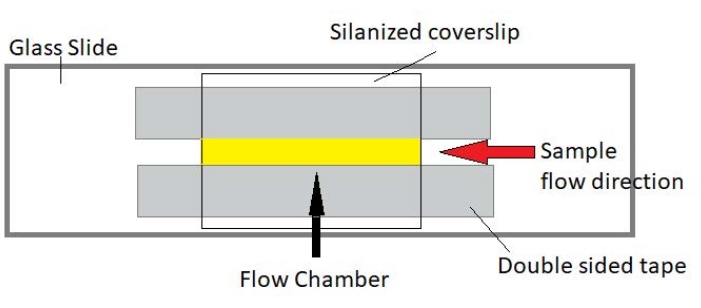
Figur 3: Samling af flowkammer. Flowkammeret er lavet ved hjælp af et glasglas, silaniseret dækglas og permanent dobbeltsidet tape. Det gule fremhævede område er den strømningssti, hvor prøven flyder og observeres. Volumenet af strømningskammeret er ~ 20 μL. Epoxy blev brugt til at forsegle enderne af kammeret for at forhindre prøven i at fordampe under langvarig billeddannelse over flere timer. Klik her for at se en større version af denne figur.
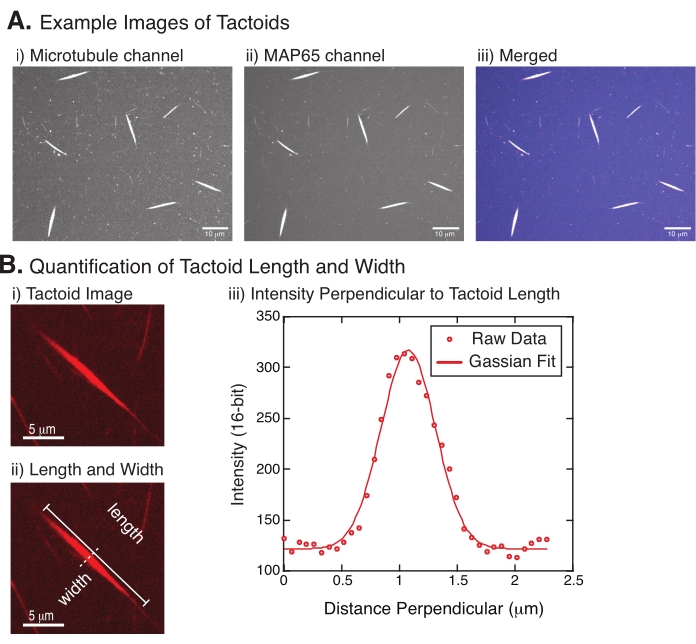
Figur 4: Tactoid billeder og længde og bredde analyse. (A) Eksempeldata for tactoider dannet som beskrevet og afbildet ved hjælp af roterende skivekonfokal, der viser (i) mikrotubuluskanalbilleddannelse rhodaminmærket tubulin ved hjælp af en 561 nm laser, (ii) GFP-MAP65-kanalbilleddannelse af GFP'en ved hjælp af en 488 nm laser og (iii) flettet overlaybillede fra mikrotubuluskanalen (magenta) og GFP-MAP65-kanalen (cyan). Overlappende områder vises som hvide og viser, at mikrotubuli og MAP65 koloniserer nøjagtigt. Skalalinje = 10 μm for alle billeder i (A). B) Kvantificering af taktoid længde og bredde. (i) Billede af et tactoid, der skal analyseres uden etiketter. Skalalinje = 5 μm. (ii) Samme billede som i (i), hvor længdemålingerne (solid linje med linjehætter) og bredde (stiplet linje) er angivet. Skalabjælke = 5 μm. (iii) Bredden blev målt ved at tage intensitetsprofilen over taktoidet ved den vinkelrette bisektor (stiplet linje) betegnet i (ii). Intensitetsprofilen var egnet til en gaussisk funktion for at afsløre amplituden og bredden af taktoidet. Klik her for at se en større version af denne figur.
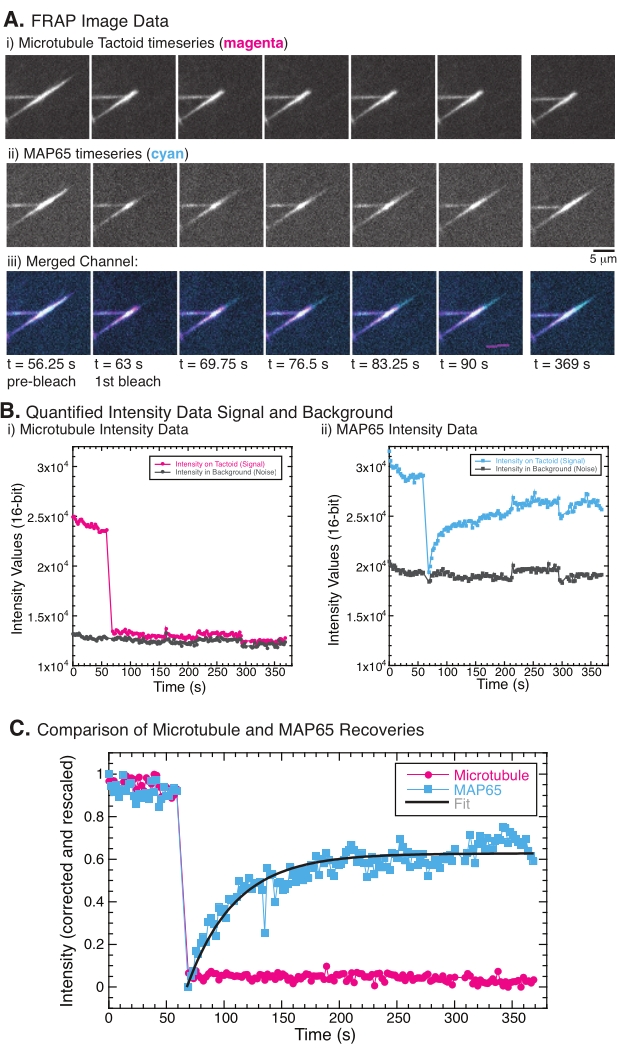
Figur 5: Repræsentative FRAP-data og analyser. (A) Mikroscopy-tidsseriedata for (i) mikrotubulus tactoid og (ii) GFP-MAP65 og (iii) overlejringsbillede af begge kanaler med mikrotubuli i magenta og GFP-MAP65 i cyan, der blev fotobleget på tidspunktet 63 s og observeret i yderligere 5 min. (B) Kvantificeret intensitet af (i ) mikrotubuluskanal i det blegede område (magentacirkler) og baggrunden (mørkegrå rande) og ii) GFP-MAP65-kanalen i det blegede område (cyankvadrationer) og baggrunden (mørkegrå firkanter). (C) Dataene blev korrigeret for baggrundsstøj og reskaleret for mikrotubuluskanalen (magentacirkler) og GFP-MAP65-kanalen (cyankvadratmeter). Mikrotubuli kommer sig ikke, men GFP-MAP65 gør og kan tilpasses (mørkegrå linje) til et stigende eksponentielt henfald for at finde amplituden og tidsskalaen for genopretning. Klik her for at se en større version af denne figur.
De metoder, der er beskrevet her, er blevet brugt i flere papirer til at skabe mikrotubulus tactoider (figur 2)22,28. Disse eksperimenter er biologisk relevante for at hjælpe med at afdække de organisatoriske principper, der styrer formen og stabiliteten af den mitotiske eller meiotiske spindel i de fleste celletyper. Derudover er mikrotubuli model flydende krystal mesogener, der kan hjælpe med at lære mere om, hvordan flydende krystaller nukleerer og vokser nematiske faser fra isotropiske faser.
Proceduren skitseret her har flere fordele ved at udforske mikrotubuli selvorganisering. For det første er det meget reproducerbart, idet det er blevet udført i laboratoriet af mange studerende, herunder gymnasieelever, med lidt forudviden eller træning, før de starter i laboratoriet. Tactoider er birefringent22, så de kan ses i transmitteret lys ud over fluorescensmikroskopi, hvilket gør denne metode tilgængelig for mange laboratorier og denne eksperimentelle procedure, der kan tilpasses uddannelsesmæssige formål ud over avanceret forskning. Endelig åbner denne proces muligheder for at fortsætte med at forstå og undersøge biologiske systemer i en fjernet, reduktionistisk tilgang, der gør det muligt for en at forstå, hvordan hver yderligere tilstand, protein eller additiv kan ændre selvorganiseringen af tactoider og måske i sidste ende spindlen. Mål for bedre biomimicry inkluderer aktivitet, fluiditet og filamentpolaritetssortering.
Der kan være flere faktorer, der påvirker eksperimentet, hvilket giver uventede resultater. Hvis tactoider f.eks. ikke dannes (figur 2), men der observeres viftelignende mønstre, er MAP65 sandsynligvis ikke til stede eller ikke bindende for mikrotubuli22,28. Dette burde også være indlysende i MAP65-fluorescenskanalen, fordi GFP-MAP65 ikke vil være bundet til mikrotubuli.
Hvis taktoiderne ikke dannes, og baggrunden vises som pletter på glasset, kan dette skyldes overfladebelægningen. Når den er udført, varer silaniseringen kun 1 måned på dækslips. Når det slides af, vil tubulinet være i stand til at binde sig til den udsatte overflade ikke-specifikt. Denne binding vil forekomme i ulige mønstre.
Hvis taktoiderne ikke dannes, og tubulinet observeres i aggregater af forskellige former og størrelser, kan dette skyldes tubulin af dårlig kvalitet. Tubulin kan centrifugeres for at fjerne indledende aggregater, der kan drive denne off-pathway aggregering i stedet for mikrotubuluspolymerisation. Hvis overfladen binder sig til tubulinet, kan det også nedbryde tubulinet i opløsningen. Lave koncentrationer af tubulin, under den kritiske koncentration for polymeriserende mikrotubuli, kan resultere i aggregater.
I FRAP-eksperimenter, hvis MAP65-kanalen ikke viser nogen genopretning (figur 5), er det muligt, at fotobleaching var fotodamaging mikrotubuli. Photodamage forårsager lokaliseret ødelæggelse af filamenterne. Dette kan kontrolleres ved undersøgelse i den transmitterede kanal. Mikrotubulus tactoider er synlige i den transmitterede kanal gennem et højt indeks for brydningsmismatch med det omgivende vand. Lysinduceret fotoskade vises som et brændemærke eller tab af kontrast i transmitteret lysbilleddannelse på det sted, hvor ROI udsættes for fotobleaching. Hvis dette sker, skal laser- eller lyseffekten reduceres for at hæmme proteinernes fotoskade.
Der var flere udfordringer i denne procedure og tilgang. Et problem er, at længdemålene i øjeblikket udføres manuelt ved at klikke på billedet. Denne metode, selvom den er ligetil, kan resultere i stor usikkerhed. Breddemålingen, der bruger tværsnittet og monteringen til en gaussisk, er en bedre metode til kvantificering af størrelsen. En lignende metode kunne anvendes i længden. Et andet problem er, at taktoiderne nogle gange kan bøje sig, fordi de er så lange og tynde. Dette gør det vanskeligere at kvantificere længden. Konturlængden kan kvantificeres ved hjælp af en segmenteret linje, men der tilføjes usikkerhed, hver gang et segment tilføjes.
Fra et videnskabeligt perspektiv har denne tilgang nogle andre udfordringer for dens anvendelse som model for flydende krystaller eller spindler. Den første udfordring har været den lange, tynde form af de taktoider, som mikrotubuli skaber (figur 3 og figur 4). Som nævnt i tidligere publikationer22 er mikrotubulus tactoider homogene taktoider, ikke bipolære. Det betyder, at mikrotubuli, der udgør formen, ikke omorienterer sig for at pege mod strukturens spidser. I stedet er alle mikrotubuli parallelle med den lange akse, og "polerne" er placeret i uendelighed. Dette er meget forskelligt fra de taktoider, der observeres for molekylære flydende krystaller eller endda for actin eller DNA, der også kan fungere som flydende krystal mesogener. I disse andre systemer er taktoiderne bipolære, og når de ses i krydsede polarisatorer, viser de de afslørende tegn på omorientering af stængerne.
En anden stor udfordring i dette system er, at mikrotubuli er ubevægelige inde i taktoidet. Dette fremgår klart af FRAP-eksperimenter og analyser, da genopretningen af mikrotubuli er meget lav. Deres faste karakter gør mikrotubulus tactoider mindre værdifulde som store flydende krystalanaloger. Den nematiske fase af en flydende krystal skal have både flydende (væske) og krystal (organiserede) egenskaber. Selvom formen virker rigtig for spindlen, gør immobiliteten systemet mindre spændende som en model mitotisk spindel. På den anden side giver dette problem mulighed for at undersøge, hvordan man kan ændre eksperimenterne for at skabe mere fluiditet i systemet.
Disse videnskabelige udfordringer giver spændende muligheder, der giver mulighed for ny viden om systemet. For at gøre mikrotubulus tactoider mere bipolære, kunne man bruge kortere mikrotubuli. Der er dog en ekstra udfordring, da mikrotubuli ikke har mange velkarakteriserede cappingproteiner til at kontrollere længden, som actin gør. Anvendelsen af kimdannelse og vækst kræver anvendelse af meget høje koncentrationer af tubulin og GMPCPP til fremstilling af korte mikrotubuli. Den høje tubulinkoncentration resulterer i et større antal filamenter i systemet, hvilket gør det vanskeligere at adskille tactoider fra hinanden. Tilføjelsen af nye mikrotubuluskapsler, såsom DARPin33, kan hjælpe med denne situation. Det andet problem med, at mikrotubuli er ubevægelige, kan afhjælpes ved tilsætning af motorproteiner, såsom kinesin-534, som er tetramerer af motorer, der anvendes i mitose. Alternativt kan kunstige dimerer af dimerer af dimerisk kinesin-1 anvendes15.
En anden måde at tilføje mere fluiditet på ville være at tillade mikrotubuli at udføre deres dynamiske ustabilitet, dyrkning og krympning af mikrotubuli. I øjeblikket er mikrotubuli, der er podet med stabile GMPCPP-filamenter og derefter gennemgår dynamisk ustabilitet, langt længere end ønsket for at danne en spindel eller taktoid, hvilket ville resultere i meget lange organisationer som fans eller bundter. Så tilføjelse af mikrotubulus dynamisk ustabilitet skal gøres omhyggeligt for at bevare den taktformede form. Tilsætningen af associerede proteiner og enzymer, der kan kontrollere længden, kan afhjælpe dette problem. For eksempel vil depolymeriserende kinesiner, såsom kinesin-1335, eller skære enzymer, som katanin36, sandsynligvis være nødvendige. Disse eksperimenter er komplekse og vanskelige, selvom de ville være meget indsigtsfulde, uanset hvad resultaterne afslører. Uanset hvilken retning fremtidige eksperimenter tager, kan platformen udviklet her til at skabe mikrotubulus tactoider udsætte ny information på det fysiske grundlag for mikrotubulusorganisation.
Forfatterne erklærer, at de ikke har nogen konkurrerende økonomiske interesser.
Forfatterne vil gerne takke alle Ross Lab-medlemmer i sommeren 2021, især K. Alice Lindsay, for deres hjælp. Dette arbejde blev støttet af et tilskud fra NSF BIO-2134215, der støttede S. Sahu, N. Goodbee, H.B. Lee og J.L. Ross. Et tilskud fra KECK Foundation (Rae Anderson, USD, lead PI) støttede delvist R. Branch og P. Chauhan
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved