
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
Auto-assemblage de tactoïdes de microtubules
Cet article présente un protocole pour la formation d’assemblages de microtubules en forme de tactoïdes en utilisant MAP65, un réticulant de microtubules à base de plantes, et PEG comme agent d’encombrement.
Le cytosquelette est responsable de l’organisation interne majeure et de la réorganisation au sein de la cellule, le tout sans gestionnaire pour diriger les changements. C’est particulièrement le cas lors de la mitose ou de la méiose, où les microtubules forment le fuseau lors de la division cellulaire. Le fuseau est la machinerie utilisée pour séparer le matériel génétique pendant la division cellulaire. Pour créer des fuseaux auto-organisés in vitro, nous avons récemment développé une technique pour reconstituer des microtubules en assemblages de type fuseau avec un ensemble minimal de protéines associées aux microtubules et d’agents d’encombrement. Plus précisément, MAP65 a été utilisé, qui est un réticulant de microtubules antiparallèles de plantes, un homologue d’Ase1 de levure et de PRC1 d’organismes de mammifères. Ce réticulant auto-organise les microtubules en assemblages auto-organisés de microtubules longs, minces et en forme de fuseau. Ces assemblages sont également similaires aux tactoïdes à cristaux liquides, et les microtubules pourraient être utilisés comme mésogènes à méso-échelle. Ici, des protocoles sont présentés pour créer ces tactoïdes de microtubules, ainsi que pour caractériser la forme des assemblages à l’aide de la microscopie à fluorescence et la mobilité des constituants en utilisant la récupération de fluorescence après photoblanchiment.
La division cellulaire par mitose est l’un des processus biologiques les plus importants pour maintenir la vie. Les filaments de microtubules, composés de dimères de tubuline, sont des éléments structurels essentiels de ce processus. La machinerie transitoire créée en métaphase lorsque les chromosomes s’alignent au centre cellulaire est appelée fuseau mitotique en raison de sa forme, qui est comme un fuseau d’un métier à tisser recouvert de fils (Figure 1A). Il est bien établi dans de nombreux organismes que les microtubules sont utilisés en métaphase pour pousser et tirer les chromosomes condensés au centre de la cellule, les aligner et les brancher à des microtubules qui les sépareront en anaphase (Figure 1B, C). Le fuseau se forme à la fois dans la méiose (Figure 1B) et la mitose (Figure 1C), créées à partir de nombreux microtubules qui se chevauchent et qui ne sont pas enroulés autour de l’axe central comme un filetage, mais parallèles à l’interface. La création de ces structures à base de microtubules nécessite des protéines associées qui se réticulent et des enzymes associées qui peuvent agir comme moteurs pour aider à pousser et à tirer les chromosomes1.
Des études sur les fuseaux méiotiques ont montré que les microtubules sont courts, dynamiques et se chevauchent dans des réseaux réticulés 2,3,4,5,6 (Figure 1Di). En raison de l’organisation physique de ces microtubules courts, le fuseau méiotique est similaire à un tactoïde à cristaux liquides (Figure 1E). En effet, il a été démontré que les fuseaux fusionnent et fusionnent, comme on pourrait s’y attendre des tactoïdes à cristaux liquides5.
De nombreuses études remontant aux années 1960 ont utilisé la fixation, les coupes en série et la microscopie électronique pour déterminer qu’il existe deux types de microtubules à l’intérieur du fuseau mitotique 7,8,9,10. Le premier type est appelé les microtubules de kinétochore, qui relient le pôle de broche au kinétochore. Le deuxième type est appelé microtubules interpolaires ou polaires, qui se développent au-delà des chromosomes et se chevauchent à la zone médiane (Figure 1Dii)8,9,10. Un troisième type est appelé les microtubules astraux, qui sont à l’extérieur de la broche et relient les pôles au bord de la cellule; ces organisations de microtubules sortent du cadre de la discussion actuelle. Il y a eu des études récentes sur l’interaction entre l’augmine6 et le complexe de cycles gamma-tubuline influençant les centres de nucléation des microtubules, ce qui a entraîné un fuseau mitotique avec des microtubules plus courts comme dans la figure 1D.
Étant donné que les microtubules sont plus longs que larges, avec un rapport d’aspect élevé et une rigidité élevée, ils sont comme des versions mises à l’échelle des molécules de cristaux liquides. En physique de la matière molle, les atomes et les molécules ont été approchés en utilisant des interactions minimales pour déduire les mécanismes physiques des transitions de phase, y compris la nucléation et la fusion des cristaux11. De même, les microtubules sont des objets à méso-échelle qui sont des versions à grande échelle de molécules de cristaux liquides, donnant un aperçu de la physique de la dynamique des cristaux liquides, y compris la nucléation et la croissance des phases nématiques à partir de phases isotropes. En outre, comme discuté ci-dessus, le fuseau méiotique présente des propriétés similaires à celles d’un tactoïde à cristaux liquides, un état nématique qui se nuclée et se développe à partir de l’état isotrope des molécules de cristaux liquides 3,4,5. Pour les tactoïdes, la nucléation et la croissance sont semblables à celles des autres cristaux (c’est-à-dire nécessitant une concentration relativement élevée de mésogènes [les molécules qui forment les cristaux liquides]). La forme unique du « fuseau » du tactoïde provient de l’alignement local des mésogènes à cristaux liquides qui s’alignent dans la phase nématique (Figure 1E). Ils ne peuvent pas former un cristal arrondi car les molécules sont très asymétriques. Compte tenu de la nature des microtubules, il n’est peut-être pas surprenant que la machinerie de broche mitotique fabriquée à partir d’une forte concentration locale de microtubules soit également de la même forme, qu’elle soit appelée tactoïde ou fuseau. Les tactoïdes peuvent être bipolaires, avec des pôles aux extrémités effilées (Figure 1Ei), ou homogènes, avec des pôles effectivement à l’infini (Figure 1Eii).
Compte tenu de l’importance de la formation du fuseau, des efforts ont été entrepris pour la formation auto-organisée du fuseau in vitro en démontrant la condensation des microtubules en faisceaux via les espèces ioniques 12,13, les agents d’encombrement qui créent des interactions d’épuisement 14,15 et les protéines de réticulation de microtubules spécifiques 13,16,17,18,19, 21. Étonnamment, bien que ces agents agissent tous pour augmenter la concentration locale de microtubules, ils se traduisent souvent par de longs faisceaux de microtubules mais pas de tactoïdes. L’une des raisons pour lesquelles ces faisceaux sont longs pourrait être que les microtubules qui les composent sont également longs. Des travaux récents utilisant des microtubules plus courts ont également rapporté des faisceaux plus longs qui ne sont pas effilés à la fin15; dans ce cas, les faisceaux sont maintenus ensemble avec des protéines motrices qui provoquent l’extension des faisceaux et, par conséquent, les rendent plus longs. Des microtubules courts avec des réticulants non extensiles sont nécessaires pour les assemblages coniques en forme de broche, comme décrit ici.
Récemment, nous avons développé une technique permettant la création de tactoïdes de microtubules à l’aide du réticulateur antiparallèle, MAP65, en présence de microtubules stables nucléants22. Les microtubules devaient être courts, mais peu de régulateurs connus de la longueur des microtubules peuvent coiffer les microtubules contre l’instabilité dynamique ou le recuit de bout en bout. Au lieu de cela, gmpcpp a été utilisé pour nucléer et stabiliser les filaments après la croissance. Cela a permis de créer une densité élevée de microtubules courts qui pouvaient s’auto-organiser en tactoïdes. Ces tactoïdes étaient homogènes lorsqu’ils étaient considérés sous biréfringence. En plus des microtubules courts, un réticulateur antiparallèle spécifique, MAP65, a été utilisé pour former les tactoïdes (Figure 2). MAP65 est une protéine végétale associée aux microtubules de la famille PRC1/Ase1 des réticulants mitotiques23. MAP65 existe sous forme de dimère, avec une forte affinité pour se lier à lui-même ainsi qu’aux microtubules24. Contrairement au fuseau méiotique et aux tactoïdes observés avec les filaments d’actine 25,26,27, qui sont bipolaires et ont les propriétés liquides des cristaux liquides, les tactoïdes de microtubules ont été observés comme étant solides 22,28.
Ici, des protocoles sont présentés pour créer les tactoïdes de microtubules et caractériser la forme des assemblages et la mobilité des constituants à l’aide de techniques basées sur la fluorescence.
REMARQUE: Sauf indication contraire, certaines parties de l’expérience peuvent être effectuées sur un banc de laboratoire tout en portant un équipement de protection approprié (gants).
1. Silanisation coverslip
REMARQUE: Les couvercles doivent être silanisés pour être utilisés avec le revêtement de brosse en polymère utilisé dans ces expériences. Il s’agit d’un traitement de silanisation hydrophobe qui permet à un copolymère séquencé avec un bloc central hydrophobe de se lier et de créer une brosse en polymère. Les étapes suivantes doivent être effectuées dans une hotte pour éviter l’exposition aux vapeurs toxiques tout en portant des gants. Le diméthyldichlorolsilane est très toxique et doit être manipulé avec le plus grand soin.
- Rincez les couvercles avec de la ddH2O, de l’éthanol à 70% et de la ddH2O dans l’ordre. Séchez-les avec des lingettes de laboratoire non pelucheuses entre chaque rinçage. Cela élimine la poussière et les particules solubles dans l’eau ou organiques de la surface avant le traitement.
- Placez les couvercles dans un rack métallique et transférez le rack à l’ozone UV (UVO). Irradier les couvercles avec de l’UVO pendant 20 minutes pour éliminer toute fluorescence de fond. Une chambre à plasma peut être utilisée à la place de l’UVO.
- À l’aide d’une pince à épiler, transférez les couvercles du rack métallique utilisé pour le traitement UVO vers un autre rack métallique utilisé pour la silanisation. N’utilisez pas les mêmes racks pour les deux, car cela provoquerait des niveaux élevés d’oxydation lors de l’application d’UVO. Pré-nettoyez les racks avec de l’eau et de l’éthanol afin qu’il ne reste aucun produit chimique résiduel provenant d’utilisations antérieures.
- Immergez le rack avec les couvercles dans un récipient contenant 100% d’acétone pendant 1 h. Rincez le récipient 3x avec de l’eau du robinet puis 3x avec ddH2O pour enlever toute l’acétone.
- Immergez le rack avec les couvercles dans de l’éthanol à 100% pendant 10 min. Rincez le récipient 3x avec de l’eau du robinet puis 3x avec du ddH2O pour éliminer tout l’éthanol.
- Immergez le rack avec les couvercles 3x dans ddH2O pendant 5 min chacun.
- Immerger le rack avec les couvercles dans 0,1 M KOH (50 mL de 1 M KOH dans 450 mL de ddH2O) pendant 15 min. Rincez le récipient 3x avec de l’eau du robinet puis 3x avec ddH2O pour enlever tout KOH.
- Immergez le rack avec des couvercles 3x dans ddH2O pendant 5 min chacun.
- Séchez à l’air libre le rack avec les couvercles pendant la nuit dans une hotte à fumée ou une hotte à flux laminaire.
- Après avoir complètement séché le rack et les couvercles, immergez-les pendant 5 min dans du diméthyldichlorosilane (DDS) à 2% pris dans un récipient différent utilisé spécifiquement pour le silane. Ne laissez rien qui n’est pas sec entrer en contact avec le silane.
- Immergez le rack et les couvercles 2x dans un récipient contenant 100% d’éthanol pendant 5 min. Rincez le récipient 3x avec de l’eau du robinet puis 3x avec ddH2O.
- Immergez le rack et les couvercles 3x dans ddH2O pendant 5 min chacun.
- Séchez à l’air libre le rack avec les couvercles pendant la nuit dans une hotte à fumée ou une hotte à flux laminaire.
- Après cette dernière étape de séchage, retransférez les couvercles dans des boîtes à couvercles à l’aide d’une pince à épiler. Ces couvercles peuvent être utilisés dans les 1-2 prochains mois. Les vieilles lames de couverture commenceront à perdre leur revêtement et devront être jetées.
2. Préparation de la tubuline
REMARQUE: La tubuline achetée se présente sous la forme d’une poudre lyophilisée non étiquetée ou étiquetée avec des fluorophores. La tubuline lyophilisée est conservée dans un congélateur à −80 °C. La procédure suivante est utilisée pour mélanger de la tubuline non étiquetée avec de la tubuline étiquetée dans un rapport bon pour la visualisation.
- Retirer du congélateur à −80 °C une aliquote de tubuline non étiquetée contenant 1 mg de poudre de tubuline lyophilisée et la conserver sur de la glace. Ajouter 200 μL de PEM-80 froid dans le tube pour porter la concentration de tubuline à 5 mg/mL. Conservez-le sur la glace pendant 10 min pour dissoudre tout le lyophilate.
- Retirer du congélateur à −80 °C une aliquote de tubuline marquée à la rhodamine contenant 20 μg de poudre de tubuline lyophilisée et la conserver sur de la glace. Ajouter 4 μL de PEM-80 froid au tube pour porter la concentration de tubuline à 5 mg/mL. Conservez-le sur la glace pendant 10 minutes pour dissoudre complètement le lyophilate.
- Une fois dissous, ajouter 100 μL de solution de tubuline remise en suspension non marquée à la solution de 4 μL de tubuline marquée à la rhodamine. Pipette 6x-7x très lentement pour mélanger. Si des agrégats sont visibles, centrifuger la tubuline solubilisée pendant 10 min à 90 000 x g pour éliminer les agrégats en jetant la pastille et en retenant le surnageant. Ce mélange de tubuline donnera ~ 4% de tubuline étiquetée.
- Congelez les 100 μL restants de tubuline non marquée dans de l’azote liquide (LN2) et conservez-les à −80 °C pour les utiliser pour d’autres mélanges de tubuline.
- Prenez le mélange de tubuline étiqueté et aliquote dans sept tubes de 15 μL chacun. Chaque aliquote peut être utilisée pour une seule chambre expérimentale. Lingotez les aliquotes restantes et stockez-les à −80 °C pour de futures expériences.
3. Purification MAP65
REMARQUE: MAP65 n’est pas disponible dans le commerce et, par conséquent, doit être purifié pour ce travail. Le protocole a déjà été élaboré dans plusieurs publications23,29.
- Transformez le plasmide MAP65 et le plasmide GFP-MAP65 en souche bactérienne BL21 pour l’expression des protéines.
- Cultiver les bactéries BL21 à une densité optique de 0,6-1 à 600 nm. Induire la production de protéines en utilisant l’opérateur de lac et développer les bactéries pendant la nuit.
- Abattez les cultures et lysez les bactéries.
- Recueillir le lysat après centrifugation et l’incuber avec des perles qui ont un ion nickel disponible pour lier l’étiquette 6x-histidine.
- Éluez la protéine à l’aide de l’imidazole et dessalez-la.
- Congelez la protéine avec de l’azote liquide et stockez-la à −80 °C pour une utilisation dans un délai de 1 an.
4. Assemblage des chambres d’écoulement
REMARQUE: Les expériences sont effectuées dans des chambres d’écoulement faites d’une lame de verre et d’un verre de couverture silanisé (Figure 3).
- Prenez une lame de verre et nettoyez-la en utilisant ddH2O, éthanol et ddH2O dans l’ordre. Sécher avec une lingette de laboratoire non pelucheuse entre chaque rinçage.
- Utilisez un morceau de ruban adhésif double face pour créer un chemin d’écoulement. À l’aide de mains gantées, coupez le ruban adhésif à ~25-30 mm de longueur. Divisez la bande dans le sens de la longueur pour créer deux bandes plus minces. Placez les deux bandes de ruban adhésif sur la glissière avec environ 5-8 mm entre elles.
REMARQUE: Comme l’épaisseur de la bande est normalisée à environ 80-100 μm, la largeur du chemin entre les morceaux de bande déterminera le volume dans la chambre. - Placez les couvercles silanisés au-dessus du chemin d’écoulement. Scellez la glissière et le couvercle sur les bandes de ruban adhésif double face en appuyant doucement sur la zone de ruban adhésif à l’arrière d’un stylo. Assurez-vous d’obtenir un bon sceau sur toute la zone; le ruban doit passer de translucide à clair lorsque le joint est bien fait.
- Retirez le ruban adhésif supplémentaire sur les bords, en ne laissant que 1 mm de l’entrée de la chambre d’écoulement en coupant le ruban avec une lame de rasoir.
- Étiquetez la chambre avec des informations sur les paramètres expérimentaux, au besoin.
5. Expériences tactoïdes
REMARQUE : Une fois que tous les réactifs et fournitures sont générés, ils peuvent être utilisés pour nucléer et polymériser les tactoïdes de microtubules dans la chambre d’écoulement.
- Collectez tous les réactifs à utiliser. Décongelez-les sur de la glace et stockez-les sur de la glace pendant que vous travaillez. Faites plusieurs chambres d’écoulement pour les expériences.
- Utilisez une chambre d’écoulement pour chaque expérience. Enduire les surfaces de la chambre d’écoulement avec une brosse en polymère en appliquant 20 μL de tensioactif copolymère à bloc non ionique à 5% (table des matériaux) dissous dans le PEM-80, avec de petites gouttes aux deux extrémités de la chambre pour empêcher la formation de bulles d’air à l’intérieur. Conservez-le dans une chambre humide (c.-à-d. boîte de Petri avec une lingette de laboratoire humide non pelucheuse) jusqu’à ce qu’il soit prêt à l’emploi (au moins 5 à 7 min).
- Dans un tube stérile, mélanger les éléments suivants pour créer le mélange tubuline-MAP : 9,5 μL de PEM-80 ; 4 μL de 10 mM GMPCPP; 4 μL de Pluronic-F127 à 5 %; 1 μL de 1M TNT; 1 μL de glucose; 2 μL de polyéthylène glycol (PEG); 12 μL de mélange de tubuline de 5 mg/mL (concentration finale de 13,6 μM) à partir de l’étape 2.5.; et 5,5 μL de stock de travail de MAP65 où 10 % est GFP-MAP65 pour la visualisation. Conserver sur la glace pendant le mélange.
REMARQUE: Il est recommandé d’utiliser une pipette à déplacement positif pour manipuler la solution de PEG visqueux. Des pipettes ordinaires peuvent être utilisées après avoir coupé la pointe pour agrandir l’ouverture; cependant, cette méthode est moins précise. - Mélanger 5x-6x par pipetage.
- Juste avant d’ajouter à la chambre, ajouter 1 μL d’une solution pré-mélangée de glucose oxydase (0,5 mg/mL) et de catalase (0,15 mg/mL) (Désoxy) dans le mélange tubuline-MAP et mélanger 7x-8x. Diviser le volume total de la solution (40 μL) en deux portions à utiliser dans des chambres séparées.
- Verser le mélange tubuline-MAP dans des chambres. Comme les chambres contiennent déjà un tensioactif copolymère non ionique, on ne peut pas ajouter plus de liquide sans éliminer l’ancien liquide. Pour ce faire, utilisez un morceau de papier filtre ou une lingette de laboratoire non pelucheuse à l’autre extrémité de la chambre pour éliminer le liquide par action capillaire.
- Une fois que l’échantillon est complètement à l’intérieur de la chambre, scellez les deux extrémités de la chambre à l’aide d’époxy de 5 min et maintenez-le à 37 °C pendant environ 30 min pour nucléer et développer des tactoïdes de microtubules.
6. Microscopie à fluorescence
- Utilisez un microscope à fluorescence pour l’imagerie des tactoïdes.
REMARQUE: La microscopie à fluorescence à réflexion interne totale ou la microscopie confocale à disque rotatif sont bonnes pour éliminer la fluorescence de fond de la tubuline libre, mais les tactoïdes sont également visibles par épifluorescence régulière et même par microscopie à lumière transmise une fois qu’ils sont formés, ce qui rend cette procédure accessible sans équipement spécialisé. - Utilisez un objectif à ouverture numérique (NA) de 1,2 NA ou plus avec un grossissement de 60x ou plus pour recueillir suffisamment de lumière en fluorescence. Ces objectifs nécessitent souvent une immersion dans de la ddH2O ou de l’huile.
- Enregistrez des images avec une caméra CMOS ou CCD. Utilisez une taille de pixel efficace sur l’appareil photo de 108 nm.
REMARQUE: La taille des pixels dépend de l’appareil photo et du grossissement utilisé, qui dans ce cas était de 60x ou 100x avec une ouverture numérique élevée (1,2 ou 1,49 NA). Des extenseurs d’image supplémentaires peuvent être utilisés avant l’appareil photo pour atteindre la taille de pixel nécessaire. - Conserver l’échantillon à 37 °C à l’aide d’une chambre environnementale réglée à cette température. Alternativement, utilisez d’autres réchauffeurs de scène, y compris des réchauffeurs de scène à air chaud et des colliers objectifs à température contrôlée avec de l’eau chaude en circulation.
- Utilisez des sources d’excitation qui sont correctes pour la fluorescence nécessaire. Pour la tubuline de rhodamine, utilisez un laser de 561 nm avec au moins 1 mW de puissance à l’échantillon, et pour GFP-MAP65, utilisez un laser de 488 nm avec au moins 1 mW de puissance à l’échantillon.
REMARQUE: Si vous utilisez la microscopie épifluoracte à champ large, utilisez un cube de filtre à rhodamine avec excitation: 540 ± 12,5 nm, dichroïque: 545 nm ± coupure de 12,5 nm et émission: 575 nm de long passage, et un cube de filtre GFP avec excitation: 480 ± 15 nm, dichroïque: 505 nm ± 15 nm de coupure et émission: 515 nm de long passe. - Prenez au moins 10 images de différentes zones pour imager plus de 100 tactoïdes. Prenez des images dans les canaux rouge et vert et enregistrez-les en tant qu’images tiff 16 bits pour analyse. Assurez-vous que la puissance d’éclairage et les temps d’exposition sont tels que l’échelle d’intensité de l’appareil photo n’est pas saturée.
7. Récupération de fluorescence après photoblanchiment (FRAP)
NOTE: Pour étudier la mobilité des constituants internes des tactoïdes, FRAP a été utilisé. FRAP fonctionne en photoblanchiment d’une partie sélectionnée de la tubuline marquée à la rhodamine et du tactoïde MAP65 marqué GFP, puis en observant la récupération de la fluorescence avec le temps dans cette région. Le taux de rétablissement dépend du renouvellement de l’espèce photoblanchi. Ce taux de renouvellement peut dépendre de la diffusion et des réactions de liaison. Pour la liaison MAP65 aux tactoïdes, les taux de change contraignants peuvent être estimés. FRAP est effectué à l’aide d’un système laser supplémentaire de 405 nm qui peut scanner le laser sous n’importe quelle forme. Il existe de nombreuses possibilités pour effectuer FRAP, y compris l’utilisation de la lampe transmise et de l’ouverture pour photographier une zone locale14.
- Sélectionnez un tactoïde isolé dans la chambre pour créer une région d’intérêt (ROI) couvrant des parties des tactoïdes et la solution environnante.
- Utilisez un microscope avec un laser supplémentaire de 405 nm pour que FRAP photoblanchisse simultanément la tubuline et le MAP65. Alternativement, une lampe lumineuse peut être utilisée à travers l’arrêt de champ de l’iris14. Ajustez empiriquement l’intensité spécifique des systèmes de photoblanchiment pour éviter d’endommager les protéines pendant le blanchiment.
- Enregistrez le tactoïde sous forme de film de série chronologique pendant 30 à 60 s avant de photoblanchiment pour obtenir des informations sur l’intensité avant l’eau de Javel. Enregistrez les canaux rouge et vert.
- Photoblanchissez le tactoïde en exposant le retour sur investissement avec les lasers ou la lampe aussi longtemps que nécessaire pour photoblanchir sans endommager le tactoïde. Déterminez l’intensité et le temps empiriquement.
- Continuez à enregistrer le film dans les deux canaux de couleur pendant 5 à 10 minutes après le photoblanchiment ou jusqu’à ce que la récupération semble avoir atteint l’équilibre.
- Inspectez visuellement le canal GFP-MAP65 pour la récupération.
8. Analyse des données
NOTE: Une analyse quantitative des images de tactoïdes a été effectuée pour en apprendre davantage sur les effets des changements environnementaux imposés par différents agents d’encombrement, les conditions ioniques et l’ajout d’autres facteurs.
- Caractérisation de la forme tactoïde
- Quantifier la longueur et la largeur des tactoïdes à partir des images rouges et vertes prises en microscopie confocale.
- Ouvrez les images à l’aide de FIJI/ImageJ.
- Si les données brutes sont prises en 16 bits, ajustez la luminosité et le contraste si nécessaire. Sélectionnez Image > Régler > luminosité et contraste pour ajuster l’image afin de pouvoir voir clairement le tactoïde. Ajustez la luminosité et le contraste sans appliquer le réglage afin de ne pas modifier accidentellement les données d’intensité.
- Une fois que les tactoïdes sont clairement visibles, sélectionnez les bons tactoïdes à mesurer (Figure 4Bi). Assurez-vous que les tactoïdes sont clairement visibles sans chevauchement avec d’autres tactoïdes ou agrégats et qu’ils ne sont pas incurvés ou pliés pour pouvoir utiliser des outils de mesure en ligne droite.
- Ensuite, vérifiez que la taille de pixel correcte est définie pour les images. Les images du microscope sont livrées avec des métadonnées sur la taille des pixels. Lorsque vous utilisez une autre caméra qui n’a pas de métadonnées ou de systèmes d’extension d’image externes pouvant modifier la taille de pixel effective attendue, ajustez manuellement la taille de pixel. Dans FIJI/ImageJ, accédez à analyser > Définir l’échelle pour définir la conversion de pixels correcte.
- À l’aide de l’outil Ligne droite de la barre d’outils de FIJI/ImageJ, cliquez sur une extrémité du tactoïde et faites glisser le curseur vers l’autre extrémité du tactoïde (Figure 4Bii). Une fois le retour sur investissement de la ligne sélectionné, sélectionnez Analyser > Mesure pour mesurer la longueur. Si la longueur n’est pas mesurée par défaut, veillez à définir la mesure pour qu’elle inclue la longueur dans la boîte de dialogue Analyser > Définir les mesures .
REMARQUE: En règle générale, lors de la mesure à l’aide de l’outil Ligne droite , il donnera la longueur et l’angle de la ligne dessinée. A titre d’exemple, la Figure 4Bii montre une ligne droite tracée sur le côté du tactoïde pour rendre ce dernier visible mais faire la mesure directement sur le tactoïde. - Après avoir effectué la mesure, utilisez l’outil Texte de la barre d’outils pour étiqueter le tactoïde. Créez une zone de texte, ajoutez une étiquette numérique et sélectionnez Modifier > Dessiner pour corriger l’étiquette dans l’image. Enregistrez l’image en tant que fichier ROI distinct.
REMARQUE: L’étiquetage et l’enregistrement de ce fichier permettent à l’enquêteur de savoir quelle mesure correspond à quel tactoïde à partir des données brutes. Assurez-vous de mesurer chaque tactoïde une fois. - Une fois les tactoïdes de l’image entière mesurés, enregistrez les données dans la fenêtre Résultats dans un fichier texte délimité par des virgules ou des tabulations (à l’aide de Fichier > Enregistrer sous) et ouvrez les données dans un tableur pour analyser les données en chiffres. Collectez toutes les données ensemble (données d’image brutes, image roi et fichier texte des résultats) dans un dossier avec une convention de dénomination appropriée pour que tout soit organisé.
REMARQUE: Bien que les mesures de longueur tactoïde soient effectuées à la main, étant donné que les largeurs tactoïdes sont étroites, il est préférable d’utiliser une méthode différente pour mesurer la largeur tactoïde (voir ci-dessous) pour réduire les erreurs de mesure. - À l’aide d’ImageJ/FIJI, tracez une zone de ligne à l’aide de l’outil Ligne droite . Tracez la ligne sous la forme d’un bisecteur perpendiculaire à l’axe long tactoïde (Figure 4Bii).
- Sélectionnez Analyser > profil de tracé pour créer le profil d’intensité du bisecteur linéaire (Figure 4Biii). Un tracé apparaîtra. Pour récupérer et enregistrer les données du tracé, sélectionnez le bouton Liste en bas à gauche ; cela génère la liste des fichiers texte des données d’intensité le long de la ligne tracée. Enregistrez le fichier texte en tant que fichier .csv ou .txt.
- Ouvrez le fichier texte dans un programme approprié tel que MatLab, Python (sciPy) ou d’autres programmes. Ajuster les données d’intensité avec une fonction gaussienne de la forme:
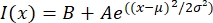 , où I(x) est la valeur en niveaux de gris le long de la longueur, x; B est le niveau de fond; A est l’amplitude du gaussien ; μ est la moyenne ou le centre du gaussien; et σ est l’écart-type du gaussien.
, où I(x) est la valeur en niveaux de gris le long de la longueur, x; B est le niveau de fond; A est l’amplitude du gaussien ; μ est la moyenne ou le centre du gaussien; et σ est l’écart-type du gaussien. - Signaler 2σ comme la largeur du tactoïde. Estimer l’intensité des microtubules dans le tactoïde en calculant la surface sous le gaussien (sans compter le fond).
REMARQUE: Si les images se situent dans la plage d’intensité linéaire de la caméra et sont prises avec le même temps d’exposition et la même intensité d’excitation, les intensités intégrées peuvent être comparées pour estimer le nombre relatif de microtubules dans le tactoïde.
- Analyse FRAP
REMARQUE: Des expériences visant à tester la mobilité des microtubules et le MAP65 ont utilisé FRAP pour enregistrer le photoblanchiment spécifique et la récupération de l’intensité due au mouvement moléculaire (Figure 5A). Les données ont été quantifiées à partir des données de la série chronologique d’images à l’aide d’ImageJ/FIJI.- Utilisez ImageJ/FIJI pour ouvrir les données du film.
- Enregistrez les piles (données de séries chronologiques) au fil du temps pour supprimer la dérive. Utilisez le plugin StackReg avec le plugin TurboReg auxiliaire; Pour obtenir des instructions sur l’utilisation des plugins, reportez-vous aux liens Web donnés dans le tableau des matériaux. Sélectionnez la traduction pour décaler la position des images et ainsi enregistrer les images.
- Une fois les images enregistrées pour supprimer la dérive, faites pivoter l’image de sorte que le tactoïde soit vertical ou horizontal dans le cadre en sélectionnant Image > Transformer > Faire pivoter. Sélectionnez l’angle de rotation et utilisez Aperçu pour déterminer si le tactoïde est suffisamment tourné. Lorsque l’aperçu indique que le tactoïde est vertical ou horizontal, sélectionnez OK pour faire pivoter toutes les images du film.
- Utilisez l’outil de sélection Rectangle de la barre d’outils pour créer une section rectangulaire sur la région photoblanchie du tactoïde. Enregistrez l’intensité intégrée de la zone de retour sur investissement pour chaque image à l’aide des piles d’images > > de mesures. Définissez le type de mesure sur Densité intégrée à l’aide de l’option Analyser > Définir les mesures. Enregistrez les données d’intensité analysées affichées dans la fenêtre Résultats sous forme de fichier texte au format .csv ou .txt en sélectionnant Fichier > Enregistrer sous.
REMARQUE : La figure 5B montre un exemple des données brutes d’intensité 16 bits mesurées pour les canaux microtubule et GFP-MAP65 dans la région de l’eau de Javel. - Comme l’intensité globale des images s’estompera globalement avec le temps en raison du photoblanchiment causé par l’imagerie, ce photoblanchiment global doit être corrigé. Pour ce faire, utilisez la même taille de retour sur investissement (étape 8.2.4.) et déplacez-la vers une région en arrière-plan de l’image où aucun microtubule ou MAP65 n’est visible. Mesurez l’intensité intégrée de la pile comme décrit à l’étape 8.2.4. Enregistrez les résultats dans un deuxième fichier texte.
REMARQUE : La figure 5B montre un exemple des données brutes d’intensité 16 bits mesurées pour les canaux microtubule et GFP-MAP65 dans la région d’arrière-plan. - Pour corriger la décoloration de l’arrière-plan, divisez l’intensité du signal sur le tactoïde par l’intensité de l’arrière-plan pour le même point de temps. Calculer Icorrigé (t) comme:
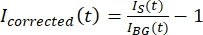 , où IS (t) (le signal) est la mesure effectuée sur la région blanchie et IBG (t) (le bruit) est la mesure effectuée sur la région de fond (Figure 5C). Cela calcule le rapport signal/bruit pour chaque image et soustrait également le bruit.
, où IS (t) (le signal) est la mesure effectuée sur la région blanchie et IBG (t) (le bruit) est la mesure effectuée sur la région de fond (Figure 5C). Cela calcule le rapport signal/bruit pour chaque image et soustrait également le bruit. - Ensuite, redimensionnez les données pour qu’elles se situent entre zéro et un en utilisant
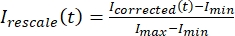 , où Imin et Imax désignent, respectivement, le minimum global et le maximum des données corrigées I sur toute la durée (Figure 5C).
, où Imin et Imax désignent, respectivement, le minimum global et le maximum des données corrigées I sur toute la durée (Figure 5C). - Ajuster ces données à une exponentielle en décomposition de la forme :
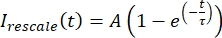 , où A est l’amplitude de la récupération et τ est l’échelle de temps de la récupération (Figure 5C).
, où A est l’amplitude de la récupération et τ est l’échelle de temps de la récupération (Figure 5C).
Avec seulement un petit nombre de composants, de dimères de tubuline et de réticulants de microtubules, des tactoïdes de microtubules peuvent se former (Figure 2A). Bien que ce protocole décrive l’incubation pour nucléer et développer des microtubules dans un incubateur, la nucléation et la croissance peuvent être observées directement au microscope (qui sont complètes en 30 minutes) (Figure 2B). La concentration de tubuline est maintenue constante à 13,6 μm et la liaison MAP65-MT à 10%.
La figure 4 représente les données réussies. Les tactoïdes doivent être visibles à la fois avec un laser de 561 nm dans le canal de la tubuline et de 488 nm dans le canal MAP65, qui se chevauchent parfaitement (Figure 4A). Un mystère du système a été que la largeur des tactoïdes ne semble pas varier sous une variété de changements expérimentaux, y compris la modification des longueurs de microtubules, de la concentration de MAP65 et des agents d’encombrement (Figure 4B)22,28. La longueur est beaucoup plus variable et dépend à la fois de la longueur des microtubules et de la concentration de MAP65 (Figure 4B)22,28.
Lors de l’exécution de FRAP, il a été observé que le signal MAP65 se rétablit, mais que le signal du microtubule ne se rétablit pas (Figure 5). La récupération dans FRAP est due à la mobilité et au mouvement des objets étiquetés et photoblanchis. Dans le cas du MAP65, les molécules assombries se dissocient et s’éloignent du microtubule et de nouvelles se déplacent dans la région (Figure 5). La liaison MAP65 est en équilibre, de sorte que le taux de liaison et de déliaison est égal (mesuré en molécules par seconde). Pour les microtubules, aucune récupération n’a été observée, ce qui implique que les microtubules ne sont pas capables de quitter le tactoïde (Figure 5A, Bi, C). De plus, aucune propagation de la région sombre n’a été observée, ce qui suggère que les microtubules sont localement immobiles et non un fluide dans la forme tactoïde.
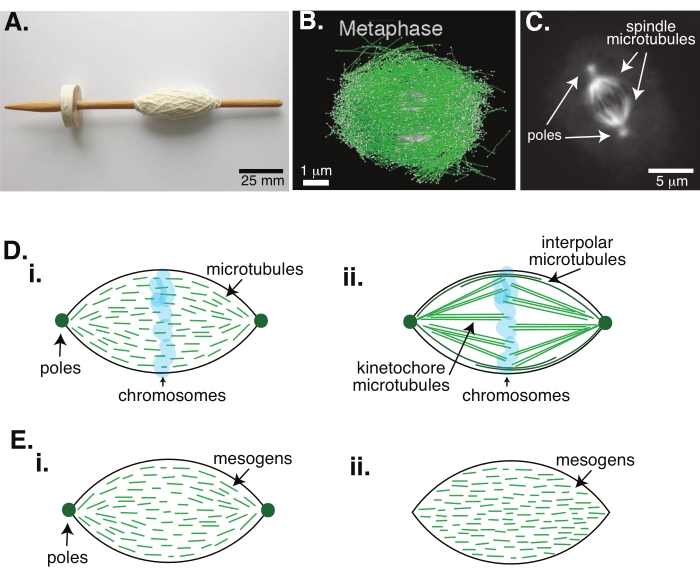
Figure 1 : Différents modèles de formation de fuseaux. Un fuseau mitotique est une machine faite de microtubules et de leurs protéines et enzymes associées qui aligne et sépare les chromosomes en deux nouvelles cellules filles pendant la division cellulaire. (A) Image d’une réplique de fuseau de chute du début du Moyen Âge avec du fil fin des Pays-Bas. Cette figure a été modifiée à partir d’une image Wikimedia par Peter van der Sluijs30. (B) Reconstruction tridimensionnelle de microtubules à différents stades de la méiose II de type sauvage. Les microtubules sont représentés en vert et les chromosomes en gris. Barre d’échelle = 1 μm. Ce chiffre a été modifié à partir de Lantzsch et al.31. (C) Image microscopique des microtubules dans un fuseau mitotique d’une cellule Sf9 en division. Les pôles du fuseau et les microtubules du fuseau sont marqués avec une protéine fluorescente verte. Barre d’échelle = 5 μm. Ce chiffre a été modifié à partir d’Advani et al.32. (D) Différents modèles d’organisation des microtubules fusiformes mitotiques et méiotiques. (i) Précédemment observés pour les fuseaux méiotiques créés à partir d’extraits d’œufs de Xenopus, les microtubules (verts) ont été déduits comme étant courts et dynamiques tout au long du fuseau. Ceci est similaire à une organisation tactoïde bipolaire à l’intérieur d’un cristal liquide. (ii) Le modèle canonique pour l’organisation des microtubules à l’intérieur d’un fuseau mitotique comporte deux types de microtubules : les microtubules interpolaires ou polaires (vert foncé) qui se réticulent à la zone médiane autour des chromosomes et les microtubules kinétochores (vert clair) qui sont regroupés et étirés du pôle au kinétochore pour pousser et tirer les chromosomes. Dans toutes les images, les chromosomes sont représentés en bleu transparent et les pôles du fuseau sont représentés en vert foncé. (E) Schémas de mésogènes (lignes vertes) dans un tactoïde à cristaux liquides pour (i) les tactoïdes bipolaires et (ii) les tactoïdes homogènes. Les tactoïdes bipolaires ont deux pôles à l’extrémité du tactoïde, et les mésogènes se réorientent pour pointer vers ces pôles. Les tactoïdes homogènes ont des pôles à l’infini, et les mésogènes ne changent pas d’orientation le long de la longueur du tactoïde. Veuillez cliquer ici pour voir une version agrandie de cette figure.
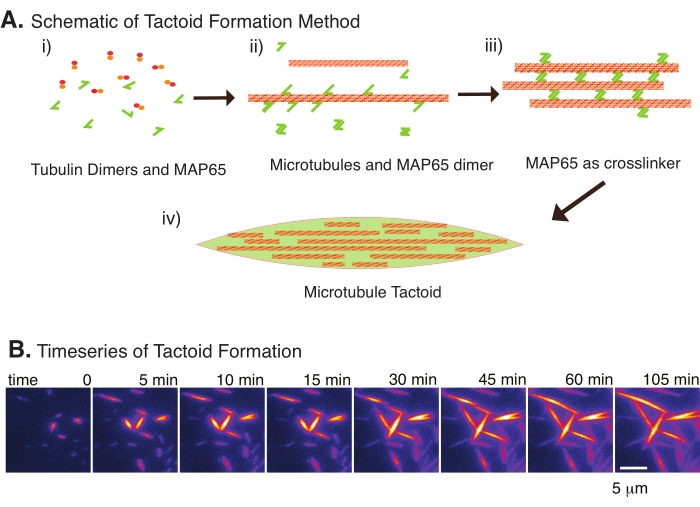
Figure 2 : Condensation des microtubules. (A) Les microtubules peuvent être regroupés et réticulés par diverses méthodes, y compris les espèces ioniques, les forces d’épuisement causées par les agents d’encombrement et les réticulants de microtubules spécifiques, tels que MAP65. i) Les dimères de tubuline et les protéines MAP65 sont mélangés pour nucléer et développer des microtubules. (ii) Les microtubules se nucléent et se développent à partir de la tubuline, et MAP65 se lie immédiatement aux microtubules, à un autre monomère MAP65, ou aux deux, et provoque un regroupement. iii) Les microtubules dans les faisceaux réticulés se nucléent et se développent. (iv) La configuration finale est un tactoïde de microtubule semblable à une broche. (B) Série chronologique de tactoïdes de microtubules se nucléant et se développant pendant 105 min. Barre d’échelle = 5 μm. Figure adaptée d’Edozie et al.22. Veuillez cliquer ici pour voir une version agrandie de cette figure.
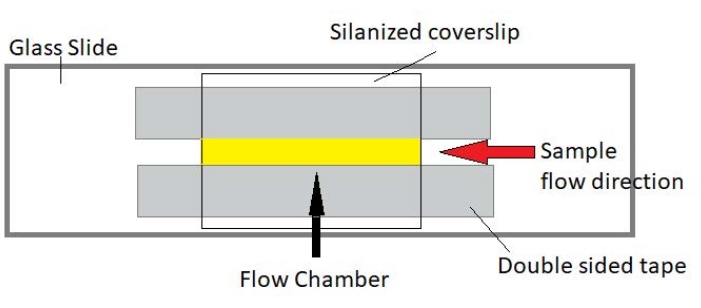
Figure 3 : Assemblage de la chambre d’écoulement. La chambre d’écoulement est fabriquée à l’aide d’une glissière en verre, d’un verre de couverture silanisé et d’un ruban adhésif double face permanent. La région surlignée en jaune est la voie d’écoulement où l’échantillon est acheminé et observé. Le volume de la chambre d’écoulement est d’environ 20 μL. L’époxy a été utilisé pour sceller les extrémités de la chambre afin d’empêcher l’échantillon de s’évaporer pendant l’imagerie à long terme pendant plusieurs heures. Veuillez cliquer ici pour voir une version agrandie de cette figure.
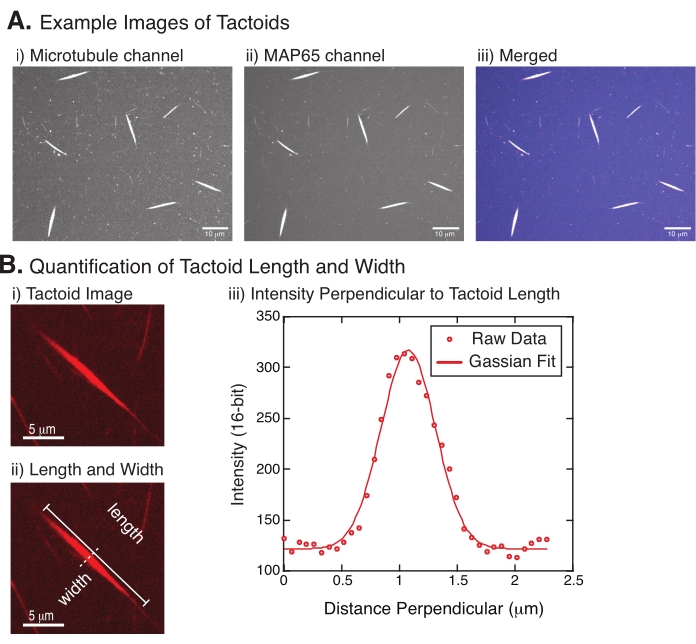
Figure 4 : Images tactoïdes et analyse de la longueur et de la largeur. (A) Exemples de données de tactoïdes formés comme décrit et imagé à l’aide d’un disque confocal en rotation montrant (i) l’imagerie du canal microtubule de la tubuline marquée à la rhodamine à l’aide d’un laser de 561 nm, (ii) l’imagerie du canal GFP-MAP65 à l’aide d’un laser de 488 nm, et (iii) l’image de superposition fusionnée du canal microtubule (magenta) et du canal GFP-MAP65 (cyan). Les régions de chevauchement sont affichées en blanc et démontrent que les microtubules et MAP65 colocalisent exactement. Barre d’échelle = 10 μm pour toutes les images en (A). (B) Quantification de la longueur et de la largeur des tactoïdes. (i) Image d’un tactoïde à analyser sans étiquettes. Barre d’échelle = 5 μm. (ii) Même image que dans (i), où les mesures de longueur (ligne continue avec capuchons de ligne) et de largeur (ligne pointillée) sont indiquées. Barre d’échelle = 5 μm. (iii) La largeur a été mesurée en prenant le profil d’intensité à travers le tactoïde au bisecteur perpendiculaire (ligne pointillée) noté en (ii). Le profil d’intensité était adapté à une fonction gaussienne pour révéler l’amplitude et la largeur du tactoïde. Veuillez cliquer ici pour voir une version agrandie de cette figure.
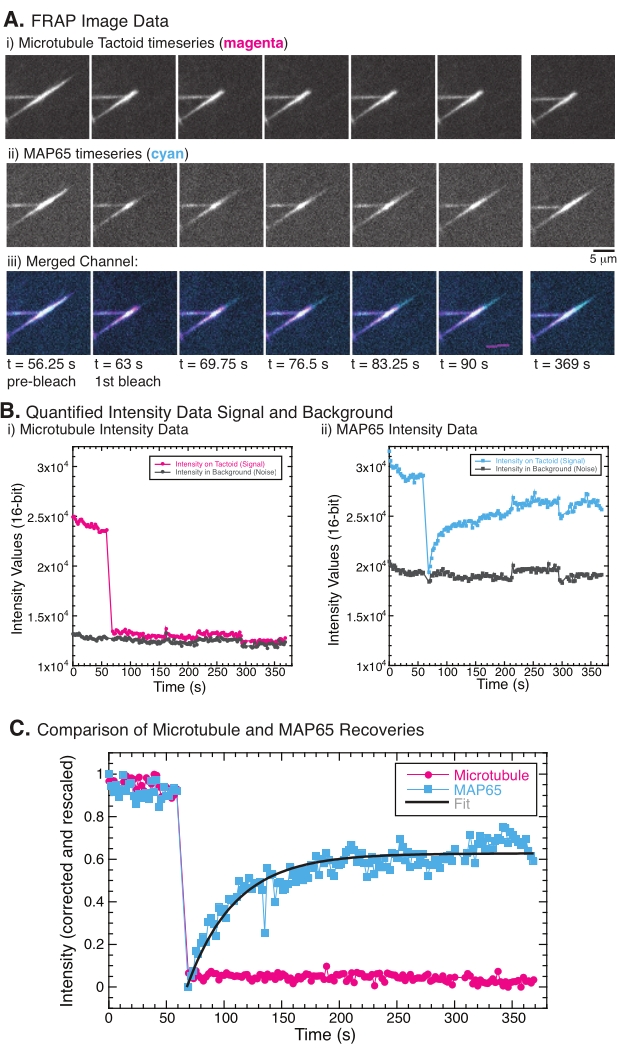
Figure 5: Données et analyses FRAP représentatives. (A) Données de série chronologique de microscopie de (i) tactoïde microtubule et (ii) GFP-MAP65, et (iii) image superposée des deux canaux avec des microtubules en magenta et GFP-MAP65 en cyan qui ont été photoblanchis au temps 63 s et observés pendant 5 minutes supplémentaires. (B) Intensité quantifiée de la (i ) canal microtubule dans la région blanchie (cercles magenta) et arrière-plan (cercles gris foncé) et ii) canal GFP-MAP65 dans la région blanchie (carrés cyan) et arrière-plan (carrés gris foncé). (C) Les données ont été corrigées pour tenir compte du bruit de fond et redimensionnées pour le canal microtubule (cercles magenta) et le canal GFP-MAP65 (carrés cyan). Les microtubules ne se rétablissent pas, mais le GFP-MAP65 le fait et peut être adapté (ligne gris foncé) à une désintégration exponentielle croissante pour trouver l’amplitude et l’échelle de temps de récupération. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Les méthodes décrites ici ont été utilisées dans plusieurs articles pour créer des tactoïdes de microtubules (Figure 2)22,28. Ces expériences sont biologiquement pertinentes pour aider à découvrir les principes organisationnels qui contrôlent la forme et la stabilité du fuseau mitotique ou méiotique dans la plupart des types de cellules. En outre, les microtubules sont des mésogènes à cristaux liquides modèles qui peuvent aider à en apprendre davantage sur la façon dont les cristaux liquides se nucléent et développent des phases nématiques à partir de phases isotropes.
La procédure décrite ici présente plusieurs avantages pour explorer l’auto-organisation des microtubules. Tout d’abord, il est hautement reproductible, ayant été effectué en laboratoire par de nombreux étudiants, y compris des lycéens, avec peu de connaissances préalables ou de formation avant de commencer dans le laboratoire. Les tactoïdes sont biréfringents22, ce qui leur permet d’être vus en lumière transmise en plus de la microscopie à fluorescence, rendant cette méthode accessible à de nombreux laboratoires et cette procédure expérimentale adaptable à des fins éducatives, en plus de la recherche haut de gamme. Enfin, ce processus ouvre des voies pour continuer à comprendre et à sonder les systèmes biologiques dans une approche réductionniste dépouillée, permettant de comprendre comment chaque condition, protéine ou additif supplémentaire peut altérer l’auto-organisation des tactoïdes et, peut-être, en fin de compte, le fuseau. Les cibles pour un meilleur biomimétisme comprennent l’activité, la fluidité et le tri de la polarité des filaments.
Plusieurs facteurs peuvent affecter l’expérience donnant des résultats inattendus. Par exemple, si les tactoïdes ne se forment pas (Figure 2) mais que des motifs en forme d’éventail sont observés, le MAP65 n’est probablement pas présent ou ne se lie pas aux microtubules22,28. Cela devrait également être évident dans le canal de fluorescence MAP65, car le GFP-MAP65 ne sera pas lié aux microtubules.
Si les tactoïdes ne se forment pas et que l’arrière-plan apparaît comme des taches sur le verre, cela pourrait être dû au revêtement de surface. Une fois effectuée, la silanisation ne dure que 1 mois sur les coverslips. Lorsqu’elle s’estompe, la tubuline pourra se lier à la surface exposée de manière non spécifique. Cette liaison se produira selon des modèles étranges.
Si les tactoïdes ne se forment pas et que la tubuline est observée dans des agrégats de différentes formes et tailles, cela pourrait être dû à une tubuline de mauvaise qualité. La tubuline peut être centrifugée pour éliminer les agrégats initiaux qui peuvent entraîner cette agrégation hors voie au lieu de la polymérisation des microtubules. Si la surface se lie à la tubuline, elle peut également épuiser la tubuline dans la solution. De faibles concentrations de tubuline, inférieures à la concentration critique pour la polymérisation des microtubules, peuvent entraîner des agrégats.
Dans les expériences FRAP, si le canal MAP65 ne montre aucune récupération (Figure 5), il est possible que le photoblanchiment ait été photodamant les microtubules. Les photodommages provoquent la destruction localisée des filaments. Cela peut être vérifié par un examen dans le canal transmis. Les tactoïdes de microtubules sont visibles dans le canal transmis à travers un indice élevé de décalage de réfraction avec l’eau environnante. Les photodommages induits par la lumière apparaîtront comme une marque de brûlure ou une perte de contraste dans l’imagerie de la lumière transmise à l’emplacement du retour sur investissement soumis au photoblanchiment. Si cela se produit, la puissance du laser ou de la lumière doit être réduite pour inhiber les photodommages des protéines.
Plusieurs défis ont été rencontrés dans cette procédure et cette approche. Un problème est que les mesures de longueur sont actuellement effectuées à la main en cliquant sur l’image. Cette méthode, bien que simple, peut entraîner une grande incertitude. La mesure de la largeur qui utilise la section transversale et l’ajustement à un gaussien est une meilleure méthode pour quantifier la taille. Une méthode similaire pourrait être utilisée pour la longueur. Un deuxième problème est que, parfois, les tactoïdes, parce qu’ils sont si longs et minces, peuvent se plier. Cela rend la quantification de la longueur plus difficile. La longueur du contour peut être quantifiée à l’aide d’une ligne segmentée, mais il y a une incertitude supplémentaire chaque fois qu’un segment est ajouté.
D’un point de vue scientifique, cette approche présente d’autres défis pour son utilisation comme modèle pour les cristaux liquides ou les fuseaux. Le premier défi a été la forme longue et mince des tactoïdes que les microtubules créent (Figure 3 et Figure 4). Comme indiqué dans des publications antérieures22, les tactoïdes microtubules sont des tactoïdes homogènes, et non bipolaires. Cela signifie que les microtubules qui composent la forme ne se réorientent pas pour pointer vers les extrémités de la structure. Au lieu de cela, tous les microtubules sont parallèles à l’axe long et les « pôles » sont situés à l’infini. Ceci est très différent des tactoïdes observés pour les cristaux liquides moléculaires ou même pour l’actine ou l’ADN qui peuvent également agir comme mésogènes à cristaux liquides. Dans ces autres systèmes, les tactoïdes sont bipolaires et, lorsqu’ils sont vus dans des polariseurs croisés, ils montrent les signes révélateurs de réorientation des tiges.
Un deuxième défi majeur dans ce système est que les microtubules sont immobiles à l’intérieur du tactoïde. Cela ressort clairement des expériences et des analyses FRAP, car la récupération des microtubules est très faible. Leur nature solide rend les tactoïdes de microtubules moins précieux que les analogues à cristaux liquides à grande échelle. La phase nématique d’un cristal liquide doit avoir à la fois des propriétés liquides (fluides) et cristallines (organisées). Bien que la forme semble appropriée pour la broche, l’immobilité rend le système moins excitant qu’un modèle de broche mitotique. D’autre part, ce numéro offre des occasions d’étudier comment on peut modifier les expériences pour créer plus de fluidité dans le système.
Ces défis scientifiques offrent des opportunités passionnantes qui permettront de nouvelles connaissances sur le système. Pour rendre les tactoïdes de microtubules plus bipolaires, on pourrait utiliser des microtubules plus courts. Il y a un défi supplémentaire, cependant, puisque les microtubules n’ont pas beaucoup de protéines de capsulage bien caractérisées pour contrôler la longueur comme le fait l’actine. L’utilisation de la nucléation et de la croissance nécessite l’utilisation de très fortes concentrations de tubuline et de GMPCPP pour fabriquer des microtubules courts. La concentration élevée de tubuline entraîne un plus grand nombre de filaments dans le système, ce qui rend plus difficile la séparation des tactoïdes les uns des autres. L’ajout de nouveaux cappers de microtubules, tels que DARPin33, peut aider à résoudre cette situation. Le deuxième problème de l’immobilité des microtubules pourrait être atténué par l’ajout de protéines motrices, telles que la kinésine-534, qui sont des tétramères de moteurs utilisés dans la mitose. Alternativement, des dimères artificiels de kinésine dimérique-1 pourraient être utilisés15.
Une autre façon d’ajouter plus de fluidité serait de permettre aux microtubules d’effectuer leur instabilité dynamique, la croissance et le rétrécissement des microtubules. Actuellement, les microtubules qui sont ensemencés avec des filaments GMPCPP stables et qui subissent ensuite une instabilité dynamique sont beaucoup plus longs que souhaité pour former un fuseau ou un tactoïde, ce qui entraînerait de très longues organisations comme des ventilateurs ou des faisceaux. Ainsi, l’ajout d’instabilité dynamique des microtubules devrait être fait avec soin pour préserver la forme tactoïde. L’ajout de protéines et d’enzymes associées qui peuvent contrôler la longueur peut atténuer ce problème. Par exemple, des kinésines dépolymérisantes, telles que la kinésine 1335, ou des enzymes de séparation, comme la katanin36, seraient probablement nécessaires. Ces expériences sont complexes et difficiles, bien qu’elles soient très perspicaces, peu importe ce que les résultats révèlent. Quelle que soit la direction que prendront les expériences futures, la plate-forme développée ici pour créer des tactoïdes de microtubules peut exposer de nouvelles informations sur la base physique de l’organisation des microtubules.
Les auteurs déclarent qu’ils n’ont pas d’intérêts financiers concurrents.
Les auteurs tiennent à remercier tous les membres du Ross Lab de l’été 2021, en particulier K. Alice Lindsay, pour leur aide. Ce travail a été soutenu par une subvention de NSF BIO-2134215 qui a soutenu S. Sahu, N. Goodbee, H.B. Lee et J.L. Ross. Une subvention de la Fondation KECK (Rae Anderson, USD, chercheur principal) a partiellement soutenu R. Branch et P. Chauhan
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved