
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
Selbstorganisation von Mikrotubuli-Taktoiden
Dieser Artikel stellt ein Protokoll für die Bildung von Mikrotubuli-Baugruppen in Form von Tactoiden unter Verwendung von MAP65, einem pflanzlichen Mikrotubuli-Vernetzer, und PEG als Crowding-Agent vor.
Das Zytoskelett ist für die wichtige interne Organisation und Reorganisation innerhalb der Zelle verantwortlich, alles ohne einen Manager, der die Veränderungen leitet. Dies ist insbesondere bei der Mitose oder Meiose der Fall, bei der die Mikrotubuli während der Zellteilung die Spindel bilden. Die Spindel ist die Maschinerie, die verwendet wird, um genetisches Material während der Zellteilung zu trennen. Um selbstorganisierte Spindeln in vitro zu erzeugen, haben wir kürzlich eine Technik entwickelt, um Mikrotubuli in spindelartige Anordnungen mit einem minimalen Satz von Mikrotubuli-assoziierten Proteinen und Crowding-Agenten zu rekonstituieren. Insbesondere wurde MAP65 verwendet, ein antiparalleler Mikrotubuli-Vernetzer aus Pflanzen, ein Homolog von Ase1 aus Hefe und PRC1 aus Säugetierorganismen. Dieser Vernetzer organisiert Mikrotubuli selbstorganisierend zu langen, dünnen, spindelartigen Mikrotubuli-selbstorganisierten Baugruppen. Diese Baugruppen ähneln auch Flüssigkristall-Tactoiden, und Mikrotubuli könnten als mesoskalige Mesogene verwendet werden. Hier werden Protokolle zur Herstellung dieser Mikrotubuli-Tactoide sowie zur Charakterisierung der Form der Baugruppen mittels Fluoreszenzmikroskopie und der Beweglichkeit der Bestandteile mittels Fluoreszenzrückgewinnung nach dem Photobleichen vorgestellt.
Die Zellteilung durch Mitose ist einer der wichtigsten biologischen Prozesse, um das Leben zu erhalten. Die Mikrotubuli-Filamente, die aus Tubulin-Dimeren bestehen, sind wesentliche Strukturelemente dieses Prozesses. Die transiente Maschinerie, die in der Metaphase entsteht, wenn sich die Chromosomen im Zellzentrum ausrichten, wird aufgrund ihrer Form, die wie eine Spindel eines mit Fäden bedeckten Webstuhls ist, als mitotische Spindel bezeichnet (Abbildung 1A). Es ist in vielen Organismen gut etabliert, dass Mikrotubuli in der Metaphase verwendet werden, um kondensierte Chromosomen in die Mitte der Zelle zu schieben und zu ziehen, sie auszurichten und an Mikrotubuli anzuschließen, die sie in Anaphase auseinanderziehen (Abbildung 1B, C). Die Spindel bildet sich sowohl in der Meiose (Abbildung 1B) als auch in der Mitose (Abbildung 1C) und entsteht aus vielen überlappenden Mikrotubuli, die nicht wie ein Gewinde um die zentrale Achse gewickelt sind, sondern parallel zur Grenzfläche verlaufen. Die Schaffung dieser mikrotubulibasierten Strukturen erfordert assoziierte Proteine, die vernetzen, und assoziierte Enzyme, die als Motoren fungieren können, um die Chromosomen zu drücken und zu ziehen1.
Studien an meiotischen Spindeln haben gezeigt, dass die Mikrotubuli in vernetzten Arrays 2,3,4,5,6 kurz, dynamisch und überlappend sind (Abbildung 1Di). Aufgrund der physikalischen Organisation dieser kurzen Mikrotubuli ähnelt die meiotische Spindel einem Flüssigkristall-Tactoid (Abbildung 1E). Tatsächlich wurde gezeigt, dass Spindeln verschmelzen und verschmelzen, wie man es von Flüssigkristall-Tactoidenerwarten würde 5.
Viele Studien aus den 1960er Jahren haben Fixierung, serielle Schnitte und Elektronenmikroskopie verwendet, um festzustellen, dass es zwei Arten von Mikrotubuli in der mitotischen Spindel 7,8,9,10 gibt. Der erste Typ wird als Kinetochor-Mikrotubuli bezeichnet, die den Spindelpol mit dem Kinetochor verbinden. Der zweite Typ wird als interpolare oder polare Mikrotubuli bezeichnet, die über die Chromosomen hinauswachsen und sich in der Mitte überlappen (Abbildung 1Dii) 8,9,10. Ein dritter Typ werden als astrale Mikrotubuli bezeichnet, die sich außerhalb der Spindel befinden und die Pole mit dem Zellrand verbinden; Diese Mikrotubuli-Organisationen liegen außerhalb des Rahmens der aktuellen Diskussion. Es gibt neuere Studien über die Wechselwirkung zwischen Augmin6 und Gamma-Tubulin-Ringkomplex, der Keimbildungszentren für Mikrotubuli beeinflusst, was zu einer mitotischen Spindel mit kürzeren Mikrotubuli wie in Abbildung 1D führt.
Da Mikrotubuli länger als breit sind, mit einem hohen Aspektverhältnis und einer hohen Steifigkeit, sind sie wie vergrößerte Versionen von Flüssigkristallmolekülen. In der Physik der weichen Materie wurden Atome und Moleküle unter Verwendung minimaler Wechselwirkungen angenähert, um die physikalischen Mechanismen von Phasenübergängen, einschließlich Keimbildung und Schmelzen von Kristallen11, abzuleiten. In ähnlicher Weise sind Mikrotubuli mesoskalige Objekte, die vergrößerte Versionen von Flüssigkristallmolekülen sind und Einblicke in die Physik der Flüssigkristalldynamik geben, einschließlich der Keimbildung und des Wachstums der nematischen Phasen aus isotropen. Darüber hinaus weist die meiotische Spindel, wie oben diskutiert, Eigenschaften wie die eines Flüssigkristall-Tactoids auf, eines nematischen Zustands, der aus dem isotropen Zustand von Flüssigkristallmolekülen 3,4,5 keimt und wächst. Für Tactoide sind Keimbildung und Wachstum wie bei anderen Kristallen (d.h. sie erfordern eine relativ hohe Konzentration an Mesogenen [den Molekülen, die Flüssigkristalle bilden]). Die einzigartige "Spindelform" des Tactoiden ergibt sich aus der lokalen Ausrichtung der Flüssigkristallmesogene, die sich in der nematischen Phase ausrichten (Abbildung 1E). Sie können keinen abgerundeten Kristall bilden, da die Moleküle stark asymmetrisch sind. Angesichts der Beschaffenheit der Mikrotubuli ist es vielleicht nicht verwunderlich, dass die mitotische Spindelmaschinerie, die aus einer hohen lokalen Konzentration von Mikrotubuli besteht, auch die gleiche Form hat, unabhängig davon, ob sie als Tactoid oder Spindel bezeichnet wird. Taktoide können bipolar sein, mit Polen an den sich verjüngenden Enden (Abbildung 1Ei), oder homogen, mit Polen effektiv im Unendlichen (Abbildung 1Eii).
Angesichts der Bedeutung der Spindelbildung wurden Bemühungen um eine selbstorganisierte Spindelbildung in vitro unternommen, indem die Kondensation von Mikrotubuli in Bündel über ionische Spezies 12,13, Crowding-Agenten, die Depletionsinteraktionen erzeugen 14,15, und spezifische Mikrotubuli-Vernetzungsproteine13,16,17,18,19 nachgewiesen wurde. 21. Obwohl diese Mittel alle daran arbeiten, die lokale Konzentration von Mikrotubuli zu erhöhen, führen sie überraschenderweise oft zu langen Mikrotubulibündeln, aber nicht zu Tactoiden. Ein Grund, warum diese Bündel lang sind, könnte sein, dass die Mikrotubuli, aus denen sie bestehen, auch lang sind. Jüngste Arbeiten mit kürzeren Mikrotubuli berichteten auch von längeren Bündeln, die sich am Ende15 nicht verjüngen; In diesem Fall werden die Bündel mit Motorproteinen zusammengehalten, die eine Verlängerung der Bündel bewirken und sie dadurch länger machen. Für konische, spindelartige Baugruppen, wie hier beschrieben, werden kurze Mikrotubuli mit nicht-extensilen Vernetzern benötigt.
Vor kurzem haben wir eine Technik entwickelt, die die Erzeugung von Mikrotubuli-Tactoiden mit dem antiparallelen Vernetzer MAP65 in Gegenwart von nukleierenden stabilen Mikrotubuliermöglicht 22. Die Mikrotubuli mussten kurz sein, aber nur wenige bekannte Regulatoren der Mikrotubuli-Länge können Mikrotubuli gegen dynamische Instabilität oder End-to-End-Glühen kappen. Stattdessen wurde GMPCPP verwendet, um die Filamente nach dem Wachstum zu keimen und zu stabilisieren. Dies ermöglichte es, eine hohe Dichte an kurzen Mikrotubuli zu erzeugen, die sich selbst in Tactoiden organisieren konnten. Diese Tactoiden waren homogen, wenn sie unter Doppelbrechung betrachtet wurden. Zusätzlich zu kurzen Mikrotubuli wurde ein spezifischer antiparalleler Vernetzer, MAP65, verwendet, um die Tactoide zu bilden (Abbildung 2). MAP65 ist ein pflanzliches Mikrotubuli-assoziiertes Protein in der PRC1/Ase1-Familie der mitotischen Vernetzer23. MAP65 existiert als Dimer, mit einer starken Affinität zur Bindung an sich selbst sowie an die Mikrotubuli24. Im Gegensatz zu den meiotischen Spindeln und Tactoiden, die mit Aktinfilamenten 25,26,27 beobachtet wurden, die bipolar sind und die flüssigkeitsähnlichen Eigenschaften von Flüssigkristallen aufweisen, wurde beobachtet, dass Mikrotubuli-Tactoide fest wie 22,28 sind.
Hier werden Protokolle zur Herstellung der Mikrotubuli-Tactoide und zur Charakterisierung der Form der Baugruppen und der Mobilität der Bestandteile mit fluoreszenzbasierten Techniken vorgestellt.
HINWEIS: Sofern nicht anders angegeben, können Teile des Experiments auf einem Labortisch mit entsprechender Schutzausrüstung (Handschuhen) durchgeführt werden.
1. Deckglas-Silanisierung
HINWEIS: Deckgläser müssen silanisiert werden, um mit der in diesen Experimenten verwendeten Polymerbürstenbeschichtung verwendet zu werden. Dies ist eine hydrophobe Silanisierungsbehandlung, die es einem Blockcopolymer mit einem hydrophoben zentralen Block ermöglicht, eine Polymerbürste zu binden und zu erzeugen. Die folgenden Schritte sollten in einem Abzug durchgeführt werden, um die Exposition gegenüber giftigen Dämpfen beim Tragen von Handschuhen zu verhindern. Dimethyldichlorsilan ist hochgiftig und muss mit größter Sorgfalt behandelt werden.
- Spülen Sie die Deckgläser mit ddH 2 O, 70% Ethanol und ddH2O in der richtigen Reihenfolge ab. Trocknen Sie sie mit fusselfreien Labortüchern zwischen jeder Spülung. Dadurch werden vor der Behandlung Staub und wasserlösliche oder organische Partikel von der Oberfläche entfernt.
- Legen Sie die Deckgläser in ein Metall-Deckglas-Haltegestell und übergeben Sie das Rack zu einer UV-Ozone (UVO) Maschine. Bestrahlen Sie die Deckgläser 20 min lang mit UVO, um jegliche Hintergrundfluoreszenz zu entfernen. Anstelle von UVO kann eine Plasmakammer verwendet werden.
- Übertragen Sie die Deckgläser mit einer Pinzette von dem für die UVO-Behandlung verwendeten Metallgestell auf ein anderes Metallgestell, das für die Silanisierung verwendet wird. Verwenden Sie nicht die gleichen Racks für beide, da dies bei der Anwendung von UVO zu hohen Oxidationsraten führt. Reinigen Sie die Racks mit Wasser und Ethanol vor, damit keine Rückstände von Chemikalien aus früheren Verwendungen übrig bleiben.
- Tauchen Sie das Gestell mit den Deckgläsern für 1 h in einen Behälter mit 100% Aceton. Spülen Sie den Behälter 3x mit Leitungswasser und dann 3x mit ddH2O ab, um das gesamte Aceton zu entfernen.
- Tauchen Sie das Rack mit den Deckgläsern für 10 min in 100% Ethanol. Spülen Sie den Behälter 3x mit Leitungswasser und dann 3x mit ddH2 O,um das gesamte Ethanol zu entfernen.
- Tauchen Sie das Rack mit den Deckgläsern 3x in ddH2O für jeweils 5 min ein.
- Tauchen Sie das Rack mit den Deckgläsern in 0,1 M KOH (50 mL von 1 M KOH in 450 mL von ddH2O) für 15 min. Spülen Sie den Behälter 3x mit Leitungswasser und dann 3x mit ddH2 O,um alle KOH zu entfernen.
- Tauchen Sie das Rack mit Deckgläsern 3x in ddH2O für jeweils 5 Minuten ein.
- Trocknen Sie das Gestell mit den Deckgläsern über Nacht in einer Dunstabzugshaube oder Laminar-Flow-Haube an der Luft.
- Nachdem Sie das Gestell und die Deckgläser vollständig getrocknet haben, tauchen Sie sie für 5 Minuten in 2% Dimethyldichlorsilan (DDS), das in einem anderen Behälter entnommen wird, der speziell für Silan verwendet wird. Lassen Sie nichts, was nicht trocken ist, mit dem Silan in Berührung kommen.
- Den Rack und die Deckgläser 2x in einen Behälter mit 100% Ethanol für 5 min tauchen. Den Behälter 3x mit Leitungswasser und dann3xmit ddH 2 O abspülen.
- Rack und Deckgläser 3x in ddH2O für jeweils 5 min eintauchen.
- Trocknen Sie das Gestell mit den Deckgläsern über Nacht in einer Dunstabzugshaube oder Laminar-Flow-Haube an der Luft.
- Nach diesem letzten Trocknungsschritt die Deckgläser mit einer Pinzette wieder in Deckglasboxen geben. Diese Deckgläser können in den nächsten 1-2 Monaten verwendet werden. Alte Deckgläser verlieren allmählich ihre Beschichtung und sollten weggeworfen werden.
2. Tubulin-Zubereitung
HINWEIS: Das gekaufte Tubulin wird als lyophilisiertes Pulver geliefert, das entweder nicht gekennzeichnet oder mit Fluorophoren gekennzeichnet ist. Lyophilisiertes Tubulin wird in einem −80 °C Gefrierschrank gelagert. Das folgende Verfahren wird verwendet, um unmarkiertes Tubulin mit markiertem Tubulin in einem Verhältnis zu mischen, das für die Visualisierung geeignet ist.
- Aus dem −80 °C Gefrierschrank ein Aliquot unmarkiertes Tubulin mit 1 mg lyophilisiertem Tubulinpulver entnehmen und auf Eis aufbewahren. Geben Sie 200 μL kaltes PEM-80 in die Röhre, um die Tubulinkonzentration auf 5 mg / ml zu bringen. Halten Sie es für 10 Minuten auf Eis, um das gesamte Lyophilat aufzulösen.
- Aus dem −80 °C Gefrierschrank ein Aliquot rhodaminmarkiertes Tubulin mit 20 μg lyophilisiertem Tubulinpulver entnehmen und auf Eis aufbewahren. Fügen Sie 4 μL kaltes PEM-80 in die Tube hinzu, um die Tubulinkonzentration auf 5 mg / ml zu bringen. Halten Sie es 10 Minuten auf Eis, um das Lyophilat vollständig aufzulösen.
- Nach der Auflösung 100 μL unmarkierte resuspendierte Tubulinlösung in die 4 μL-Lösung von Rhodamin-markiertem Tubulin geben. Pipette 6x-7x sehr langsam zu mischen. Wenn Aggregate sichtbar sind, zentrifugieren Sie das gelöste Tubulin für 10 min bei 90.000 x g , um Aggregate zu entfernen, indem Sie das Pellet entsorgen und den Überstand zurückhalten. Diese Tubulinmischung führt zu ~ 4% markiertem Tubulin.
- Die restlichen 100 μL unmarkiertes Tubulin fallen lassen zu flüssigem Stickstoff (LN2) einfrieren und bei −80 °C lagern, um es für zusätzliche Tubulinmischungen zu verwenden.
- Nehmen Sie die markierte Tubulinmischung und Aliquot in sieben Röhrchen mit jeweils 15 μL. Jedes Aliquot kann für eine einzelne Versuchskammer verwendet werden. Die verbleibenden Aliquots fallen und für zukünftige Experimente bei −80 °C lagern.
3. MAP65-Reinigung
HINWEIS: MAP65 ist nicht im Handel erhältlich und muss daher für diese Arbeit gereinigt werden. Das Protokoll wurde bereits in mehreren Veröffentlichungenausgearbeitet 23,29.
- Wandeln Sie das MAP65-Plasmid und das GFP-MAP65-Plasmid in BL21-Bakterienstamm für die Proteinexpression um.
- BL21-Bakterien auf eine optische Dichte von 0,6-1 bei 600 nm züchten. Induzieren Sie die Proteinproduktion mit dem Lac-Operator und züchten Sie die Bakterien über Nacht.
- Pelletieren Sie die Kulturen und lysieren Sie die Bakterien.
- Sammeln Sie das Lysat nach der Zentrifugation und inkubieren Sie es mit Perlen, die ein Nickelion zur Verfügung haben, um den 6x-Histidin-Tag zu binden.
- Eluieren Sie das Protein mit Imidazol und entsalzen Sie es.
- Das Protein mit flüssigem Stickstoff einfrieren und bei −80 °C lagern, um es innerhalb von 1 Jahr zu verwenden.
4. Montage von Durchflusskammern
HINWEIS: Die Experimente werden in Durchflusskammern aus einem Glasobjektträger und silanisiertem Deckglas durchgeführt (Abbildung 3).
- Nehmen Sie einen Glasschieber und reinigen Sie ihn mit ddH 2 O, Ethanol und ddH2Oin der richtigen Reihenfolge. Trocknen Sie mit einem fusselfreien Labortuch zwischen jeder Spülung.
- Verwenden Sie ein Stück doppelseitiges Klebeband, um einen Fließpfad zu erstellen. Schneiden Sie das Band mit behandschuhten Händen auf eine Länge von ~ 25-30 mm. Teilen Sie das Band der Länge nach auf, um zwei dünnere Streifen zu erhalten. Legen Sie die beiden Bandstreifen mit ca. 5-8 mm zwischen ihnen auf den Schlitten.
HINWEIS: Da die Dicke des Bandes auf etwa 80-100 μm standardisiert ist, bestimmt die Breite des Weges zwischen den Bandstücken das Volumen in der Kammer. - Legen Sie die silanisierten Deckgläser auf den Fließweg. Verschließen Sie den Schlitten und das Deckglas mit den doppelseitigen Klebestreifen, indem Sie mit der Rückseite eines Stiftes vorsichtig auf den Bandbereich drücken. Stellen Sie sicher, dass Sie eine gute Abdichtung über das gesamte Gebiet erhalten. Das Band sollte sich von durchscheinend zu klar drehen, wenn die Dichtung gut gemacht ist.
- Entfernen Sie das zusätzliche Klebeband an den Kanten, so dass nur 1 mm vom Eingang der Durchflusskammer entfernt sind, indem Sie das Band mit einer Rasierklinge schneiden.
- Beschriften Sie die Kammer bei Bedarf mit Informationen zu den experimentellen Parametern.
5. Taktoide Experimente
HINWEIS: Sobald alle Reagenzien und Vorräte erzeugt sind, können sie verwendet werden, um Mikrotubuli-Tactoide in der Durchflusskammer zu keimen und zu polymerisieren.
- Sammeln Sie alle Reagenzien, die verwendet werden sollen. Tauen Sie sie auf Eis auf und lagern Sie sie während der Arbeit auf Eis. Machen Sie mehrere Durchflusskammern für die Experimente.
- Verwenden Sie für jedes Experiment eine Durchflusskammer. Beschichten Sie die Flusskammeroberflächen mit einer Polymerbürste, indem Sie in 20 μL 5% nichtionisches Blockcopolymer-Tensid (Tabelle der Materialien) in PEM-80 gelöst sind, mit kleinen Tropfen an beiden Enden der Kammer, um die Bildung von Luftblasen im Inneren zu verhindern. Bewahren Sie diese in einer feuchten Kammer (d. H. Petrischale mit einem nassen, fusselfreien Labortuch) auf, bis sie gebrauchsfertig ist (mindestens 5-7 min).
- Mischen Sie in einem sterilen Röhrchen Folgendes, um die Tubulin-MAP-Mischung zu erhalten: 9,5 μL PEM-80; 4 μL von 10 mM GMPCPP; 4 μL von 5% Pluronic-F127; 1 μL 1M DTT; 1 μL Glukose; 2 μL Polyethylenglykol (PEG); 12 μL 5 mg/ml Tubulinmischung (13,6 μM Endkonzentration) aus Schritt 2.5.; und 5,5 μL Arbeitsmaterial von MAP65, wobei 10% GFP-MAP65 zur Visualisierung sind. Halten Sie sich während des Mixens auf Eis.
HINWEIS: Es wird empfohlen, eine Verdrängerpipette für die Handhabung der viskosen PEG-Lösung zu verwenden. Regelmäßige Pipetten können nach dem Schneiden der Spitze verwendet werden, um die Öffnung größer zu machen; Diese Methode ist jedoch weniger genau. - 5x-6x durch Pipettieren mischen.
- Kurz vor der Zugabe in die Kammer 1 μL einer vorgemischten Lösung aus Glucoseoxidase (0,5 mg/ml) und Katalase (0,15 mg/ml) (Desoxy) in die Tubulin-MAP-Mischung geben und 7x-8x mischen. Teilen Sie das Gesamtvolumen der Lösung (40 μL) in zwei Teile, die in separaten Kammern verwendet werden.
- Gießen Sie die Tubulin-MAP-Mischung in Kammern. Da sich in den Kammern bereits nichtionisches Blockcopolymer-Tensid befindet, kann nicht mehr Flüssigkeit hinzugefügt werden, ohne die alte Flüssigkeit zu entfernen. Verwenden Sie dazu ein Stück Filterpapier oder ein fusselfreies Labortuch am anderen Ende der Kammer, um Flüssigkeit durch Kapillarwirkung zu entfernen.
- Sobald sich die Probe vollständig in der Kammer befindet, versiegeln Sie die beiden Enden der Kammer mit 5-minütigem Epoxidharz und halten Sie sie bei 37 ° C für ~ 30 Minuten, um Mikrotubuli-Tactoide zu keimen und zu züchten.
6. Fluoreszenzmikroskopie
- Verwenden Sie ein Fluoreszenzmikroskop zur Abbildung der Tactoide.
HINWEIS: Totale interne Reflexionsfluoreszenzmikroskopie oder konfokale Mikroskopie der rotierenden Scheibe sind gut, um Hintergrundfluoreszenz aus freiem Tubulin zu entfernen, aber Tactoide sind auch durch regelmäßige Epifluoreszenz und sogar Durchlichtmikroskopie sichtbar, sobald sie gebildet sind, so dass dieses Verfahren ohne spezielle Ausrüstung zugänglich ist. - Verwenden Sie ein numerisches Aperturobjektiv (NA) von 1,2 NA oder höher mit einer Vergrößerung von 60x oder höher, um genügend Licht in Fluoreszenz zu sammeln. Diese Objektive erfordern oft das Eintauchen in ddH2O oder Öl.
- Nehmen Sie Bilder mit CMOS- oder CCD-Kameras auf. Verwenden Sie eine effektive Pixelgröße auf der Kamera von 108 nm.
HINWEIS: Die Pixelgröße hängt von der Kamera und der verwendeten Vergrößerung ab, die in diesem Fall 60x oder 100x mit einer hohen numerischen Blende (1,2 oder 1,49 NA) betrug. Zusätzliche Bildexpander können vor der Kamera verwendet werden, um die benötigte Pixelgröße zu erreichen. - Halten Sie die Probe bei 37 °C mit einer auf diese Temperatur eingestellten Umgebungskammer. Alternativ können Sie andere Bühnenheizungen verwenden, einschließlich Heißluft-Bühnenheizungen und objektiv temperierte Halsbänder mit zirkulierendem warmem Wasser.
- Verwenden Sie Anregungsquellen, die für die benötigte Fluoreszenz korrekt sind. Für Rhodamintubuli verwenden Sie einen 561-nm-Laser mit einer Leistung von mindestens 1 mW an der Probe und für GFP-MAP65 einen 488-nm-Laser mit einer Leistung von mindestens 1 mW an der Probe.
HINWEIS: Wenn Sie eine Weitfeld-Epifluoreszenzmikroskopie verwenden, verwenden Sie einen Rhodaminfilterwürfel mit Anregung: 540 ± 12,5 nm, dichroitisch: 545 nm ± 12,5 nm Cut-off und Emission: 575 nm langer Durchgang und einen GFP-Filterwürfel mit Anregung: 480 ± 15 nm, dichroitisch: 505 nm ± 15 nm Grenzwert und Emission: 515 nm langer Durchgang. - Nehmen Sie mindestens 10 Bilder von verschiedenen Bereichen auf, um über 100 Tactoiden abzubilden. Nehmen Sie Bilder sowohl im roten als auch im grünen Kanal auf und speichern Sie sie zur Analyse als 16-Bit-TIFF-Bilder. Stellen Sie sicher, dass die Beleuchtungsleistung und die Belichtungszeiten so sind, dass die Intensitätsskala für die Kamera nicht gesättigt ist.
7. Fluoreszenzrückgewinnung nach Photobleaching (FRAP)
ANMERKUNG: Um die Mobilität der internen Bestandteile der Tactoiden zu untersuchen, wurde FRAP verwendet. FRAP funktioniert, indem es einen ausgewählten Teil des mit Rhodamin markierten Tubulins und des GFP-markierten MAP65-Tactoids photobleicht und dann die Rückgewinnung der Fluoreszenz mit der Zeit in dieser Region beobachtet. Die Geschwindigkeit der Erholung hängt vom Umsatz der Arten ab, die photogebleicht werden. Diese Umschlagshäufigkeit kann von Diffusions- und Bindungsreaktionen abhängen. Für MAP65, das für die Tactoide verbindlich ist, können verbindliche Wechselkurse geschätzt werden. FRAP wird mit einem zusätzlichen 405-nm-Lasersystem durchgeführt, das den Laser in jeder Form scannen kann. Es gibt viele Möglichkeiten, FRAP durchzuführen, einschließlich der Verwendung der Durchlichtlampe und der Blende, um einen lokalen Bereich14 zu photobleichen.
- Wählen Sie einen isolierten Taktoiden in der Kammer aus, um einen Bereich von Interesse (ROI) zu erstellen, der Teile der Tactoide und der umgebenden Lösung abdeckt.
- Verwenden Sie ein Mikroskop mit einem zusätzlichen 405-nm-Laser für FRAP, um sowohl das Tubulin als auch MAP65 gleichzeitig zu photobleichen. Alternativ kann eine helle Lampe durch den Feldanschlag der Iris14 verwendet werden. Passen Sie die spezifische Intensität der Photobleichsysteme empirisch an, um eine Beschädigung der Proteine während des Bleichens zu vermeiden.
- Nehmen Sie den Taktoid als Zeitreihenfilm für 30-60 s auf, bevor Sie Photobleichen machen, um Informationen über die Intensität vor dem Bleichen zu erhalten. Nehmen Sie sowohl den roten als auch den grünen Kanal auf.
- Photobleichen Sie den Tactoid aus, indem Sie den ROI entweder mit den Lasern oder der Lampe so lange wie nötig belichten, um den Tactoid zu beschädigen, ohne den Tactoid zu beschädigen. Bestimmen Sie die Intensität und Zeit empirisch.
- Nehmen Sie den Film in beiden Farbkanälen für 5-10 Minuten nach dem Photobleichen oder bis die Erholung ein Gleichgewicht erreicht zu haben scheint, auf.
- Überprüfen Sie den GFP-MAP65-Kanal visuell auf Wiederherstellung.
8. Datenanalyse
HINWEIS: Eine quantitative Analyse der Bilder von Tactoiden wurde durchgeführt, um mehr über die Auswirkungen von Umweltveränderungen zu erfahren, die durch verschiedene Crowding-Agenten, ionische Bedingungen und die Hinzufügung anderer Faktoren verursacht werden.
- Charakterisierung der taktischen Form
- Quantifizieren Sie die Länge und Breite der Tactoide aus den roten und grünen Bildern, die mit konfokaler Mikroskopie aufgenommen wurden.
- Öffnen Sie die Bilder mit FIJI/ImageJ.
- Wenn Rohdaten in 16-Bit aufgenommen werden, passen Sie bei Bedarf die Helligkeit und den Kontrast an. Wählen Sie Bild > Passen Sie > Helligkeit und Kontrast an , um das Bild so anzupassen, dass der Taktoid klar sehen kann. Passen Sie die Helligkeit und den Kontrast an, ohne die Einstellung anzuwenden, um die Intensitätsdaten nicht versehentlich zu ändern.
- Sobald die Tactoide deutlich sichtbar sind, wählen Sie gute zu messende Tactoide aus (Abbildung 4Bi). Stellen Sie sicher, dass die Tactoide deutlich sichtbar sind, ohne sich mit anderen Tactoiden oder Aggregaten zu überlappen, und dass sie nicht gekrümmt oder gebogen sind, um geradlinige Messwerkzeuge verwenden zu können.
- Überprüfen Sie als nächstes, ob die richtige Pixelgröße für die Bilder eingestellt ist. Die Mikroskopbilder enthalten Metadaten über die Pixelgröße. Wenn Sie eine andere Kamera ohne Metadaten oder externe Bilderweiterungssysteme verwenden, die die erwartete effektive Pixelgröße ändern können, passen Sie die Pixelgröße manuell an. Gehen Sie in FIJI/ImageJ zum Analyze > Set Scale , um die richtige Pixelkonvertierung festzulegen.
- Klicken Sie mit dem Werkzeug "Gerade Linie" in der Symbolleiste in FIJI/ImageJ auf ein Ende des Taktoids und ziehen Sie den Cursor an das andere Ende des Taktoids (Abbildung 4Bii). Sobald der Linien-ROI ausgewählt ist, wählen Sie Analyze > Measure (Analysieren Messen ), um die Länge zu messen. Wenn die Länge nicht standardmäßig gemessen wird, stellen Sie sicher, dass die Messung im Dialogfeld Analysieren > Festlegen von Messungen die Länge einschließt.
HINWEIS: Wenn Sie mit dem Werkzeug " Gerade Linie " messen, werden in der Regel die Länge und der Winkel der gezeichneten Linie angegeben. Als Beispiel zeigt Abbildung 4Bii eine gerade Linie, die zur Seite des Taktoids gezeichnet wird, um letzteren sichtbar zu machen, aber die Messung direkt auf dem Taktoid durchzuführen. - Verwenden Sie nach der Messung das Textwerkzeug in der Symbolleiste, um den Taktoid zu beschriften. Erstellen Sie ein Textfeld, fügen Sie eine Zahlenbeschriftung hinzu, und wählen Sie " Bearbeiten" > "Zeichnen ", um die Beschriftung im Bild zu fixieren. Speichern Sie das Bild als separate ROI-Datei.
HINWEIS: Durch die Beschriftung und Speicherung dieser Datei kann der Prüfer anhand der Rohdaten wissen, welche Messung welchem Taktoid entspricht. Stellen Sie sicher, dass Sie jeden Taktoiden einmal messen. - Nachdem die Tactoiden für das gesamte Bild gemessen wurden, speichern Sie die Daten im Ergebnisfenster in einer durch Kommas oder Tabulatoren getrennten Textdatei (mithilfe von Datei > Speichern unter) und öffnen Sie die Daten in einem Tabellenkalkulationsprogramm, um die Daten in Zahlen zu analysieren. Sammeln Sie alle Daten zusammen (Rohbilddaten, ROI-Bild und Textdatei der Ergebnisse) in einem Ordner mit einer geeigneten Namenskonvention, um alles organisiert zu halten.
HINWEIS: Obwohl die taktischen Längenmessungen von Hand durchgeführt werden, ist es angesichts der Tatsache, dass die taktischen Breiten schmal sind, besser, eine andere Methode zur Messung der taktischen Breite (siehe unten) anzuwenden, um den Messfehler zu reduzieren. - Zeichnen Sie mit ImageJ/FIJI einen Linienbereich mit dem Geradeaus-Werkzeug . Zeichnen Sie die Linie als senkrechten Bisektor zur taktoiden Längsachse (Abbildung 4Bii).
- Wählen Sie > Plotprofil analysieren (Analyze Plot Profile ), um das Intensitätsprofil des linearen Bisektors zu erstellen (Abbildung 4Biii). Eine Handlung wird angezeigt. Um die Daten aus dem Diagramm abzurufen und zu speichern, wählen Sie die Schaltfläche Liste unten links aus. Dadurch wird die Textdateiliste der Intensitätsdaten entlang der Länge der gezeichneten Linie generiert. Speichern Sie die Textdatei als .csv oder .txt Datei.
- Öffnen Sie die Textdatei in einem passenden Programm wie MatLab, Python (sciPy) oder anderen Programmen. Passen Sie die Intensitätsdaten mit einer Gaußschen Funktion der Form an:
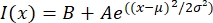 , wobei I(x) der Graustufenwert entlang der Länge x ist; B ist die Hintergrundebene; A ist die Amplitude des Gaußschen; μ ist der Mittelwert oder das Zentrum des Gaußschen; und σ ist die Standardabweichung der Gaußschen Abweichung.
, wobei I(x) der Graustufenwert entlang der Länge x ist; B ist die Hintergrundebene; A ist die Amplitude des Gaußschen; μ ist der Mittelwert oder das Zentrum des Gaußschen; und σ ist die Standardabweichung der Gaußschen Abweichung. - Bericht 2σ als Breite des Tactoids. Schätzen Sie die Intensität der Mikrotubuli im Taktoid, indem Sie die Fläche unter dem Gaußschen berechnet (ohne Hintergrund).
HINWEIS: Wenn die Bilder innerhalb des linearen Intensitätsbereichs der Kamera liegen und mit der gleichen Belichtungszeit und Anregungsintensität aufgenommen werden, können integrierte Intensitäten verglichen werden, um die relative Anzahl der Mikrotubuli im Taktoid zu schätzen.
- FRAP-Analyse
HINWEIS: Experimente zum Testen der Beweglichkeit der Mikrotubuli und des MAP65 verwendeten FRAP, um das spezifische Photobleichen und die Wiederherstellung der Intensität aufgrund molekularer Bewegung aufzuzeichnen (Abbildung 5A). Die Daten wurden aus den Bildzeitreihendaten mit ImageJ/FIJI quantifiziert.- Verwenden Sie ImageJ/FIJI, um die Filmdaten zu öffnen.
- Registrieren Sie die Stapel (Zeitreihendaten) im Laufe der Zeit, um die Drift zu entfernen. Verwenden Sie das StackReg-Plugin zusammen mit dem zusätzlichen TurboReg-Plugin. Anweisungen zur Verwendung der Plugins finden Sie unter den Weblinks in der Tabelle der Materialien. Wählen Sie die Übersetzung, um die Position der Rahmen zu verschieben und so die Bilder zu registrieren.
- Sobald die Bilder registriert wurden, um Drift zu entfernen, drehen Sie das Bild so, dass der Taktoid entweder vertikal oder horizontal im Rahmen ist, indem Sie Bild > Transformieren > Drehen auswählen. Wählen Sie den zu drehenden Winkel aus und verwenden Sie die Vorschau , um festzustellen, ob der Takt ausreichend gedreht ist. Wenn in der Vorschau angezeigt wird, dass der Taktoid entweder vertikal oder horizontal ist, wählen Sie OK, um alle Bilder im Film zu drehen.
- Verwenden Sie das Rechteckauswahlwerkzeug in der Symbolleiste, um einen rechteckigen Schnitt über dem photogebleichten Bereich des Taktoids zu erstellen. Zeichnen Sie die integrierte Intensität des ROI-Bereichs für jeden Frame mit Image > Stacks > Measure Stack auf. Legen Sie den Messtyp mit Analyze > Set Measurements auf Integrated Density (Integrierte Dichte) fest. Speichern Sie die analysierten Intensitätsdaten, die im Ergebnisfenster angezeigt werden, als Textdatei im .csv- oder .txt-Format, indem Sie Datei > Speichern unter auswählen.
HINWEIS: Abbildung 5B zeigt ein Beispiel für die rohen 16-Bit-Intensitätsdaten, die für die Mikrotubuli- und GFP-MAP65-Kanäle im Bleichbereich gemessen wurden. - Da die Gesamtintensität der Bilder im Laufe der Zeit aufgrund der durch Bildgebung verursachten Photobleiche weltweit verblassen wird, muss diese globale Photobleiche korrigiert werden. Verwenden Sie dazu dieselbe ROI-Größe (Schritt 8.2.4.) und verschieben Sie sie in einen Bereich im Hintergrund des Bildes, in dem keine Mikrotubuli oder MAP65 sichtbar sind. Messen Sie die integrierte Intensität des Stacks, wie in Schritt 8.2.4 beschrieben. Speichern Sie die Ergebnisse als zweite Textdatei.
HINWEIS: Abbildung 5B zeigt ein Beispiel für die rohen 16-Bit-Intensitätsdaten, die für die Mikrotubuli- und GFP-MAP65-Kanäle im Hintergrundbereich gemessen wurden. - Um das Ausbleichen des Hintergrunds zu korrigieren, dividieren Sie die Signalintensität auf dem Taktoid durch die Hintergrundintensität für denselben Zeitpunkt. Berechnen Sie I korrigiert (t) als:
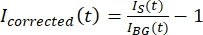 , wobei I S (t) (das Signal) die Messung am gebleichten Bereich und IBG (t) (das Rauschen) die Messung am Hintergrundbereich ist (Abbildung 5C). Dies berechnet das Signal-Rausch-Verhältnis für jeden Frame und subtrahiert auch das Rauschen.
, wobei I S (t) (das Signal) die Messung am gebleichten Bereich und IBG (t) (das Rauschen) die Messung am Hintergrundbereich ist (Abbildung 5C). Dies berechnet das Signal-Rausch-Verhältnis für jeden Frame und subtrahiert auch das Rauschen. - Skalieren Sie dann die Daten so neu, dass sie zwischen Null und eins liegen, wobei
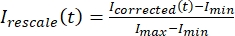 I min und I max das globale Minimum bzw. Maximum der von Ikorrigierten Daten über die gesamte Zeit bezeichnen (Abbildung 5C).
I min und I max das globale Minimum bzw. Maximum der von Ikorrigierten Daten über die gesamte Zeit bezeichnen (Abbildung 5C). - Passen Sie diese Daten an ein zerfallendes Exponential der Form an:
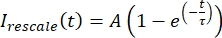 , wobei A die Amplitude der Wiederherstellung und τ die Zeitskala der Wiederherstellung ist (Abbildung 5C).
, wobei A die Amplitude der Wiederherstellung und τ die Zeitskala der Wiederherstellung ist (Abbildung 5C).
Mit nur einer kleinen Anzahl von Komponenten, Tubulindimeren und Mikrotubuli-Vernetzern können sich Mikrotubuli-Tactoide bilden (Abbildung 2A). Obwohl dieses Protokoll die Inkubation beschreibt, um Mikrotubuli in einem Inkubator zu keimen und zu züchten, können die Keimbildung und das Wachstum direkt unter dem Mikroskop beobachtet werden (die innerhalb von 30 Minuten abgeschlossen sind) (Abbildung 2B). Die Tubulinkonzentration wird konstant bei 13,6 μm und MAP65-MT-Bindung bei 10% gehalten.
Abbildung 4 stellt erfolgreiche Daten dar. Die Tactoide sollten sowohl mit einem 561-nm-Laser im Tubulinkanal als auch mit 488 nm im MAP65-Kanal sichtbar sein, die sich perfekt überlappen (Abbildung 4A). Ein Rätsel des Systems war, dass die Breite der Tactoide unter einer Vielzahl von experimentellen Veränderungen nicht zu variieren scheint, einschließlich der Änderung der Mikrotubuli-Längen, der MAP65-Konzentration und der Crowding-Agenten (Abbildung 4B) 22,28. Die Länge ist viel variabler und hängt sowohl von den Mikrotubuli-Längen als auch von der MAP65-Konzentration ab (Abbildung 4B)22,28.
Bei der Durchführung von FRAP wurde beobachtet, dass sich das MAP65-Signal erholt, das Mikrotubuli-Signal jedoch nicht (Abbildung 5). Die Erholung in FRAP ist auf die Mobilität und Bewegung der markierten und photogebleichten Objekte zurückzuführen. Im Falle des MAP65 dissoziieren die abgedunkelten Moleküle und bewegen sich vom Mikrotubuli weg und neue bewegen sich in die Region (Abbildung 5). Die MAP65-Bindung befindet sich im Gleichgewicht, so dass die Bindungs- und Unbindungsrate gleich ist (gemessen in Molekülen pro Sekunde). Für die Mikrotubuli wurde keine Erholung beobachtet, was darauf hindeutet, dass die Mikrotubuli nicht in der Lage sind, den Taktus zu verlassen (Abbildung 5A, Bi, C). Darüber hinaus wurde keine Ausbreitung der abgedunkelten Region beobachtet, was darauf hindeutet, dass die Mikrotubuli lokal unbeweglich sind und keine Flüssigkeit innerhalb der Taktoidform.
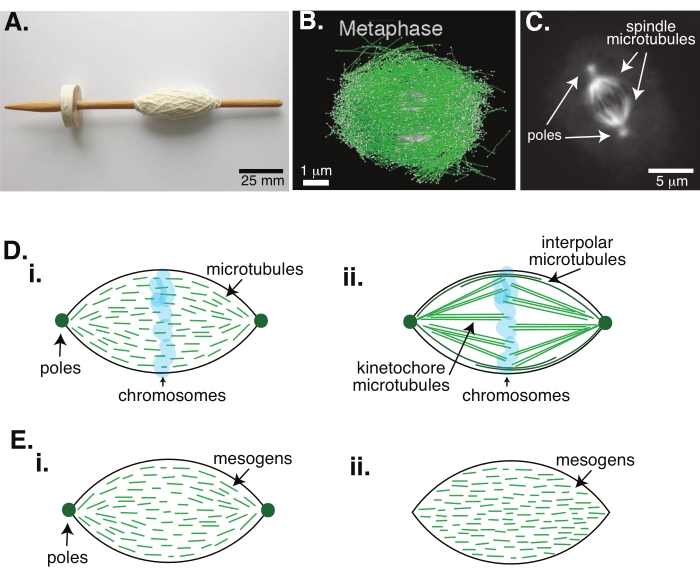
Abbildung 1: Verschiedene Modelle der Spindelbildung. Eine mitotische Spindel ist eine Maschine aus Mikrotubuli und den damit verbundenen Proteinen und Enzymen, die die Chromosomen während der Zellteilung zu den beiden neuen Tochterzellen ausrichtet und trennt. (A) Bild einer frühmittelalterlichen Fallspindelreplik mit feinem Garn aus den Niederlanden. Diese Figur wurde von einem Wikimedia-Bild von Peter van derSluijs 30 modifiziert. (B) Dreidimensionale Rekonstruktion von Mikrotubuli in verschiedenen Stadien der Wildtyp-Meiose II. Mikrotubuli sind grün und Chromosomen grau dargestellt. Maßstabsleiste = 1 μm. Diese Figur wurde von Lantzsch et al.31 modifiziert. (C) Mikroskopische Aufnahme der Mikrotubuli in einer mitotischen Spindel einer sich teilenden Sf9-Zelle. Die Spindelpole und die Mikrotubuli der Spindel sind mit einem grün fluoreszierenden Protein markiert. Maßstabsleiste = 5 μm. Diese Figur wurde von Advani et al.32 modifiziert. (D) Verschiedene Modelle, wie mitotische und meiotische Spindelmikrotubuli organisiert sind. (i) Zuvor wurde für meiotische Spindeln, die aus Xenopus-Ei-Extrakten hergestellt wurden, beobachtet, dass die Mikrotubuli (grün) in der gesamten Spindel kurz und dynamisch sind. Dies ist vergleichbar mit einer bipolaren taktoiden Organisation in einem Flüssigkristall. (ii) Das kanonische Modell für die Mikrotubuli-Organisation innerhalb einer mitotischen Spindel hat zwei Arten von Mikrotubuli: interpolare oder polare Mikrotubuli (dunkelgrün), die sich in der Mitte um die Chromosomen vernetzen, und Kinetochor-Mikrotubuli (hellgrün), die vom Pol zum Kinetochor gebündelt und gestreckt werden, um die Chromosomen zu drücken und zu ziehen. In allen Bildern sind die Chromosomen in transparentem Blau und die Spindelpole dunkelgrün dargestellt. (E) Schematische Darstellung von Mesogenen (grüne Linien) in einem Flüssigkristall-Tactoid für (i) bipolare und (ii) homogene Taktoide. Bipolare Tactoide haben zwei Pole am Ende des Tactoids, und die Mesogene orientieren sich neu, um auf diese Pole zu zeigen. Homogene Tactoide haben Pole im Unendlichen, und die Mesogene ändern nicht die Ausrichtung entlang der Länge des Tactoids. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
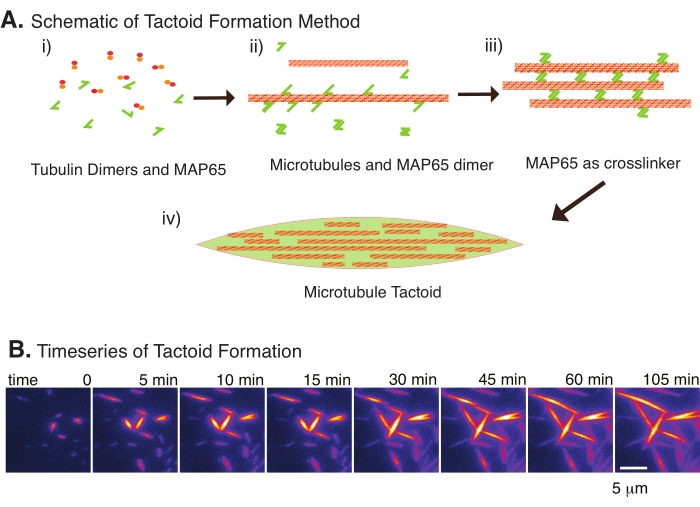
Abbildung 2: Kondensation von Mikrotubuli. (A) Mikrotubuli können durch eine Vielzahl von Methoden gebündelt und vernetzt werden, einschließlich ionischer Spezies, Erschöpfungskräfte, die durch Crowding-Agenten verursacht werden, und spezifischer Mikrotubuli-Vernetzer wie MAP65. (i) Tubulin-Dimere und MAP65-Proteine werden gemischt, um Mikrotubuli zu keimen und zu züchten. (ii) Mikrotubuli keimen und wachsen aus dem Tubulin, und MAP65 bindet sofort an Mikrotubuli, ein anderes MAP65-Monomer oder beides und verursacht eine Bündelung. (iii) Mikrotubuli in den vernetzten Bündeln keimen und wachsen. (iv) Die endgültige Konfiguration ist ein Mikrotubuli-Tactoid ähnlich einer Spindel. (B) Zeitreihe von Mikrotubuli-Tactoiden, die über 105 min keimen und wachsen. Maßstabsleiste = 5 μm. Abbildung nach Edozie et al.22. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
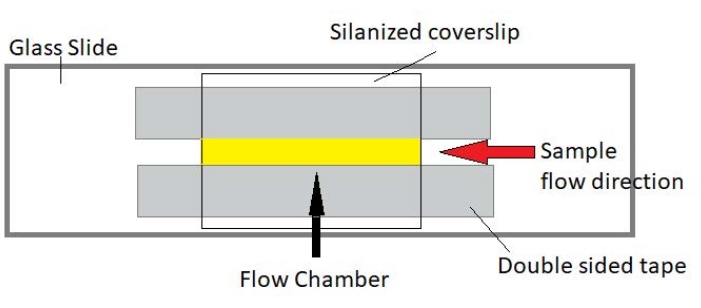
Abbildung 3: Baugruppe der Durchflusskammer Die Durchflusskammer besteht aus einem Glasobjektträger, silanisiertem Deckglas und permanentem doppelseitigem Klebeband. Der gelb hervorgehobene Bereich ist der Fließweg, auf dem die Probe fließt und beobachtet wird. Das Volumen der Durchflusskammer beträgt ~20 μL. Epoxidharz wurde verwendet, um die Enden der Kammer zu versiegeln, um zu verhindern, dass die Probe während der Langzeitbildgebung über mehrere Stunden verdampft. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
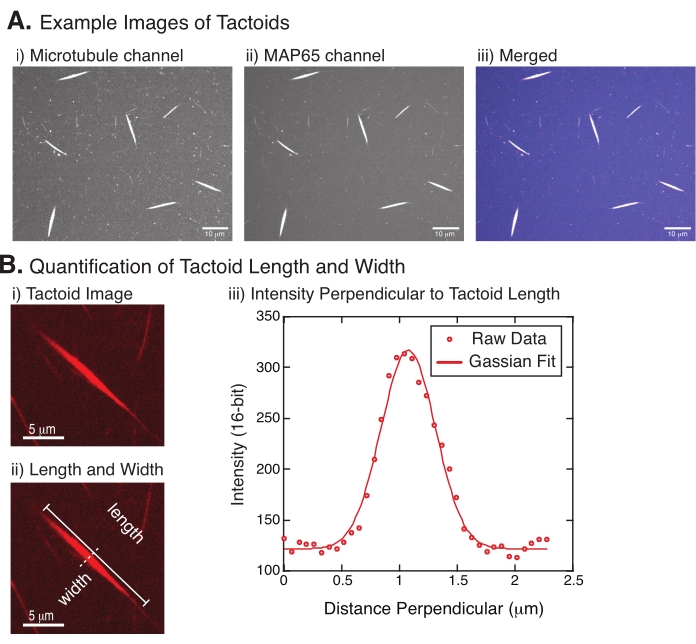
Abbildung 4: Taktische Bilder und Längen- und Breitenanalyse. (A) Beispieldaten von Tactoiden, die wie beschrieben gebildet und mit konfokaler Spinnscheibe abgebildet wurden, zeigen (i) Mikrotubulikanal-Bildgebung von Rhodamin-markiertem Tubulin mit einem 561-nm-Laser, (ii) GFP-MAP65-Kanalabbildung des GFP mit einem 488-nm-Laser und (iii) zusammengeführtes Overlay-Bild aus dem Mikrotubuli-Kanal (Magenta) und GFP-MAP65-Kanal (Cyan). Überlappungsbereiche werden weiß dargestellt und zeigen, dass die Mikrotubuli und MAP65 exakt kolokalisieren. Maßstabsleiste = 10 μm für alle Bilder in (A). (B) Quantifizierung der Tactoidlänge und -breite. (i) Bild eines zu analysierenden Tactoiden ohne Markierungen. Maßstabsleiste = 5 μm. (ii) Gleiches Bild wie in (i), wo die Längenmessungen (durchgezogene Linie mit Zeilenkappen) und Breite (gestrichelte Linie) angegeben sind. Skalenbalken = 5 μm. (iii) Die Breite wurde gemessen, indem das Intensitätsprofil über den Taktoid am senkrechten Bisektor (gestrichelte Linie) gemessen wurde, der in (ii) bezeichnet wird. Das Intensitätsprofil wurde an eine Gaußsche Funktion angepasst, um die Amplitude und Breite des Tactoiden aufzudecken. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
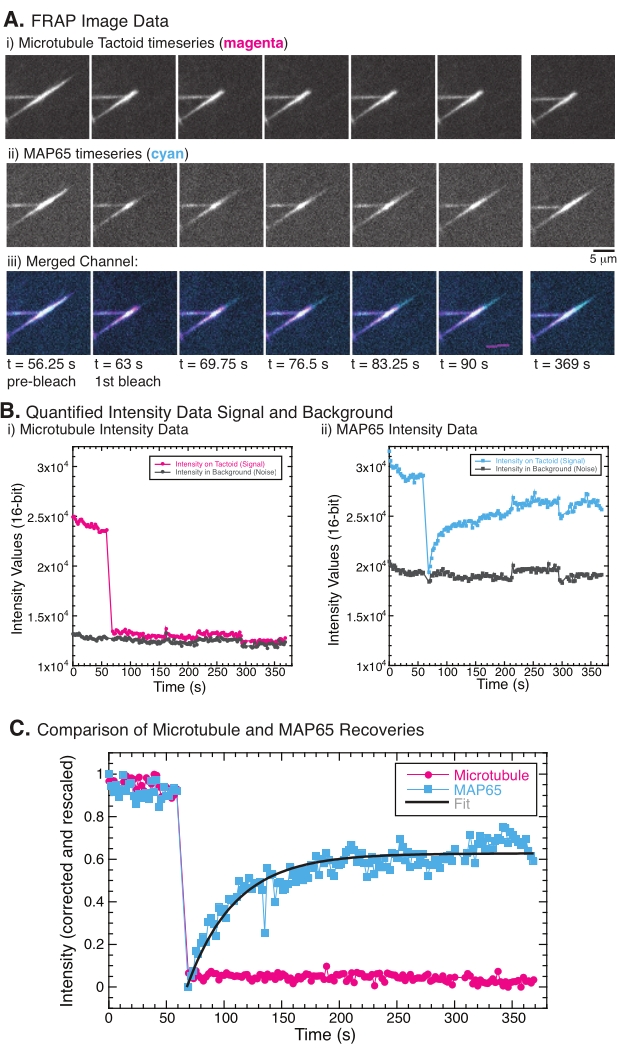
Abbildung 5: Repräsentative FRAP-Daten und -Analysen. (A) Mikroskopie-Zeitreihendaten von (i) Mikrotubuli-Tactoid und (ii) GFP-MAP65 und (iii) Overlay-Bild beider Kanäle mit Mikrotubuli in Magenta und GFP-MAP65 in Cyan, die zum Zeitpunkt von 63 s photogebleicht und für weitere 5 min beobachtet wurden. (B) Quantifizierte Intensität der (i ) Mikrotubuli-Kanal im gebleichten Bereich (Magenta-Kreise) und im Hintergrund (dunkelgraue Kreise) und (ii) GFP-MAP65-Kanal im gebleichten Bereich (cyanfarbene Quadrate) und im Hintergrund (dunkelgraue Quadrate). (C) Die Daten wurden um das Hintergrundrauschen korrigiert und für den Mikrotubuli-Kanal (Magenta-Kreise) und den GFP-MAP65-Kanal (Cyan-Quadrate) neu skaliert. Die Mikrotubuli erholen sich nicht, aber der GFP-MAP65 tut es und kann (dunkelgraue Linie) an einen steigenden exponentiellen Zerfall angepasst werden, um die Amplitude und die Zeitskala der Erholung zu finden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
Die hier beschriebenen Methoden wurden in mehreren Arbeiten zur Erzeugung von Mikrotubuli-Tactoiden verwendet (Abbildung 2)22,28. Diese Experimente sind biologisch relevant, um die Organisationsprinzipien aufzudecken, die die Form und Stabilität der mitotischen oder meiotischen Spindel in den meisten Zelltypen kontrollieren. Darüber hinaus sind Mikrotubuli Modell-Flüssigkristallmesogene, die helfen können, mehr darüber zu erfahren, wie Flüssigkristalle keimen und nematische Phasen aus isotropen Phasen wachsen lassen.
Das hier beschriebene Verfahren hat mehrere Vorteile für die Erforschung der Mikrotubuli-Selbstorganisation. Erstens ist es sehr reproduzierbar, da es im Labor von vielen Studenten, einschließlich Gymnasiasten, mit wenig Vorwissen oder Training durchgeführt wurde, bevor es im Labor begann. Die Taktoide sind doppelbrechend22, so dass sie zusätzlich zur Fluoreszenzmikroskopie im Durchlichtlicht betrachtet werden können, wodurch diese Methode vielen Labors zugänglich ist und dieses experimentelle Verfahren neben der High-End-Forschung auch für Bildungszwecke angepasst werden kann. Schließlich eröffnet dieser Prozess Wege, um biologische Systeme in einem reduzierten, reduktionistischen Ansatz weiter zu verstehen und zu untersuchen, so dass man verstehen kann, wie jede zusätzliche Bedingung, jedes Protein oder jeder Zusatzstoff die Selbstorganisation von Tactoiden und vielleicht letztendlich die Spindel verändern kann. Ziele für eine bessere Biomimikry sind Aktivität, Fließfähigkeit und Filamentpolaritätssortierung.
Es kann mehrere Faktoren geben, die das Experiment beeinflussen und unerwartete Ergebnisse liefern. Wenn sich beispielsweise die Tactoide nicht bilden (Abbildung 2), aber fächerartige Muster beobachtet werden, ist der MAP65 wahrscheinlich nicht vorhanden oder bindet nicht an die Mikrotubuli22,28. Dies sollte auch im MAP65-Fluoreszenzkanal offensichtlich sein, da der GFP-MAP65 nicht an die Mikrotubuli gebunden ist.
Wenn sich die Tactoide nicht bilden und der Hintergrund als Flecken auf dem Glas erscheint, könnte dies an der Oberflächenbeschichtung liegen. Einmal durchgeführt, dauert die Silanisierung nur 1 Monat auf Deckgläsern. Wenn es nachlässt, kann sich das Tubulin unspezifisch an die exponierte Oberfläche binden. Diese Bindung erfolgt in ungeraden Mustern.
Wenn sich die Tactoide nicht bilden und das Tubulin in Aggregaten verschiedener Formen und Größen beobachtet wird, könnte dies auf Tubulin von schlechter Qualität zurückzuführen sein. Tubulin kann zentrifugiert werden, um anfängliche Aggregate zu entfernen, die diese Off-Pathway-Aggregation anstelle der Mikrotubuli-Polymerisation antreiben können. Wenn die Oberfläche an das Tubulin bindet, kann sie auch das Tubulin in der Lösung abbauen. Niedrige Konzentrationen von Tubulin, unterhalb der kritischen Konzentration für die Polymerisation von Mikrotubuli, können zu Aggregaten führen.
Wenn der MAP65-Kanal in FRAP-Experimenten keine Erholung zeigt (Abbildung 5), ist es möglich, dass die Photobleiche die Mikrotubuli photoschädlich gemacht hat. Lichtschäden verursachen die lokalisierte Zerstörung der Filamente. Dies kann durch Untersuchung im Sendekanal überprüft werden. Mikrotubuli-Tactoide sind im übertragenen Kanal durch einen hohen Index der Brechungsfehlanpassung mit dem umgebenden Wasser sichtbar. Lichtinduzierte Lichtschäden treten als Brandmarke oder Kontrastverlust in der Durchlichtbildgebung am Ort des ROI auf, der einer Photobleiche unterzogen wird. Tritt dies ein, muss die Laser- oder Lichtleistung reduziert werden, um die Lichtschädigung der Proteine zu hemmen.
Bei diesem Verfahren und Ansatz gab es mehrere Herausforderungen. Ein Problem ist, dass die Längenmessungen derzeit von Hand durch Klicken auf das Bild durchgeführt werden. Diese Methode ist zwar unkompliziert, kann aber zu einer hohen Unsicherheit führen. Die Breitenmessung, die den Querschnitt und die Anpassung an einen Gaußschen Wert verwendet, ist eine bessere Methode zur Quantifizierung der Größe. Eine ähnliche Methode könnte für die Länge verwendet werden. Ein zweites Problem ist, dass sich die Tactoide, weil sie so lang und dünn sind, manchmal biegen können. Dies erschwert die Quantifizierung der Länge. Die Konturlänge kann mit einer segmentierten Linie quantifiziert werden, aber jedes Mal, wenn ein Segment hinzugefügt wird, kommt es zu einer zusätzlichen Unsicherheit.
Aus wissenschaftlicher Sicht hat dieser Ansatz einige andere Herausforderungen für seine Verwendung als Modell für Flüssigkristalle oder Spindeln. Die erste Herausforderung war die lange, dünne Form der Tactoide, die Mikrotubuli erzeugen (Abbildung 3 und Abbildung 4). Wie in früheren Veröffentlichungen22 erwähnt, sind Mikrotubuli-Tactoide homogene Tactoide, keine bipolaren. Dies bedeutet, dass die Mikrotubuli, aus denen die Form besteht, nicht neu ausgerichtet werden, um auf die Spitzen der Struktur zu zeigen. Stattdessen befinden sich alle Mikrotubuli parallel zur Längsachse und die "Pole" befinden sich im Unendlichen. Dies unterscheidet sich stark von den Tactoiden, die für molekulare Flüssigkristalle oder sogar für Aktin oder DNA beobachtet wurden, die auch als Flüssigkristallmesogene wirken können. In diesen anderen Systemen sind die Tactoiden bipolar und zeigen, wenn sie in gekreuzten Polarisatoren betrachtet werden, die verräterischen Anzeichen einer Neuorientierung der Stäbe.
Eine zweite große Herausforderung in diesem System besteht darin, dass die Mikrotubuli im Inneren des Tactoiden unbeweglich sind. Dies geht aus FRAP-Experimenten und -Analysen hervor, da die Rückgewinnung der Mikrotubuli sehr gering ist. Ihre feststoffartige Natur macht Mikrotubuli-Tactoide weniger wertvoll als großflächige Flüssigkristallanaloga. Die nematische Phase eines Flüssigkristalls sollte sowohl flüssige (flüssige) als auch kristalline (organisierte) Eigenschaften haben. Obwohl die Form für die Spindel richtig erscheint, macht die Unbeweglichkeit das System weniger aufregend als eine mitotische Modellspindel. Auf der anderen Seite bietet dieses Problem Möglichkeiten zu untersuchen, wie man die Experimente modifizieren kann, um mehr Flüssigkeit im System zu schaffen.
Diese wissenschaftlichen Herausforderungen bieten spannende Möglichkeiten, die neue Erkenntnisse über das System ermöglichen. Um die Mikrotubuli-Tactoide bipolarer zu machen, könnte man kürzere Mikrotubuli verwenden. Es gibt jedoch eine zusätzliche Herausforderung, da Mikrotubuli nicht viele gut charakterisierte Capping-Proteine haben, um die Länge wie Aktin zu kontrollieren. Die Verwendung von Keimbildung und Wachstum erfordert die Verwendung sehr hoher Konzentrationen von Tubulin und GMPCPP, um kurze Mikrotubuli herzustellen. Die hohe Tubulinkonzentration führt zu einer größeren Anzahl von Filamenten im System, was es schwieriger macht, Tactoide voneinander zu trennen. Das Hinzufügen neuer Mikrotubuli-Verschließer, wie DARPin33, kann in dieser Situation helfen. Das zweite Problem, dass die Mikrotubuli unbeweglich sind, könnte durch die Zugabe von Motorproteinen wie Kinesin-534, die Tetramere von Motoren sind, die in der Mitose verwendet werden, gemildert werden. Alternativ könnten künstliche Dimere von dimerem Kinesin-115 verwendet werden.
Eine andere Möglichkeit, mehr Flüssigkeit hinzuzufügen, wäre, den Mikrotubuli zu erlauben, ihre dynamische Instabilität, das Wachstum und Schrumpfen von Mikrotubuli, durchzuführen. Derzeit sind Mikrotubuli, die mit stabilen GMPCPP-Filamenten ausgesät werden und dann eine dynamische Instabilität erfahren, viel länger als gewünscht, um eine Spindel oder einen Tactoid zu bilden, was zu sehr langen Organisationen wie Fans oder Bündeln führen würde. Das Hinzufügen von dynamischer Instabilität des Mikrotubuli müsste also sorgfältig durchgeführt werden, um die taktoide Form zu erhalten. Die Zugabe von assoziierten Proteinen und Enzymen, die die Länge kontrollieren können, kann dieses Problem mildern. Zum Beispiel wäre wahrscheinlich eine Depolymerisation von Kinesinen wie Kinesin-1335 oder die Trennung von Enzymen wie Katanin36 erforderlich. Diese Experimente sind komplex und schwierig, obwohl sie sehr aufschlussreich wären, egal was die Ergebnisse zeigen. Unabhängig davon, in welche Richtung zukünftige Experimente gehen, kann die hier entwickelte Plattform zur Herstellung von Mikrotubuli-Tactoiden neue Informationen über die physikalische Grundlage der Mikrotubuli-Organisation freilegen.
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen haben.
Die Autoren möchten allen Mitgliedern des Ross Lab im Sommer 2021, insbesondere K. Alice Lindsay, für ihre Hilfe danken. Diese Arbeit wurde durch einen Zuschuss von NSF BIO-2134215 unterstützt, der S. Sahu, N. Goodbee, HB Lee und J.L. Ross unterstützte. Ein Zuschuss der KECK Foundation (Rae Anderson, USD, Lead PI) unterstützte teilweise R. Branch und P. Chauhan
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved