
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
Auto-assemblaggio di microtubuli tactoidi
Questo articolo presenta un protocollo per la formazione di assemblaggi di microtubuli a forma di tactoidi utilizzando MAP65, un reticolante di microtubuli a base vegetale e PEG come agente di affollamento.
Il citoscheletro è responsabile della principale organizzazione interna e della riorganizzazione all'interno della cellula, il tutto senza un manager che diriga i cambiamenti. Questo è particolarmente vero durante la mitosi o la meiosi, dove i microtubuli formano il fuso durante la divisione cellulare. Il mandrino è il macchinario utilizzato per separare il materiale genetico durante la divisione cellulare. Verso la creazione di fusi auto-organizzati in vitro, abbiamo recentemente sviluppato una tecnica per ricostituire i microtubuli in assemblaggi simili a mandrini con un set minimo di proteine associate ai microtubuli e agenti di affollamento. Nello specifico, è stato utilizzato MAP65, che è un reticolante microtubulo antiparallelo dalle piante, un omologo di Ase1 dal lievito e PRC1 da organismi di mammiferi. Questo reticolante auto-organizza i microtubuli in gruppi auto-organizzati di microtubuli lunghi, sottili e simili a mandrini. Questi assemblaggi sono anche simili ai tactoidi a cristalli liquidi e i microtubuli potrebbero essere usati come mesogeni a mesoscala. Qui vengono presentati protocolli per la creazione di questi tactoidi microtubuli, nonché per caratterizzare la forma degli assiemi utilizzando la microscopia a fluorescenza e la mobilità dei costituenti utilizzando il recupero della fluorescenza dopo il fotosbiancamento.
La divisione cellulare tramite mitosi è uno dei processi biologici più importanti per sostenere la vita. I filamenti di microtubuli, composti da dimeri di tubulina, sono elementi strutturali essenziali di questo processo. Il meccanismo transitorio creato in metafase quando i cromosomi si allineano nel centro cellulare è chiamato fuso mitotico a causa della sua forma, che è come un fuso di un telaio coperto di fili (Figura 1A). È ben noto in molti organismi che i microtubuli sono usati nella metafase per spingere e tirare i cromosomi condensati nel centro della cellula, allineandoli e collegandoli a microtubuli che li separeranno in anafase (Figura 1B, C). Il mandrino si forma sia in meiosi (Figura 1B) che in mitosi (Figura 1C), creato da molti microtubuli sovrapposti non avvolti attorno all'asse centrale come filo ma che corrono paralleli all'interfaccia. La creazione di queste strutture basate su microtubuli richiede proteine associate che si reticolano e enzimi associati che possono agire come motori per aiutare a spingere e tirare i cromosomi1.
Studi sui fusi meiotici hanno dimostrato che i microtubuli sono corti, dinamici e sovrapposti in array reticolati 2,3,4,5,6 (Figura 1Di). A causa dell'organizzazione fisica di questi microtubuli corti, il fuso meiotico è simile a un tactoide a cristalli liquidi (Figura 1E). In effetti, è stato dimostrato che i mandrini si fondono e si fondono, come ci si aspetterebbe dai tactoidi a cristalli liquidi5.
Molti studi risalenti al 1960 hanno utilizzato la fissazione, le sezioni seriali e la microscopia elettronica per determinare che ci sono due tipi di microtubuli all'interno del fuso mitotico 7,8,9,10. Il primo tipo è chiamato microtubuli di chinetocore, che collegano il polo del fuso al chinetocore. Il secondo tipo è chiamato microtubuli interpolari o polari, che crescono oltre i cromosomi e si sovrappongono nella zona centrale (Figura 1Dii)8,9,10. Un terzo tipo è chiamato microtubuli astrali, che si trovano all'esterno del fuso e collegano i poli al bordo cellulare; queste organizzazioni di microtubuli sono al di fuori dell'ambito della discussione in corso. Ci sono stati studi recenti sull'interazione tra augmin6 e complesso dell'anello gamma-tubulina che influenza i centri di nucleazione per i microtubuli, risultando in un fuso mitotico con microtubuli più corti come nella Figura 1D.
Poiché i microtubuli sono più lunghi di quanto siano larghi, con un elevato rapporto di aspetto e un'elevata rigidità, sono come versioni in scala di molecole a cristalli liquidi. Nella fisica della materia soffice, atomi e molecole sono stati approssimati usando interazioni minime per dedurre i meccanismi fisici delle transizioni di fase, tra cui la nucleazione e la fusione dei cristalli11. Allo stesso modo, i microtubuli sono oggetti a mesoscala che sono versioni in scala di molecole a cristalli liquidi, fornendo approfondimenti sulla fisica della dinamica dei cristalli liquidi, compresa la nucleazione e la crescita delle fasi nematiche da quelle isotrope. Inoltre, come discusso sopra, il fuso meiotico mostra proprietà come quelle di un tactoide a cristalli liquidi, uno stato nematico che nuclea e cresce dallo stato isotropo delle molecole a cristalli liquidi 3,4,5. Per i tactoidi, la nucleazione e la crescita sono come quelle di altri cristalli (cioè, richiedono una concentrazione relativamente alta di mesogeni [le molecole che formano i cristalli liquidi]). L'unica forma a "mandrino" del tactoide deriva dall'allineamento locale dei mesogeni a cristalli liquidi che si allineano nella fase nematica (Figura 1E). Non possono formare un cristallo arrotondato perché le molecole sono altamente asimmetriche. Data la natura dei microtubuli, forse non sorprende che anche il macchinario del fuso mitotico costituito da un'alta concentrazione locale di microtubuli sia della stessa forma, sia che si chiami tactoide o fuso. I tactoidi possono essere bipolari, con poli alle estremità affusolate (Figura 1Ei), o omogenei, con poli effettivamente all'infinito (Figura 1Eii).
Data l'importanza della formazione del fuso, sono stati avviati sforzi verso la formazione di mandrino auto-organizzata in vitro dimostrando la condensazione dei microtubuli in fasci attraverso specie ioniche12,13, agenti di affollamento che creano interazioni di deplezione14,15 e specifiche proteine reticolanti di microtubuli 13,16,17,18,19, 21. Sorprendentemente, sebbene questi agenti lavorino tutti per aumentare la concentrazione locale di microtubuli, spesso si traducono in lunghi fasci di microtubuli ma non tactoidi. Uno dei motivi per cui questi fasci sono lunghi potrebbe essere che anche i microtubuli che li compongono sono lunghi. Lavori recenti che utilizzano microtubuli più corti hanno anche riportato fasci più lunghi che non sono affusolati alla fine15; in questo caso, i fasci sono tenuti insieme con proteine motorie che causano l'estensione dei fasci e, quindi, li rendono più lunghi. I microtubuli corti con reticolanti non estensibili sono necessari per assemblaggi conici simili a mandrini, come descritto qui.
Recentemente, abbiamo sviluppato una tecnica per consentire la creazione di tactoidi microtubuli utilizzando il reticolante antiparallelo, MAP65, in presenza di microtubuli stabili nucleanti22. I microtubuli dovevano essere corti, ma pochi regolatori noti della lunghezza dei microtubuli possono tappare i microtubuli contro l'instabilità dinamica o la ricottura end-to-end. Invece, GMPCPP è stato utilizzato per nucleare e stabilizzare i filamenti dopo la crescita. Ciò ha permesso di creare un'alta densità di microtubuli corti che potrebbero auto-organizzarsi in tactoidi. Questi tactoidi erano omogenei se visti sotto birifrangenza. Oltre ai microtubuli corti, uno specifico reticolante antiparallelo, MAP65, è stato impiegato per formare i tactoidi (Figura 2). MAP65 è una proteina associata a microtubuli vegetali della famiglia PRC1/Ase1 dei reticolanti mitotici23. MAP65 esiste come dimero, con una forte affinità di legarsi a se stesso così come ai microtubuli24. A differenza del fuso meiotico e dei tactoidi osservati con i filamenti di actina 25,26,27, che sono bipolari e hanno le proprietà liquide dei cristalli liquidi, i tactoidi dei microtubuli sono stati osservati essere solidicome 22,28.
Qui vengono presentati i protocolli per la creazione dei tactoidi dei microtubuli e la caratterizzazione della forma degli assiemi e della mobilità dei costituenti utilizzando tecniche basate sulla fluorescenza.
NOTA: Salvo diversa indicazione, parti dell'esperimento possono essere eseguite su un banco di laboratorio indossando adeguati dispositivi di protezione (guanti).
1. Silanizzazione coverslip
NOTA: I coverslip devono essere silanizzati per essere utilizzati con il rivestimento a spazzola polimerica impiegato in questi esperimenti. Si tratta di un trattamento di silanizzazione idrofobica che consente a un copolimero a blocchi con un blocco centrale idrofobo di legare e creare una spazzola polimerica. I seguenti passaggi devono essere eseguiti in una cappa aspirante per prevenire l'esposizione a vapori tossici mentre si indossano i guanti. Il dimetildiclorolosilano è altamente tossico e deve essere maneggiato con la massima cura.
- Risciacquare le coperture con ddH2O, etanolo al 70% e ddH2O in ordine. Asciugarli con salviette da laboratorio prive di lanugine tra ogni risciacquo. Questo rimuove la polvere e le particelle idrosolubili o organiche dalla superficie prima del trattamento.
- Posizionare i coverslip in un rack metallico e trasferire il rack su una macchina UV-Ozone (UVO). Irradiare i coverslip con UVO per 20 minuti per rimuovere qualsiasi fluorescenza di fondo. Una camera al plasma può essere utilizzata al posto di UVO.
- Utilizzando una pinzetta, trasferire i coverslip dal rack metallico utilizzato per il trattamento UVO a un diverso rack metallico utilizzato per la silanizzazione. Non utilizzare gli stessi rack per entrambi, in quanto causerà alti livelli di ossidazione quando viene applicato UVO. Pre-pulire i rack con acqua ed etanolo in modo che non rimangano sostanze chimiche residue da usi precedenti.
- Immergere il rack con i coverslips in un contenitore con acetone al 100% per 1 ora. Risciacquare il contenitore 3x con acqua di rubinetto e poi 3x con ddH2O per rimuovere tutto l'acetone.
- Immergere il rack con i coverslips in etanolo al 100% per 10 min. Risciacquare il contenitore 3x con acqua di rubinetto e poi 3x con ddH2O per rimuovere tutto l'etanolo.
- Immergere il rack con i coverslip 3x in ddH2O per 5 min ciascuno.
- Immergere il rack con i coverslips in 0,1 M KOH (50 mL di 1 M KOH in 450 mL di ddH2O) per 15 min. Risciacquare il contenitore 3x con acqua di rubinetto e poi 3x con ddH2O per rimuovere tutto il KOH.
- Immergere il rack con coverslips 3x in ddH2O per 5 min ciascuno.
- Asciugare all'aria il rack con i coperchi durante la notte in una cappa aspirante o in una cappa a flusso laminare.
- Dopo aver asciugato completamente il rack e i coverslip, immergerli per 5 minuti nel 2% di dimetildiclorosilano (DDS) preso in un contenitore diverso che viene utilizzato specificamente per il silano. Non lasciare che nulla che non sia asciutto entri in contatto con il silano.
- Immergere il rack e i coverslips 2x in un contenitore con etanolo al 100% per 5 minuti. Risciacquare il contenitore 3x con acqua di rubinetto e poi 3x con ddH2O.
- Immergere il rack e i coverslip 3x in ddH2O per 5 minuti ciascuno.
- Asciugare all'aria il rack con i coperchi durante la notte in una cappa aspirante o in una cappa a flusso laminare.
- Dopo questa fase finale di asciugatura, trasferire nuovamente i coverslip in scatole coverslip usando una pinzetta. Questi coverslip possono essere utilizzati nei prossimi 1-2 mesi. I vecchi coverslip inizieranno a perdere il loro rivestimento e dovrebbero essere scartati.
2. Preparazione della tubulina
NOTA: La tubulina acquistata si presenta come una polvere liofilizzata che non è etichettata o etichettata con fluorofori. La tubulina liofilizzata viene conservata in un congelatore a -80 °C. La seguente procedura viene utilizzata per miscelare la tubulina non etichettata con la tubulina etichettata in un rapporto che è buono per la visualizzazione.
- Rimuovere dal congelatore a -80 °C un'aliquota di tubulina non etichettata contenente 1 mg di tubulina liofilizzata in polvere e tenerla sul ghiaccio. Aggiungere 200 μL di PEM-80 freddo al tubo per portare la concentrazione di tubulina a 5 mg/mL. Tenerlo sul ghiaccio per 10 minuti per sciogliere tutto il liofilo.
- Rimuovere dal congelatore a -80 °C un'aliquota di tubulina marcata con rodamina contenente 20 μg di tubulina liofilizzata in polvere e tenerla sul ghiaccio. Aggiungere 4 μL di PEM-80 freddo al tubo per portare la concentrazione di tubulina a 5 mg/mL. Tenerlo sul ghiaccio per 10 minuti per sciogliere completamente il liofilato.
- Una volta sciolto, aggiungere 100 μL di soluzione di tubulina risospesa non marcata alla soluzione da 4 μL di tubulina marcata con rodamina. Pipetta 6x-7x molto lentamente da mescolare. Se gli aggregati sono visibili, centrifugare la tubulina solubilizzata per 10 minuti a 90.000 x g per rimuovere gli aggregati scartando il pellet e trattenendo il surnatante. Questa miscela di tubuline si tradurrà in ~ 4% di tubulina etichettata.
- Congelare a goccia i restanti 100 μL di tubulina non etichettata in azoto liquido (LN2) e conservarla a -80 °C per essere utilizzata per ulteriori miscele di tubuline.
- Prendere la miscela di tubuline etichettate e l'aliquota in sette tubi da 15 μL ciascuno. Ogni aliquota può essere utilizzata per una singola camera sperimentale. Congelare a goccia le aliquote rimanenti e conservarle a -80 °C per esperimenti futuri.
3. Purificazione MAP65
NOTA: MAP65 non è disponibile in commercio e, pertanto, deve essere purificato per questo lavoro. Il protocollo è stato elaborato in precedenza in diverse pubblicazioni23,29.
- Trasforma il plasmide MAP65 e il plasmide GFP-MAP65 in un ceppo batterico BL21 per l'espressione proteica.
- Far crescere i batteri BL21 a una densità ottica di 0,6-1 a 600 nm. Indurre la produzione di proteine utilizzando l'operatore lac e far crescere i batteri durante la notte.
- Pellet le colture e lisare i batteri.
- Raccogliere il lisato dopo la centrifugazione e incubarlo con perline che hanno uno ione nichel disponibile per legare il tag 6x-istidina.
- Eluire la proteina usando imidazolo e dissalarla.
- Congelare la proteina con azoto liquido e conservarla a -80 °C per l'uso entro 1 anno.
4. Assemblaggio delle camere di flusso
NOTA: Gli esperimenti vengono eseguiti in camere di flusso costituite da un vetrino e vetro di copertura silanizzato (Figura 3).
- Prendi un vetrino e puliscilo usando ddH2O, etanolo e ddH2O in ordine. Asciugare con una salvietta da laboratorio priva di lanugine tra ogni risciacquo.
- Utilizzare un pezzo di nastro biadesivo per creare un percorso di flusso. Usando le mani guantate, tagliare il nastro a ~ 25-30 mm di lunghezza. Dividere il nastro longitudinalmente per creare due strisce più sottili. Posizionare le due strisce di nastro sulla diapositiva con circa 5-8 mm tra di loro.
NOTA: poiché lo spessore del nastro è standardizzato a circa 80-100 μm, la larghezza del percorso tra i pezzi di nastro determinerà il volume nella camera. - Posizionate i coverslip silanizzati sopra il percorso del flusso. Sigillare la diapositiva e il coverslip sulle strisce di nastro biadesivo premendo delicatamente sulla regione del nastro con il retro di una penna. Assicurati di ottenere una buona tenuta su tutta l'area; il nastro dovrebbe passare da traslucido a trasparente quando il sigillo è fatto bene.
- Rimuovere il nastro extra sui bordi, lasciando solo 1 mm dall'ingresso della camera di flusso tagliando il nastro con una lama di rasoio.
- Etichettare la camera con informazioni sui parametri sperimentali, se necessario.
5. Esperimenti tactoidi
NOTA: Una volta generati tutti i reagenti e le forniture, possono essere utilizzati per nucleare e polimerizzare i tactoidi dei microtubuli nella camera di flusso.
- Raccogliere tutti i reagenti da utilizzare. Scongelali sul ghiaccio e conservali sul ghiaccio mentre lavori. Crea diverse camere di flusso per gli esperimenti.
- Utilizzare una camera di flusso per ogni esperimento. Rivestire le superfici della camera di flusso con una spazzola polimerica facendo scorrere 20 μL di tensioattivo copolimero a blocchi non ionici al 5% (Table of Materials) disciolto in PEM-80, con piccole gocce ad entrambe le estremità della camera per prevenire la formazione di bolle d'aria all'interno. Tenerlo in una camera umida (cioè una capsula di Petri con una salvietta da laboratorio bagnata senza lanugine) fino al momento dell'uso (almeno 5-7 minuti).
- In un tubo sterile, mescolare quanto segue per creare la miscela Tubulin-MAP: 9,5 μL di PEM-80; 4 μL di 10 mM GMPCPP; 4 μL del 5% Pluronic-F127; 1 μL di 1M DTT; 1 μL di glucosio; 2 μL di polietilenglicole (PEG); 12 μL di miscela di tubulina da 5 mg/mL (concentrazione finale di 13,6 μM) dalla fase 2.5.; e 5,5 μL di stock di lavoro di MAP65 dove il 10% è GFP-MAP65 per la visualizzazione. Tenere sul ghiaccio durante la miscelazione.
NOTA: si consiglia di utilizzare una pipetta a spostamento positivo per la gestione della soluzione PEG viscosa. Le pipette regolari possono essere utilizzate dopo aver tagliato la punta per ingrandire l'apertura; tuttavia, questo metodo è meno accurato. - Mescolare 5x-6x mediante pipettaggio.
- Poco prima di aggiungere alla camera, aggiungere 1 μL di una soluzione premiscelata di glucosio ossidasi (0,5 mg / mL) e catalasi (0,15 mg / mL) (Deoxy) nella miscela Tubulin-MAP e mescolare 7x-8x. Dividere il volume totale della soluzione (40 μL) in due porzioni da utilizzare in camere separate.
- Far scorrere la miscela Tubulin-MAP in camere. Poiché le camere contengono già un tensioattivo copolimero a blocchi non ionici, non è possibile aggiungere più liquido senza rimuovere il vecchio liquido. Per fare questo, utilizzare un pezzo di carta da filtro o una salvietta da laboratorio priva di lanugine all'altra estremità della camera per rimuovere il liquido tramite azione capillare.
- Una volta che il campione è completamente all'interno della camera, sigillare le due estremità della camera usando 5 minuti di resina epossidica e tenerlo a 37 °C per ~ 30 minuti per nucleare e far crescere i tactoidi dei microtubuli.
6. Microscopia a fluorescenza
- Utilizzare un microscopio a fluorescenza per l'imaging dei tactoidi.
NOTA: La microscopia a fluorescenza a riflessione interna totale o la microscopia confocale a disco rotante sono buone per rimuovere la fluorescenza di fondo dalla tubulina libera, ma i tactoidi sono visibili anche mediante epifluorescenza regolare e persino microscopia a luce trasmessa una volta formati, rendendo questa procedura accessibile senza attrezzature specializzate. - Utilizzare un obiettivo di apertura numerica (NA) di 1,2 NA o superiore con un ingrandimento di 60x o superiore per raccogliere abbastanza luce in fluorescenza. Questi obiettivi spesso richiedono l'immersione in ddH2O o olio.
- Registra immagini con la fotocamera CMOS o CCD. Utilizzare una dimensione effettiva dei pixel sulla fotocamera di 108 nm.
NOTA: la dimensione dei pixel dipende dalla fotocamera e dall'ingrandimento utilizzato, che in questo caso era 60x o 100x con un'apertura numerica elevata (1,2 o 1,49 NA). Ulteriori espansori di immagine possono essere utilizzati prima della fotocamera per ottenere la dimensione dei pixel necessaria. - Mantenere il campione a 37 °C utilizzando una camera ambientale impostata su questa temperatura. In alternativa, utilizzare altri riscaldatori a stadi, tra cui riscaldatori ad aria calda e collari a temperatura controllata obiettiva con acqua calda circolante.
- Utilizzare fonti di eccitazione corrette per la fluorescenza necessaria. Per la tubulina rodamina, utilizzare un laser a 561 nm con almeno 1 mW di potenza al campione e, per GFP-MAP65, utilizzare un laser a 488 nm con almeno 1 mW di potenza al campione.
NOTA: Se si utilizza la microscopia a epifluorescenza a campo largo, utilizzare un cubo di filtro rodaminico con eccitazione: 540 ± 12,5 nm, dicroico: 545 nm ± cut-off di 12,5 nm ed emissione: passaggio lungo 575 nm e un cubo filtrante GFP con eccitazione: 480 ± 15 nm, dicroico: cut-off di 505 nm ± 15 nm ed emissione: passaggio lungo 515 nm. - Scatta almeno 10 immagini di diverse aree per visualizzare oltre 100 tactoidi. Scatta immagini in entrambi i canali rosso e verde e salvale come immagini tiff a 16 bit per l'analisi. Assicurarsi che la potenza di illuminazione e i tempi di esposizione siano tali che la scala di intensità per la fotocamera non sia satura.
7. Recupero della fluorescenza dopo fotosbiancamento (FRAP)
NOTA: Per studiare la mobilità dei costituenti interni dei tactoidi, è stato utilizzato FRAP. FRAP funziona fotosbiancando una parte selezionata di tubulina marcata con rodamina e tactoide MAP65 con etichetta GFP e quindi osservando il recupero della fluorescenza con il tempo in quella regione. Il tasso di recupero dipende dal turnover della specie fotosbiancata. Questo tasso di turnover può dipendere dalla diffusione e dalle reazioni di legame. Per MAP65 che si lega ai tactoidi, è possibile stimare i tassi di cambio vincolanti. Il FRAP viene eseguito utilizzando un ulteriore sistema laser a 405 nm in grado di scansionare il laser in qualsiasi forma. Ci sono molte possibilità per eseguire FRAP, tra cui l'utilizzo della lampada trasmessa e dell'apertura per fotosbiancare un'area locale14.
- Selezionare un tactoide isolato nella camera per creare una regione di interesse (ROI) che copra parti dei tactoidi e la soluzione circostante.
- Utilizzare un microscopio con un ulteriore laser da 405 nm per FRAP per fotosbiancare contemporaneamente sia la tubulina che map65. In alternativa, una lampada luminosa può essere utilizzata attraverso l'arresto del campo dell'iride14. Regolare empiricamente l'intensità specifica dei sistemi di fotosbiancamento per evitare di danneggiare le proteine durante lo sbiancamento.
- Registra il tactoide come un filmato di serie temporali per 30-60 s prima del fotosbiancamento per ottenere informazioni sull'intensità prima della candeggina. Registrare entrambi i canali rosso e verde.
- Fotosbiancare il tactoide esponendo il ROI con i laser o la lampada per tutto il tempo necessario per la fotosbiancheria senza danneggiare il tactoide. Determina empiricamente l'intensità e il tempo.
- Continuare a registrare il filmato in entrambi i canali di colore per 5-10 minuti dopo il fotosbiancamento o fino a quando il recupero sembra aver raggiunto l'equilibrio.
- Ispezionare visivamente il canale GFP-MAP65 per il ripristino.
8. Analisi dei dati
NOTA: L'analisi quantitativa delle immagini dei tactoidi è stata eseguita per conoscere gli effetti dei cambiamenti ambientali imposti attraverso diversi agenti di affollamento, condizioni ioniche e l'aggiunta di altri fattori.
- Caratterizzazione della forma tactoide
- Quantificare la lunghezza e la larghezza dei tactoidi dalle immagini rosse e verdi scattate con la microscopia confocale.
- Apri le immagini usando FIJI/ImageJ.
- Se i dati grezzi vengono acquisiti a 16 bit, regolare la luminosità e il contrasto, se necessario. Selezionare Immagine > Regola > luminosità e contrasto per regolare l'immagine in modo da poter vedere chiaramente il tactoide. Regola la luminosità e il contrasto senza applicare l'impostazione in modo da non alterare accidentalmente i dati di intensità.
- Una volta che i tactoidi sono chiaramente visibili, selezionare buoni tactoidi da misurare (Figura 4Bi). Assicurarsi che i tactoidi siano chiaramente visibili senza sovrapposizioni con altri tactoidi o aggregati e non siano curvi o piegati per poter utilizzare strumenti di misurazione rettilinei.
- Quindi, controlla che sia impostata la dimensione dei pixel corretta per le immagini. Le immagini del microscopio vengono fornite con metadati sulla dimensione dei pixel. Quando si utilizza una fotocamera diversa che non dispone di metadati o sistemi di espansione delle immagini esterni in grado di modificare la dimensione effettiva dei pixel prevista, regolare manualmente la dimensione dei pixel. In FIJI/ImageJ, vai a Analizza > Imposta scala per impostare la conversione dei pixel corretta.
- Utilizzando lo strumento Linea retta dalla barra degli strumenti in FIJI/ImageJ, fare clic su un'estremità del tactoide e trascinare il cursore sull'altra estremità del tactoide (Figura 4Bii). Una volta selezionato il ROI della linea, selezionare Analizza > Misura per misurare la lunghezza. Se la lunghezza non viene misurata per impostazione predefinita, assicurarsi di impostare la misurazione in modo da includere la lunghezza nella finestra di dialogo Analizza > Imposta misure .
NOTA: in genere, quando si esegue la misurazione con lo strumento Linea retta , vengono forniti la lunghezza e l'angolo della linea disegnata. Ad esempio, la Figura 4Bii mostra una linea retta disegnata sul lato del tactoide per rendere visibile quest'ultimo ma effettuare la misurazione direttamente sul tactoide. - Dopo aver effettuato la misurazione, utilizzare lo strumento testo nella barra degli strumenti per etichettare il tactoide. Creare una casella di testo, aggiungere un'etichetta numerica e selezionare Modifica > Disegna per fissare l'etichetta nell'immagine. Salvare l'immagine come file ROI separato.
NOTA: l'etichettatura e il salvataggio di questo file consente allo sperimentatore di sapere quale misurazione corrisponde a quale tactoide dai dati grezzi. Assicurati di misurare ogni tactoide una volta. - Dopo aver misurato i tactoidi per l'intera immagine, salvare i dati nella finestra Risultati in un file di testo delimitato da virgole o tabulazioni (utilizzando File > Salva con nome) e aprire i dati in un programma di fogli di calcolo per analizzare i dati in numeri. Raccogli tutti i dati insieme (dati immagine grezzi, immagine ROI e file di testo dei risultati) in una cartella con una convenzione di denominazione appropriata per mantenere tutto organizzato.
NOTA: Sebbene le misurazioni della lunghezza del tactoide siano eseguite a mano, dato che le larghezze dei tactoidi sono strette, è meglio utilizzare un metodo diverso per misurare la larghezza del tactoide (vedi sotto) per ridurre l'errore di misurazione. - Utilizzando ImageJ/FIJI, disegnate una regione di linee utilizzando lo strumento Linea retta . Disegnare la linea come una bisettrice perpendicolare all'asse lungo tactoide (Figura 4Bii).
- Selezionate Analizza profilo > plottaggio per creare il profilo di intensità della bisettrice lineare (Figura 4Biii). Apparirà un grafico. Per recuperare e salvare i dati dal plottaggio, selezionare il pulsante Elenco in basso a sinistra; questo genera l'elenco dei file di testo dei dati di intensità lungo la lunghezza della linea disegnata. Salvare il file di testo come file di .csv o .txt.
- Apri il file di testo in un programma adatto come MatLab, Python (sciPy) o altri programmi. Adatta i dati di intensità con una funzione gaussiana della forma:
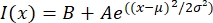 , dove I(x) è il valore in scala di grigi lungo la lunghezza, x; B è il livello di fondo; A è l'ampiezza del gaussiano; μ è la media o il centro del gaussiano; e σ è la deviazione standard del gaussiano.
, dove I(x) è il valore in scala di grigi lungo la lunghezza, x; B è il livello di fondo; A è l'ampiezza del gaussiano; μ è la media o il centro del gaussiano; e σ è la deviazione standard del gaussiano. - Il rapporto 2σ come larghezza del tactoide. Stimare l'intensità dei microtubuli nel tactoide calcolando l'area sotto il gaussiano (escluso lo sfondo).
NOTA: se le immagini rientrano nell'intervallo di intensità lineare della fotocamera e vengono scattate con lo stesso tempo di esposizione e intensità di eccitazione, è possibile confrontare le intensità integrate per stimare il numero relativo di microtubuli nel tactoide.
- Analisi FRAP
NOTA: Esperimenti per testare la mobilità dei microtubuli e del MAP65 hanno utilizzato FRAP per registrare il fotosbiancamento specifico e il recupero dell'intensità dovuta al movimento molecolare (Figura 5A). I dati sono stati quantificati dai dati delle serie temporali dell'immagine utilizzando ImageJ / FIJI.- Usa ImageJ/FIJI per aprire i dati del filmato.
- Registrare gli stack (dati delle serie temporali) nel tempo per rimuovere la deriva. Utilizzare il plugin StackReg insieme al plugin ausiliario TurboReg ; per istruzioni sull'utilizzo dei plugin, fare riferimento ai collegamenti web indicati nella Tabella dei materiali. Selezionare la traduzione per spostare la posizione dei fotogrammi e quindi registrare le immagini.
- Una volta registrate le immagini per rimuovere la deriva, ruotare l'immagine in modo che il tactoide sia verticale o orizzontale nella cornice selezionando Immagine > Trasforma > Ruota. Selezionate l'angolo di rotazione e utilizzate Anteprima per determinare se il tactoide è ruotato abbastanza. Quando l'anteprima mostra che il tactoide è verticale o orizzontale, selezionare OK per ruotare tutte le immagini del filmato.
- Utilizzate lo strumento di selezione Rettangolo nella barra degli strumenti per creare una sezione rettangolare sulla regione fotosbiancata del tactoide. Registra l'intensità integrata dell'area ROI per ogni fotogramma utilizzando Image > Stacks > Measure Stack. Impostare il tipo di misura su Densità integrata utilizzando Analizza > Imposta misurazioni. Salvare i dati di intensità analizzati visualizzati nella finestra Risultati come file di testo in formato .csv o .txt selezionando File > Salva con nome.
NOTA: la Figura 5B mostra un esempio dei dati grezzi di intensità a 16 bit misurati per i canali microtubulo e GFP-MAP65 nella regione della candeggina. - Poiché l'intensità complessiva delle immagini svanirà nel tempo a livello globale a causa del fotosbiancamento causato dall'imaging, questo fotosbiancamento globale deve essere corretto. A tale scopo, utilizzare la stessa dimensione del ROI (passaggio 8.2.4) e spostarla in un'area sullo sfondo dell'immagine in cui non sono visibili microtubuli o MAP65. Misurare l'intensità integrata dello stack come descritto nel passaggio 8.2.4. Salvare i risultati come secondo file di testo.
NOTA: la Figura 5B mostra un esempio dei dati grezzi sull'intensità a 16 bit misurati per i canali microtubulo e GFP-MAP65 nella regione di sfondo. - Per correggere lo sbiadimento dello sfondo, dividere l'intensità del segnale sul tactoide per l'intensità di sfondo per lo stesso punto temporale. Calcola Icorretto (t) come:
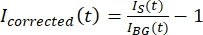 , dove IS (t) (il segnale) è la misurazione effettuata sulla regione sbiancata e IBG (t) (il rumore) è la misurazione effettuata sulla regione di fondo (Figura 5C). Questo calcola il rapporto segnale-rumore per ogni fotogramma e sottrae anche il rumore.
, dove IS (t) (il segnale) è la misurazione effettuata sulla regione sbiancata e IBG (t) (il rumore) è la misurazione effettuata sulla regione di fondo (Figura 5C). Questo calcola il rapporto segnale-rumore per ogni fotogramma e sottrae anche il rumore. - Quindi, ridimensionare i dati per variare tra zero e uno usando
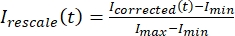 , dove Imin e Imax denotano, rispettivamente, il minimo globale e il massimo dei dati corretti I per tutto il tempo (Figura 5C).
, dove Imin e Imax denotano, rispettivamente, il minimo globale e il massimo dei dati corretti I per tutto il tempo (Figura 5C). - Adatta questi dati a un esponenziale decadente della forma:
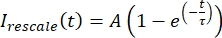 , dove A è l'ampiezza del recupero e τ è la scala temporale del recupero (Figura 5C).
, dove A è l'ampiezza del recupero e τ è la scala temporale del recupero (Figura 5C).
Con solo un piccolo numero di componenti, dimeri di tubulina e reticolatori di microtubuli, possono formarsi tactoidi di microtubuli (Figura 2A). Sebbene questo protocollo descriva l'incubazione per nucleare e far crescere microtubuli in un incubatore, la nucleazione e la crescita possono essere osservate direttamente al microscopio (che sono complete entro 30 minuti) (Figura 2B). La concentrazione di tubulina è mantenuta costante a 13,6 μm e il legame MAP65-MT al 10%.
La Figura 4 rappresenta i dati riusciti. I tactoidi dovrebbero essere visibili sia con un laser a 561 nm nel canale della tubulina che con 488 nm nel canale MAP65, che si sovrappongono perfettamente l'uno con l'altro (Figura 4A). Un mistero del sistema è stato che la larghezza dei tactoidi non sembra variare sotto una varietà di cambiamenti sperimentali, tra cui la modifica delle lunghezze dei microtubuli, la concentrazione di MAP65 e gli agenti di affollamento (Figura 4B) 22,28. La lunghezza è molto più variabile e dipende sia dalla lunghezza dei microtubuli che dalla concentrazione di MAP65 (Figura 4B)22,28.
Durante l'esecuzione di FRAP, è stato osservato che il segnale MAP65 si riprende, ma il segnale dei microtubuli non si riprende (Figura 5). Il recupero in FRAP è dovuto alla mobilità e al movimento degli oggetti etichettati e fotosbiancati. Nel caso del MAP65, le molecole oscurate si dissociano e si allontanano dal microtubulo e quelle nuove si spostano nella regione (Figura 5). Il legame MAP65 è in equilibrio, quindi il tasso di legame e svincolo è uguale (misurato in molecole al secondo). Per i microtubuli, non è stato osservato alcun recupero che implica che i microtubuli non sono in grado di lasciare il tactoide (Figura 5A, Bi, C). Inoltre, non è stata osservata alcuna diffusione della regione oscurata, suggerendo che i microtubuli sono localmente immobili e non un fluido all'interno della forma tactoide.
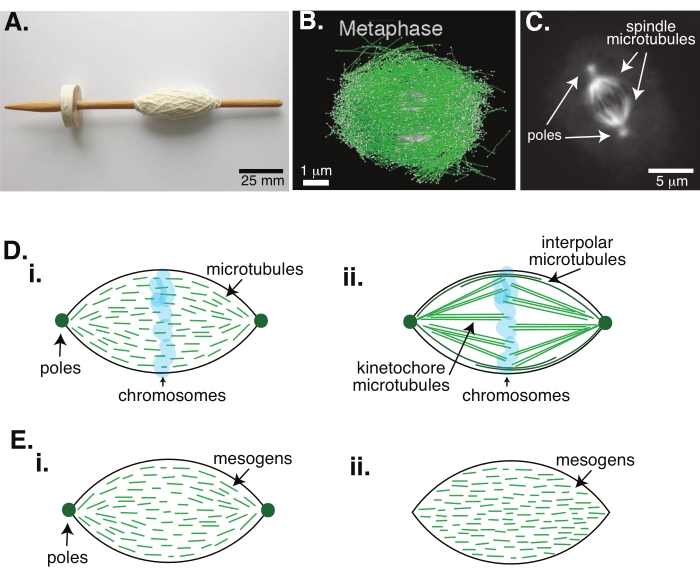
Figura 1: Diversi modelli di formazione del mandrino. Un fuso mitotico è una macchina fatta di microtubuli e delle loro proteine ed enzimi associati che allinea e separa i cromosomi nelle due nuove cellule figlie durante la divisione cellulare. (A) Immagine di una replica del fuso a goccia dell'alto Medioevo con filati pregiati provenienti dai Paesi Bassi. Questa figura è stata modificata da un'immagine Wikimedia di Peter van der Sluijs30. (B) Ricostruzione tridimensionale di microtubuli in diversi stadi della meiosi wild-type II. I microtubuli sono mostrati in verde e i cromosomi sono mostrati in grigio. Barra della scala = 1 μm. Questa cifra è stata modificata da Lantzsch et al.31. (C) Immagine al microscopio dei microtubuli in un fuso mitotico di una cellula Sf9 in divisione. I poli del fuso e i microtubuli del fuso sono etichettati con una proteina fluorescente verde. Barra di scala = 5 μm. Questa cifra è stata modificata da Advani et al.32. (D) Diversi modelli di come sono organizzati i microtubuli del fuso mitotico e meiotico. (i) Precedentemente osservati per i fusi meiotici creati da estratti di uova di Xenopus, i microtubuli (verdi) sono stati dedotti come corti e dinamici in tutto il fuso. Questo è simile a un'organizzazione tactoide bipolare all'interno di un cristallo liquido. (ii) Il modello canonico per l'organizzazione dei microtubuli all'interno di un fuso mitotico ha due tipi di microtubuli: microtubuli interpolari o polari (verde scuro) che si reticolano nella zona centrale attorno ai cromosomi e microtubuli di chinetocore (verde chiaro) che sono raggruppati e allungati dal polo al chinetocore per spingere e tirare i cromosomi. In tutte le immagini, i cromosomi sono mostrati in blu trasparente e i poli del fuso sono rappresentati in verde scuro. (E) Schemi di mesogeni (linee verdi) in un tactoide a cristalli liquidi per (i) tactoidi bipolari e (ii) omogenei. I tactoidi bipolari hanno due poli all'estremità del tactoide e i mesogeni si riorientano per puntare a quei poli. I tactoidi omogenei hanno poli all'infinito e i mesogeni non cambiano orientamento lungo la lunghezza del tactoide. Fare clic qui per visualizzare una versione più grande di questa figura.
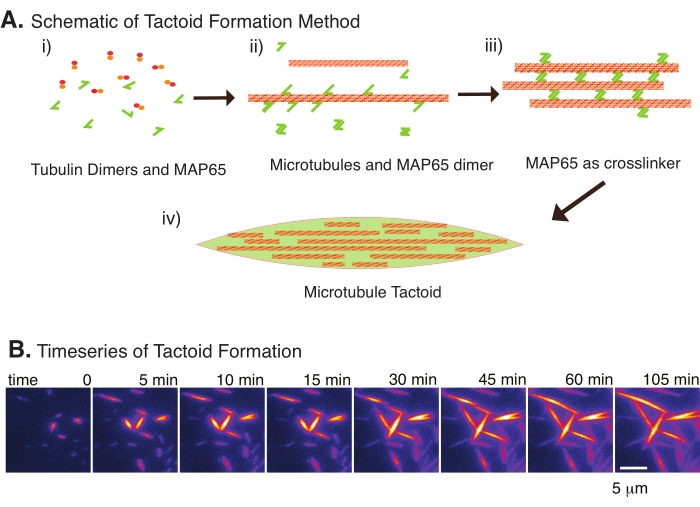
Figura 2: Condensazione dei microtubuli. (A) I microtubuli possono essere raggruppati e reticolati con una varietà di metodi, tra cui specie ioniche, forze di esaurimento causate da agenti di affollamento e specifici reticolanti di microtubuli, come MAP65. i) I dimeri di tubulina e le proteine MAP65 sono miscelati per nucleare e far crescere microtubuli. (ii) I microtubuli nucleano e crescono dalla tubulina, e MAP65 si lega immediatamente ai microtubuli, un altro monomero MAP65 o entrambi e causa il raggruppamento. iii) I microtubuli nei fasci reticolati nucleano e crescono. (iv) La configurazione finale è un tactoide microtubulico simile a un mandrino. (B) Serie temporale di tactoidi microtubulici che nucleano e crescono oltre 105 min. Barra di scala = 5 μm. Figura adattata da Edozie et al.22. Fare clic qui per visualizzare una versione più grande di questa figura.
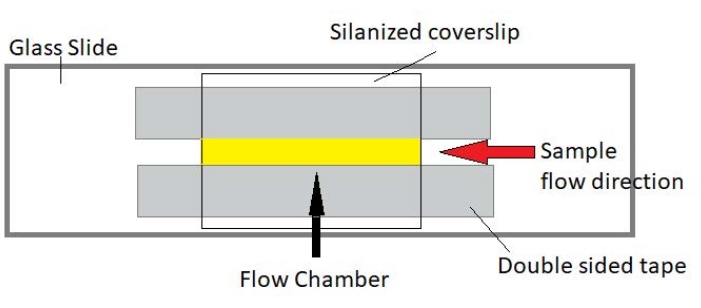
Figura 3: Assemblaggio della camera di flusso. La camera di flusso è realizzata utilizzando un vetrino, vetro di copertura silanizzato e nastro biadesivo permanente. La regione evidenziata in giallo è il percorso del flusso in cui il campione viene fluito e osservato. Il volume della camera di flusso è di ~ 20 μL. La resina epossidica è stata utilizzata per sigillare le estremità della camera per evitare che il campione evaporasse durante l'imaging a lungo termine per diverse ore. Fare clic qui per visualizzare una versione più grande di questa figura.
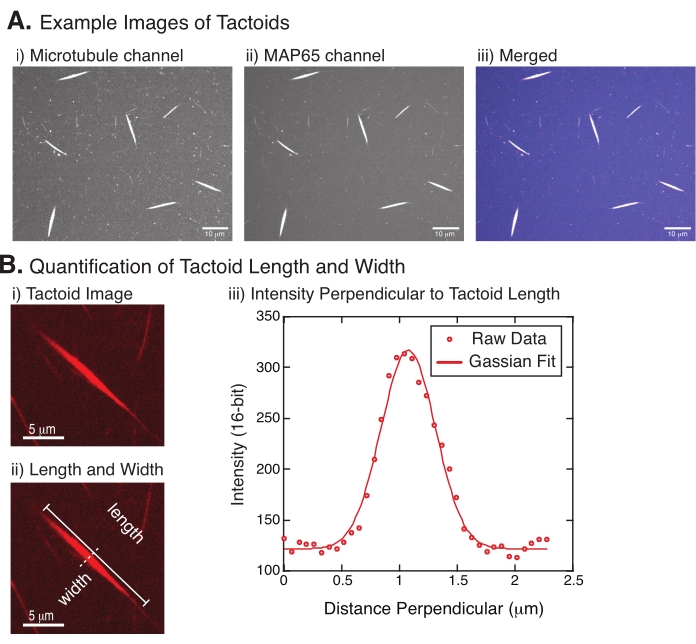
Figura 4: Immagini tactoidi e analisi di lunghezza e larghezza. (A) Dati di esempio di tactoidi formati come descritto e ripresi utilizzando la confocale del disco rotante che mostra (i) l'imaging del canale dei microtubuli tubulina marcata con rodamina utilizzando un laser a 561 nm, (ii) il canale GFP-MAP65 che visualizza la GFP utilizzando un laser a 488 nm e (iii) l'immagine di sovrapposizione unita dal canale dei microtubuli (magenta) e dal canale GFP-MAP65 (ciano). Le regioni di sovrapposizione vengono visualizzate come bianche e dimostrano che i microtubuli e MAP65 si colocalizzano esattamente. Barra di scala = 10 μm per tutte le immagini in (A). (B) Quantificazione della lunghezza e della larghezza dei tactoidi. (i) Immagine di un tactoide da analizzare senza etichette. Barra della scala = 5 μm. (ii) Stessa immagine del punto (i), dove sono indicate le misure di lunghezza (linea continua con tappi di linea) e larghezza (linea tratteggiata). Barra della scala = 5 μm. (iii) La larghezza è stata misurata prendendo il profilo di intensità attraverso il tactoide alla bisettrice perpendicolare (linea tratteggiata) indicata in (ii). Il profilo di intensità era adatto a una funzione gaussiana per rivelare l'ampiezza e la larghezza del tactoide. Fare clic qui per visualizzare una versione più grande di questa figura.
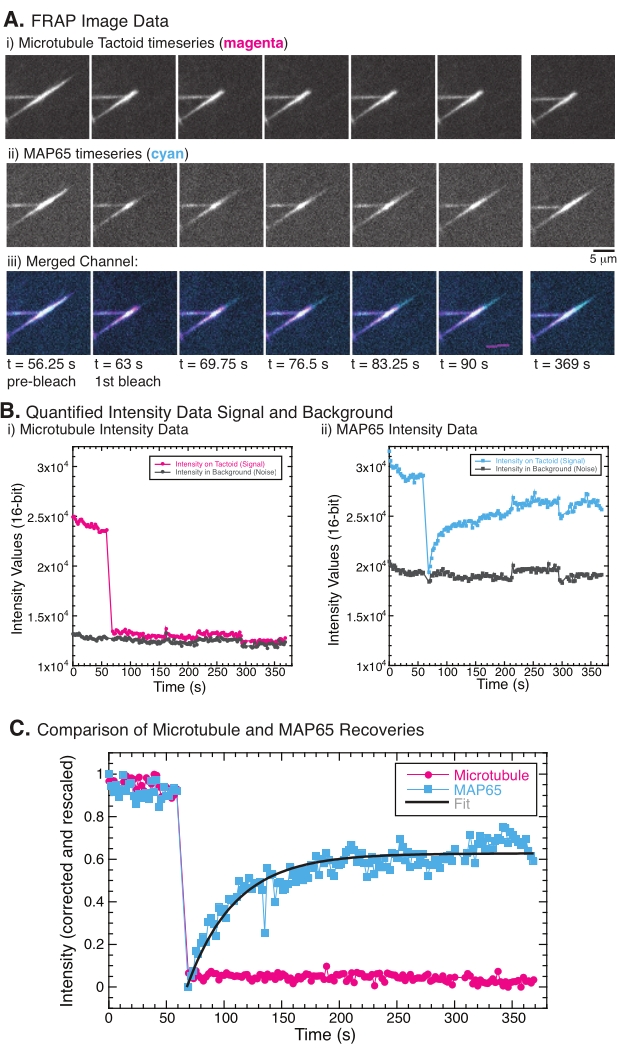
Figura 5: Dati FRAP rappresentativi e analisi. (A) Dati delle serie temporali al microscopio di (i) microtubuli tactoidi e (ii) GFP-MAP65 e (iii) immagine sovrapposta di entrambi i canali con microtubuli in magenta e GFP-MAP65 in ciano che sono stati fotosbiancati al tempo 63 s e osservati per altri 5 minuti. (B) Intensità quantificata del (i) ) canale microtubulico nella regione sbiancata (cerchi magenta) e lo sfondo (occhiaie grigio scuro) e (ii) canale GFP-MAP65 nella regione sbiancata (quadrati ciano) e lo sfondo (quadrati grigio scuro). (C) I dati sono stati corretti per il rumore di fondo e riscalati per il canale dei microtubuli (cerchi magenta) e il canale GFP-MAP65 (quadrati ciano). I microtubuli non recuperano, ma il GFP-MAP65 lo fa e può essere adattato (linea grigio scuro) a un crescente decadimento esponenziale per trovare l'ampiezza e la scala temporale del recupero. Fare clic qui per visualizzare una versione più grande di questa figura.
I metodi qui descritti sono stati utilizzati in diversi articoli per creare tactoidi microtubuli (Figura 2)22,28. Questi esperimenti sono biologicamente rilevanti per aiutare a scoprire i principi organizzativi che controllano la forma e la stabilità del fuso mitotico o meiotico nella maggior parte dei tipi di cellule. Inoltre, i microtubuli sono mesogeni a cristalli liquidi modello che possono aiutare a saperne di più su come i cristalli liquidi nucleano e fanno crescere fasi nematiche da fasi isotrope.
La procedura qui descritta ha diversi vantaggi per esplorare l'auto-organizzazione dei microtubuli. In primo luogo, è altamente riproducibile, essendo stato eseguito in laboratorio da molti studenti, compresi gli studenti delle scuole superiori, con poca preconoscenza o formazione prima di iniziare in laboratorio. I tactoidi sono birifrangenti22, consentendo loro di essere visualizzati in luce trasmessa oltre alla microscopia a fluorescenza, rendendo questo metodo accessibile a molti laboratori e questa procedura sperimentale adattabile a scopi educativi, oltre alla ricerca di fascia alta. Infine, questo processo apre strade per continuare a comprendere e sondare i sistemi biologici in un approccio riduzionista e ridotto, consentendo di capire come ogni condizione aggiuntiva, proteina o additivo può alterare l'auto-organizzazione dei tactoidi e, forse, in definitiva, del fuso. Gli obiettivi per una migliore biomimetica includono attività, fluidità e ordinamento della polarità del filamento.
Ci possono essere diversi fattori che influenzano l'esperimento dando risultati inaspettati. Ad esempio, se i tactoidi non si formano (Figura 2) ma si osservano modelli simili a ventagli, è probabile che il MAP65 non sia presente o non si leghi ai microtubuli22,28. Questo dovrebbe essere evidente anche nel canale di fluorescenza MAP65 perché il GFP-MAP65 non sarà legato ai microtubuli.
Se i tactoidi non si formano e lo sfondo appare come macchie sul vetro, ciò potrebbe essere dovuto al rivestimento superficiale. Una volta eseguita, la silanizzazione dura solo 1 mese su coverslips. Quando svanisce, la tubulina sarà in grado di legarsi alla superficie esposta in modo non specifico. Questa associazione si verificherà in modelli dispari.
Se i tactoidi non si formano e la tubulina viene osservata in aggregati di varie forme e dimensioni, ciò potrebbe essere dovuto alla tubulina di scarsa qualità. La tubulina può essere centrifugata per rimuovere gli aggregati iniziali che possono guidare questa aggregazione fuori percorso invece della polimerizzazione dei microtubuli. Se la superficie è legata alla tubulina, può anche esaurire la tubulina nella soluzione. Basse concentrazioni di tubulina, al di sotto della concentrazione critica per la polimerizzazione dei microtubuli, possono provocare aggregati.
Negli esperimenti FRAP, se il canale MAP65 non mostra alcun recupero (Figura 5), è possibile che il fotosbiancamento stesse fotodannando i microtubuli. Il fotodanno provoca la distruzione localizzata dei filamenti. Questo può essere verificato mediante esame nel canale trasmesso. I tactoidi dei microtubuli sono visibili nel canale trasmesso attraverso un alto indice di mancata corrispondenza della rifrazione con l'acqua circostante. Il fotodanno indotto dalla luce apparirà come un segno di bruciatura o perdita di contrasto nell'imaging della luce trasmessa nella posizione del ROI sottoposto a fotosbiancamento. Se ciò accade, la potenza del laser o della luce deve essere ridotta per inibire il fotodanno delle proteine.
Ci sono state diverse sfide incontrate in questa procedura e approccio. Un problema è che le misure di lunghezza vengono attualmente eseguite a mano facendo clic sull'immagine. Questo metodo, sebbene semplice, può causare un'elevata incertezza. La misurazione della larghezza che utilizza la sezione trasversale e l'adattamento a un gaussiano è un metodo migliore per quantificare la dimensione. Un metodo simile potrebbe essere impiegato per la lunghezza. Un secondo problema è che, a volte, i tactoidi, perché sono così lunghi e sottili, possono piegarsi. Ciò rende più difficile quantificare la lunghezza. La lunghezza del contorno può essere quantificata utilizzando una linea segmentata, ma c'è un'incertezza aggiuntiva ogni volta che viene aggiunto un segmento.
Da un punto di vista scientifico, questo approccio ha alcune altre sfide per il suo uso come modello per cristalli liquidi o mandrini. La prima sfida è stata la forma lunga e sottile dei tactoidi che i microtubuli creano (Figura 3 e Figura 4). Come notato nelle precedenti pubblicazioni22, i tactoidi dei microtubuli sono tactoidi omogenei, non bipolari. Ciò significa che i microtubuli che compongono la forma non si riorientano per puntare verso le punte della struttura. Invece, tutti i microtubuli sono paralleli all'asse lungo e i "poli" si trovano all'infinito. Questo è molto diverso dai tactoidi osservati per i cristalli liquidi molecolari o anche per l'actina o il DNA che possono anche agire come mesogeni a cristalli liquidi. In questi altri sistemi, i tactoidi sono bipolari e, se visti in polarizzatori incrociati, mostrano i segni rivelatori di riorientamento delle aste.
Una seconda grande sfida in questo sistema è che i microtubuli sono immobili all'interno del tactoide. Questo è chiaro dagli esperimenti e dalle analisi FRAP poiché il recupero dei microtubuli è molto basso. La loro natura solida rende i tactoidi dei microtubuli meno preziosi degli analoghi a cristalli liquidi su larga scala. La fase nematica di un cristallo liquido dovrebbe avere proprietà sia liquide (fluide) che cristalline (organizzate). Sebbene la forma sembri giusta per il mandrino, l'immobilità rende il sistema meno eccitante come un mandrino mitotico modello. D'altra parte, questo numero offre l'opportunità di indagare su come è possibile modificare gli esperimenti per creare più fluidità nel sistema.
Queste sfide scientifiche offrono interessanti opportunità che consentiranno nuove conoscenze sul sistema. Per rendere i tactoidi dei microtubuli più bipolari, si potrebbero usare microtubuli più corti. C'è un'ulteriore sfida, tuttavia, dal momento che i microtubuli non hanno molte proteine di tappatura ben caratterizzate per controllare la lunghezza come fa l'actina. L'uso della nucleazione e della crescita richiede l'uso di concentrazioni molto elevate di tubulina e GMPCPP per produrre microtubuli corti. L'alta concentrazione di tubulina si traduce in un numero maggiore di filamenti nel sistema, il che rende più difficile separare i tactoidi l'uno dall'altro. L'aggiunta di nuovi tappi di microtubuli, come DARPin33, può aiutare in questa situazione. Il secondo problema dell'immobilità dei microtubuli potrebbe essere mitigato dall'aggiunta di proteine motorie, come la kinesin-534, che sono tetrameri di motori utilizzati nella mitosi. In alternativa, i dimeri artificiali di kinesin-1 dimerica potrebbero essere usati15.
Un altro modo per aggiungere più fluidità sarebbe quello di consentire ai microtubuli di eseguire la loro instabilità dinamica, la crescita e il restringimento dei microtubuli. Attualmente, i microtubuli che vengono seminati con filamenti GMPCPP stabili e quindi subiscono instabilità dinamica sono molto più lunghi di quanto desiderato per formare un fuso o un tactoide, il che si tradurrebbe in organizzazioni molto lunghe come ventilatori o fasci. Quindi, l'aggiunta di instabilità dinamica dei microtubuli dovrebbe essere fatta con attenzione per preservare la forma tattile. L'aggiunta di proteine ed enzimi associati in grado di controllare la lunghezza può mitigare questo problema. Ad esempio, sarebbero probabilmente necessarie kinesine depolimerizzanti, come la kinesin-1335, o enzimi di taglio, come lakatanina 36. Questi esperimenti sono complessi e difficili, anche se sarebbero molto perspicaci, indipendentemente da ciò che i risultati rivelano. Qualunque sia la direzione degli esperimenti futuri, la piattaforma sviluppata qui per la creazione di tactoidi microtubuli può esporre nuove informazioni sulla base fisica dell'organizzazione dei microtubuli.
Gli autori dichiarano di non avere interessi finanziari concorrenti.
Gli autori desiderano ringraziare tutti i membri del Ross Lab dell'estate 2021, in particolare K. Alice Lindsay, per il loro aiuto. Questo lavoro è stato supportato da una sovvenzione di NSF BIO-2134215 che ha supportato S. Sahu, N. Goodbee, H.B. Lee e J.L. Ross. Una sovvenzione della KECK Foundation (Rae Anderson, USD, lead PI) ha parzialmente sostenuto R. Branch e P. Chauhan
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved