
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
微小管タクトイドの自己組織化
この記事では、植物ベースの微小管架橋剤であるMAP65およびPEGをクラウディング剤として使用して、タクトイドの形状の微小管アセンブリを形成するためのプロトコルを提示する。
細胞骨格は、細胞内の主要な内部組織化および再編成を担うが、すべて変更を指示するマネージャーがいない。これは、有糸分裂または減数分裂の間に特に当てはまり、微小管が細胞分裂中に紡錘体を形成する。スピンドルは、細胞分裂中に遺伝物質を分離するために使用される機械です。 in vitroで自己組織化されたスピンドルを作るために、我々は最近、微小管関連タンパク質とクラウディング剤の最小セットで微小管をスピンドル状の集合体に再構成する技術を開発しました。具体的には、植物由来の逆平行微小管架橋剤であるMAP65、酵母由来のAse1および哺乳動物由来のPRC1のホモログを用いた。この架橋剤は、微小管を長くて細い紡錘形の微小管自己組織化集合体に自己組織化する。これらのアセンブリも液晶タクトイドに似ており、微小管はメソスケールのメソゲンとして使用することができる。ここでは、これらの微小管タクトイドを作成するためのプロトコル、ならびに蛍光顕微鏡法を用いたアセンブリの形状およびフォトブリーチング後の蛍光回収を用いた構成成分の移動度を特徴付けるためのプロトコルが提示される。
有糸分裂による細胞分裂は、生命を維持するための最も重要な生物学的プロセスの1つです。チューブリン二量体で構成される微小管フィラメントは、このプロセスに不可欠な構造要素である。染色体が細胞中心に整列している中期に作られた一過性機構は、その形状から有糸分裂紡錘と呼ばれ、糸で覆われた織機の紡錘体のようなものです(図1A)。微小管が中期に使用され、凝縮した染色体を細胞の中心に押し込んだり引いたりして、それらを整列させ、分裂後期に引き離す微小管に接続することは、多くの生物にわたって十分に確立されています(図1B、C)。紡錘体は減数分裂(図1B)と有糸分裂(図1C)の両方で形成され、糸のように中心軸の周りに巻き付けられていないが界面に平行に走る多くの重なり合う微小管から形成される。これらの微小管ベースの構造を作成するには、架橋する関連タンパク質と、染色体1を押したり引いたりするのに役立つモーターとして働くことができる関連酵素が必要です。
減数分裂スピンドルの研究は、微小管が短く、動的であり、架橋アレイ2、3、4、5、6において重なり合っていることを示している(図1Di)。これらの短い微小管の物理的組織のために、減数分裂紡錘体は液晶タクトイドに似ています(図1E)。実際、スピンドルは、液晶タクトイド5から予想されるように、合体して結合することが示されている。
1960年代にさかのぼる多くの研究は、固定、連続切片、および電子顕微鏡を用いて、有糸分裂紡錘体7,8,9,10の内側に2種類の微小管があることを確認しました。最初のタイプはキネトコレ微小管と呼ばれ、スピンドルポールをキネトコレに接続します。第2のタイプは、染色体を過ぎて成長し、中間ゾーンで重なり合う極性間または極性微小管と呼ばれる(図1Dii)8、9、10。第3のタイプは、紡錘体の外側にあり、極を細胞縁に接続するアストラル微小管と呼ばれる。これらの微小管組織は、現在の議論の範囲外である。オーグミン6とガンマチューブリン環複合体の相互作用が微小管の核生成中心に影響を与え、図1Dのようなより短い微小管を有する有糸分裂紡錘体をもたらす最近の研究がある。
微小管は幅よりも長く、高いアスペクト比と高い剛性を持つため、液晶分子のスケールアップ版のようなものです。ソフトマター物理学では、原子や分子は、核生成や結晶の融解を含む相転移の物理的メカニズムを推測するために、最小限の相互作用を使用して近似されています11。同様に、微小管は、液晶分子のスケールアップされたバージョンであるメソスケールの物体であり、等方性相からのネマチック相の核生成および成長を含む、液晶ダイナミクスの物理学への洞察を与える。また、上述したように、減数分裂スピンドルは、液晶タクトイドのような性質、液晶分子3、4、5の等方性状態から核形成して成長するネマチック状態を示す。タクトイドの場合、核生成と成長は他の結晶の核生成と成長に似ています(つまり、比較的高濃度のメソゲン[液晶を形成する分子]が必要です)。タクトイドのユニークな「スピンドル」形状は、ネマチック相に整列する液晶メソゲンの局所的な配向から来ています(図1E)。分子は非常に非対称であるため、丸みを帯びた結晶を形成することはできません。微小管の性質を考えると、局所的な高濃度の微小管から作られた有糸分裂紡錘体機構も、それがタクトイドまたは紡錘と呼ばれるかどうかにかかわらず、同じ形状であることはおそらく驚くべきことではない。タクトイドは、先細りの端に極を持つ双極性(図1Ei)または均質で、極が無限大に効果的にある(図1Eii)ことができます。
紡錘体形成の重要性に鑑み、イオン種12、13、枯渇相互作用を生じるクラウディング剤14、15、および特定の微小管架橋タンパク質13、16、17、18、19を介して微小管縮合を束にすることを実証することにより、インビトロでの自己組織化紡錘体形成に向けた努力が進められており、驚くべきことに、これらの薬剤はすべて微小管の局所濃度を増加させるために働くが、それらはしばしば長い微小管束をもたらすが、タクトイドは生じない。これらの束が長い理由の1つは、それらを構成する微小管も長いことであると考えられる。より短い微小管を用いた最近の研究では、末尾15で先細りになっていないより長い束も報告された。この場合、バンドルは、バンドルの伸長を引き起こすモータータンパク質と一緒に保持され、それによって、それらをより長くする。ここで説明するように、非伸張架橋剤を備えた短い微小管は、テーパー状の紡錘形のアセンブリに必要です。
最近、我々は、核形成安定微小管22の存在下で、反平行架橋剤MAP65を用いて微小管タクトイドの作成を可能にする技術を開発した。微小管は短くする必要がありましたが、微小管の長さの既知のレギュレーターで、微小管を動的不安定性またはエンドツーエンドのアニーリングに対してキャップできるものはほとんどありません。代わりに、GMPCPPを使用して、成長後のフィラメントを核形成および安定化させた。これにより、タクトイドに自己組織化できる高密度の短い微小管を作成することができました。これらのタクトイドは、複屈折下で見ると均質であった。短い微小管に加えて、特定の反平行架橋剤MAP65がタクトイドを形成するために採用された(図2)。MAP65は、有糸分裂架橋剤23のPRC1/Ase1ファミリーにおける植物微小管関連タンパク質である。MAP65は二量体として存在し、微小管24と同様にそれ自体に結合する強い親和性を有する。アクチンフィラメント25,26,27で観察された減数分裂紡錘体およびタクトイドは双極性であり、液晶の液体様特性を有するが、微小管タクトイドは固体状であることが観察されている22,28。
ここでは、微小管タクトイドを作成し、蛍光ベースの技術を使用してアセンブリの形状と構成成分の移動性を特徴付けるためのプロトコルが提示されています。
注:特に明記されていない限り、実験の一部は、適切な保護具(手袋)を着用したまま、ラボベンチで行うことができます。
1. カバースリップシラナイゼーション
メモ:カバースリップは、これらの実験で使用したポリマーブラシコーティングで使用するためにシラン化する必要があります。これは、疎水性中央ブロックを有するブロックコポリマーが結合してポリマーブラシを作成することを可能にする疎水性シラン化処理である。次の手順は、手袋を着用している間に有毒な蒸気にさらされるのを防ぐために、ヒュームフードで実行する必要があります。ジメチルジクロロルシランは非常に毒性が高く、細心の注意を払って取り扱わなければなりません。
- カバースリップをddH2O、70%エタノール、ddH2Oで順番にすすいでください。各すすぎの間に糸くずのない実験室用ワイプでそれらを乾燥させます。これは、処理前に表面からほこりや水溶性または有機粒子を除去する。
- カバースリップを金属製のカバースリップ保持ラックに置き、ラックを UV-Ozone (UVO) マシンに移します。カバースリップにUVOを20分間照射し、バックグラウンド蛍光を除去します。UVOの代わりにプラズマチャンバを使用することができます。
- ピンセットを使用して、カバースリップをUVO処理に使用する金属ラックからシラン処理に使用する別の金属ラックに移します。UVOを適用すると高レベルの酸化を引き起こすため、両方に同じラックを使用しないでください。ラックを水とエタノールで事前に清掃して、以前の使用による残留化学物質が残らないようにします。
- カバースリップの入ったラックを、アセトン 100% の入った容器に 1 時間浸します。容器を水道水で3xすすぎ、次にddH2Oで3xですすぎ、アセトンをすべて除去します。
- カバースリップの入ったラックを 100% エタノールに 10 分間浸します。容器を水道水で3xすすぎ、次にddH2Oで3xすすぎ、すべてのエタノールを除去します。
- カバースリップ 3x でラックを ddH2O に 5 分間浸します。
- カバースリップでラックを 0.1 M KOH (1 M KOH 50 mL と ddH2O 450 mL) に 15 分間浸します。容器を水道水で3xすすぎ、次にddH2Oで3xすすぎ、すべてのKOHを除去します。
- カバースリップ 3x でラックを ddH2O に 5 分間浸します。
- カバースリップでラックをヒュームフードまたは層流フードで一晩空気乾燥させます。
- ラックとカバースリップを完全に乾燥させた後、シラン専用の別の容器に入れた2%ジメチルジクロロシラン(DDS)に5分間浸漬します。乾燥していないものをシランに接触させないでください。
- ラックとカバースリップを100%エタノールの入った容器に2倍に5分間浸します。容器を水道水で3xすすぎ、次にddH2Oで3xすすいでください。
- ラックとカバースリップを ddH 2 O に3xずつ 5 分間浸します。
- カバースリップでラックをヒュームフードまたは層流フードで一晩空気乾燥させます。
- この最後の乾燥ステップの後、ピンセットを使用してカバースリップをカバースリップボックスに戻します。これらのカバースリップは、今後1〜2ヶ月で使用できます。古いカバースリップはコーティングを失い始め、廃棄する必要があります。
2. チューブリン製剤
注:購入したチューブリンは、標識されていないか、蛍光色素で標識された凍結乾燥粉末として提供されます。凍結乾燥チューブリンは−80°Cの冷凍庫に貯蔵される。次の手順は、標識されていないチューブリンと標識されたチューブリンを可視化に適した比率で混合するために使用されます。
- 凍結乾燥チューブリン粉末1mgを含む標識されていないチューブリンのアリコートを−80°Cの冷凍庫から取り出し、氷上に保管する。チューブに200 μLの冷たいPEM-80を加え、チューブリン濃度を5 mg/mLにします。すべての凍結乾燥物を溶解するために10分間氷の上に保管してください。
- 凍結乾燥チューブリン粉末20μgを含むローダミン標識チューブリンのアリコート1個を−80°Cの冷凍庫から取り出し、氷上に保管する。チューブに4 μLの冷たいPEM-80を加え、チューブリン濃度を5 mg/mLにします。凍結乾燥剤を完全に溶解するために10分間氷の上に保管してください。
- 溶解したら、100 μLの非標識再懸濁チューブリン溶液をローダミン標識チューブリンの4 μL溶液に加える。ピペット6x-7xを非常にゆっくりと混合します。凝集体が見える場合は、可溶化チューブリンを90,000 x g で10分間遠心分離し、ペレットを捨てて上清を保持することによって凝集体を除去します。このチューブリン混合は、〜4%の標識チューブリンをもたらすであろう。
- 残りの100 μLの非標識チューブリンを液体窒素(LN2)にドロップフリーズし、追加のチューブリン混合物に使用するために−80°Cで保存します。
- 標識されたチューブリンミックスとアリコートを、それぞれ15μLの7本のチューブに入れます。各アリコートは、単一の実験チャンバーに使用することができる。残りのアリコートをドロップフリーズし、将来の実験のために−80°Cで保存する。
3. MAP65 精製
注:MAP65は市販されていないため、この作業のために精製する必要があります。このプロトコルは、以前にいくつかの出版物23,29で詳述されている。
- MAP65プラスミドおよびGFP-MAP65プラスミドをBL21細菌株に形質転換し、タンパク質発現を行う。
- BL21細菌を600nmで0.6-1の光学密度まで増殖させる。lacオペレータを使用してタンパク質生産を誘導し、細菌を一晩増殖させる。
- 培養物をペレット化し、細菌を溶解する。
- 遠心分離後に溶解液を収集し、6x-ヒスチジンタグを結合するために利用可能なニッケルイオンを有するビーズでインキュベートする。
- イミダゾールを使用してタンパク質を溶出し、それを脱塩します。
- タンパク質を液体窒素でドロップフリーズし、1年以内に使用するために−80°Cで保存してください。
4. フローチャンバの組み立て
注:実験は、スライドガラスとシラン処理カバーガラスで作られたフローチャンバで行われます(図3)。
- スライドガラスを取り、ddH2O、エタノール、ddH2Oを順番に使って掃除します。各すすぎの間に糸くずの出ない実験室用ワイプで乾燥させます。
- 両面テープを使用して流路を作成します。手袋をはめた手を使って、テープを約25〜30mmの長さにカットします。テープを縦に分割して、2 つの薄いストリップを作成します。2 つのテープ ストリップをスライドの上に置き、その間に約 5 ~ 8 mm を置きます。
メモ:テープの厚さは約80〜100μmに標準化されているため、テープ片間のパスの幅によってチャンバ内の体積が決まります。 - シラン処理されたカバースリップを流路の上に置きます。スライドとカバースリップを両面テープストリップに密封するには、ペンの背面でテープ領域を軽く押します。エリア全体で良いシールをしてください。シールがうまく作られると、テープは半透明から透明に変わるはずです。
- 縁の余分なテープを取り外し、カミソリの刃でテープを切断して、フローチャンバの入り口からわずか1mmを残します。
- 必要に応じて、実験パラメータに関する情報をチャンバーにラベル付けします。
5. タクトイド実験
注:すべての試薬と供給が生成されると、フローチャンバ内の微小管タクトイドを核形成および重合するために使用できます。
- 使用する試薬をすべて集めます。氷の上でそれらを解凍し、作業中に氷の上にそれらを保存します。実験のためにいくつかのフローチャンバを作ります。
- 実験ごとに1つのフローチャンバを使用します。PEM-80に溶解した5%非イオン性ブロック共重合体界面活性剤(材料表)を20μLで流すことによって、チャンバの両端に小さな滴を流してフローチャンバ表面をポリマーブラシでコーティングし、内部の気泡の形成を防止します。使用する準備ができるまで(少なくとも5〜7分)、湿気の多いチャンバー(すなわち、濡れた糸くずのない実験室用ワイプ付きのペトリ皿)に保管してください。
- 滅菌チューブ中で、以下を混合してTubulin-MAPミックスを作成する:9.5μLのPEM-80;4 μL の 10 mM GMPCPP;4μLの5%プルロニック-F127;1 μL の 1M DTT;1μLのグルコース;2μLのポリエチレングリコール(PEG);ステップ2.5からの12 μLの5 mg/mLチューブリンミックス(13.6 μM最終濃度)。MAP65の5.5μLの作業ストック(10%は視覚化のためのGFP-MAP65)です。混合しながら氷の上を保ちます。
注:粘性PEG溶液の取り扱いには、正の変位ピペットを使用することをお勧めします。通常のピペットは、開口部を大きくするために先端を切断した後に使用することができます。ただし、この方法は精度が低くなります。 - ピペッティングで5x-6xを混ぜる。
- チャンバーに添加する直前に、グルコースオキシダーゼ(0.5 mg/mL)とカタラーゼ(0.15 mg/mL)(デオキシ)の予備混合溶液1 μLをTubulin-MAPミックスに加え、7x-8xを混合します。溶液の全容量(40 μL)を2つの部分に分割し、別々のチャンバーで使用します。
- チューブリン-MAPミックスをチャンバーに流します。チャンバーにはすでに非イオン性ブロック共重合界面活性剤が含まれているため、古い液体を取り除かずにより多くの液体を追加することはできません。これを行うには、チャンバーのもう一方の端にあるろ紙または糸くずの出ない実験室用ワイプを使用して、毛細管現象によって液体を除去します。
- サンプルがチャンバ内に完全に収まったら、5分間エポキシを使用してチャンバの両端を密閉し、37°Cで約30分間保持して微小管タクトイドを核形成および成長させます。
6. 蛍光顕微鏡
- タクトイドをイメージングするために蛍光顕微鏡を使用してください。
注:全反射蛍光顕微鏡または回転ディスク共焦点顕微鏡は、遊離チューブリンからバックグラウンド蛍光を除去するのに適していますが、タクトイドは、形成された後に通常の落射蛍光および透過光顕微鏡によっても見えるため、この手順は特別な装置なしでアクセス可能です。 - 開口数(NA)の対物レンズを1.2NA以上、倍率60倍以上で使用して、蛍光で十分な光を集めます。これらの目標は、多くの場合、ddH2Oまたはオイルのいずれかに浸漬する必要があります。
- CMOSまたはCCDカメラで画像を記録します。108 nm のカメラで有効ピクセル サイズを使用します。
メモ:ピクセルサイズは、カメラと使用する倍率によって異なりますが、この場合は60倍または100倍で、大口径(1.2または1.49 NA)でした。カメラの前に追加の画像エキスパンダーを使用して、必要なピクセルサイズを実現できます。 - この温度に設定した環境チャンバーを用いて試料を37°Cに保った。あるいは、熱風ステージヒーターや温水を循環させる客観的な温度制御カラーなど、他のステージヒーターを採用してください。
- 必要な蛍光に適した励起源を使用してください。ローダミンチューブリンの場合は、サンプルで少なくとも1mWのパワーを持つ561nmレーザーを使用し、GFP-MAP65の場合は、サンプルで少なくとも1mWのパワーを持つ488nmレーザーを使用します。
メモ:広視野落射蛍光顕微鏡を使用する場合は、励起:540 ± 12.5 nm、ダイクロイック:545 nm ± 12.5 nmカットオフ、発光:575 nmロングパス、励起:480 ± 15 nm、ダイクロイック:505 nm±15 nmカットオフ、および発光:515 nmロングパスのローダミンフィルターキューブを使用してください。 - 異なる領域の少なくとも10枚の画像を撮影して、100枚以上のタクトイドを画像化します。赤チャンネルと緑チャンネルの両方で画像を撮影し、分析のために16ビットTIFF画像として保存します。照明パワーと露出時間が、カメラの強度スケールが飽和していないことを確認してください。
7. フォトブリーチング後の蛍光回復(FRAP)
注:タクトイドの内部構成要素の移動性を調べるために、FRAPが使用された。FRAPは、ローダミン標識チューブリンおよびGFP標識MAP65タクトイドの選択された部分をフォトブリーチングし、その領域における時間とともに蛍光の回復を観察することによって機能する。回復速度は、光漂白される種の回転に依存する。このターンオーバー速度は、拡散および結合反応に依存し得る。タクトイドへのMAP65結合について、結合交換速度を推定することができる。FRAPは、任意の形状のレーザーをスキャンできる追加の405nmレーザーシステムを使用して実行されます。FRAPを行うには、局所領域14をフォト漂白するために透過ランプおよび絞りを使用することを含む多くの可能性がある。
- チャンバ内の孤立したタクトイドを選択して、タクトイドの一部と周囲の溶液を覆う関心領域(ROI)を作成します。
- FRAP用の追加の405nmレーザーを備えた顕微鏡を使用して、チューブリンとMAP65の両方を同時に光漂白します。あるいは、明るいランプを、虹彩14のフィールドストップを介して使用することができる。フォトブリーチングシステムの特定の強度を経験的に調整して、ブリーチング中のタンパク質の損傷を回避します。
- フォトブリーチングの前に30〜60秒間、タクトイドを時系列ムービーとして記録して、漂白剤の前の強度に関する情報を取得します。赤と緑のチャンネルの両方を録音します。
- タクトイドを損傷することなく光漂白するために必要な限りレーザーまたはランプのいずれかでROIを露光することによってタクトイドをフォトブリーチする。強度と時間を経験的に決定します。
- フォトブリーチング後5〜10分間、または回復が平衡に達したと思われるまで、両方のカラーチャンネルでムービーを録画し続けます。
- GFP-MAP65チャネルのリカバリを目視で確認します。
8. データ解析
注:タクトイドの画像の定量的分析は、異なるクラウディング剤、イオン条件、および他の要因の付加を介して課せられる環境変化の影響について学ぶために実施された。
- タクトイド形状特性評価
- 共焦点顕微鏡で撮影した赤と緑の画像からタクトイドの長さと幅を定量化します。
- フィジー/イメージJを使用して画像を開きます。
- 生データを16ビットで取得する場合は、必要に応じて明るさとコントラストを調整します。[ 画像]>[明るさとコントラスト> 調整]を選択して、タクタイドをはっきりと見ることができるように画像を調整します。誤って強度データを変更しないように、設定を適用せずに明るさとコントラストを調整してください。
- タクトイドがはっきりと見えたら、測定する良いタクトイドを選択します(図4Bi)。タクトイドが他のタクトイドや凝集体と重なることなくはっきりと見え、直線測定ツールを使用できるように湾曲したり曲がったりしていないことを確認します。
- 次に、画像に正しいピクセルサイズが設定されていることを確認します。顕微鏡画像には、ピクセルサイズに関するメタデータが付属しています。メタデータを持たない別のカメラや、予想される有効ピクセルサイズを変更できる外部画像拡張システムを使用する場合は、ピクセルサイズを手動で調整します。FIJI/ImageJ で、[ 分析] > [スケールの設定] に移動して、正しいピクセル変換を設定します。
- FIJI/ImageJのツールバーの 直線 ツールを使用して、タクトイドの一方の端をクリックし、カーソルをタクトイドのもう一方の端にドラッグします(図4Bii)。ラインROIを選択したら、[ 分析]>[測定] を選択して長さを測定します。長さがデフォルトで測定されない場合は、[分析] または [測定値の設定] ダイアログ ボックスで長さを含めるように 測定>設定 してください。
メモ: 通常、[ 直線] ツールを使用して測定する場合、描画される線の長さと角度が表示されます。例として、 図4Bii は、タクトイドを見えるようにするためにタクトイドの側面に描かれた直線を示していますが、タクトイド上で直接測定を行います。 - 測定後、ツールバーの テキスト ツールを使用してタクトイドにラベルを付けます。テキスト ボックスを作成し、数値ラベルを追加し、[ 編集] > [描画] を選択して、ラベルをイメージに固定します。イメージを別のROIファイルとして保存します。
注: このファイルにラベルを付けて保存すると、研究者は、どの測定値が生データのどのタクトイドに対応しているかを知ることができます。各タクトイドを一度測定してください。 - 画像全体のタクトイドが測定されたら、「 結果 」ウィンドウのデータをコンマ区切りまたはタブ区切りのテキスト・ファイルに保存し (「 ファイル」または「名前を付けて保存」を使用)、スプレッドシート・プログラムでデータ>開いてデータを数値に解析します。すべてのデータ (生の画像データ、ROI 画像、および結果のテキスト ファイル) をまとめて、適切な命名規則を持つフォルダーに集めて、すべてを整理します。
注:タクチイドの長さの測定は手作業で行われますが、タクトイドの幅が狭いため、測定誤差を減らすために別のタクトイド幅の測定方法(下記参照)を使用することをお勧めします。 - ImageJ/FIJIを使用して、 直線ツールを使用して線 領域を描画します。タクトイドの長軸に垂直な二等分線として線を引きます(図4Bii)。
- プロットプロファイル>分析を選択して、線形二等分線の強度プロファイルを作成します(図4Biii)。プロットが表示されます。プロットからデータを取得して保存するには、左下のリストボタンを選択します。これにより、描画された線の長さに沿った強度データのテキストファイルリストが生成されます。テキスト ファイルを.csvファイルまたは.txtファイルとして保存します。
- MatLab、Python (sciPy)、またはその他のプログラムなどの適切なプログラムでテキストファイルを開きます。強度データを次の形式のガウス関数で適合します。
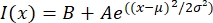 ここで、I(x) は長さ x に沿ったグレースケール値です。B はバックグラウンド レベルです。A はガウスの振幅です。μはガウスの平均または中心です。σはガウスの標準偏差です。
ここで、I(x) は長さ x に沿ったグレースケール値です。B はバックグラウンド レベルです。A はガウスの振幅です。μはガウスの平均または中心です。σはガウスの標準偏差です。 - 報告書2は、タクトイドの幅としてσ する。タクトイド内の微小管の強度を推定するには、ガウス下の面積(背景を含まない)を計算します。
注:画像がカメラの線形強度範囲内にあり、同じ露光時間と励起強度で撮影された場合、積分強度を比較して、タクトイド内の微小管の相対数を推定することができます。
- フラップ解析
注:微小管およびMAP65の移動度を試験する実験では、FRAPを使用して、分子運動による強度の特異的なフォトブリーチングおよび回復を記録した(図5A)。データは、ImageJ/FIJIを用いて画像時系列データから定量化した。- ImageJ/FIJIを使用して、ムービーデータを開きます。
- 時間の経過とともにスタック (時系列データ) を登録して、ドリフトを除去します。 StackRegプラグイン を補助的な TurboReg プラグインと一緒に使用してください。プラグインの使用方法については、 資料表に記載されているウェブリンクを参照してください。平行移動を選択すると、フレームの位置がシフトされ、画像が登録されます。
- ドリフトを除去するために画像が登録されたら、[画像]または[変換]または[回転]を選択して、タクトイドがフレーム内で垂直または水平 になるように画像>回転>ます。回転する角度を選択し、「 プレビュー」 を使用して、タクトイドが十分に回転しているかどうかを確認します。プレビューでタクトイドが垂直または水平のいずれかであることが示されたら、「 OK」 を選択してムービー内のすべての画像を回転させます。
- ツールバーの長方形選択ツールを使用して、タクトイドのフォトブリーチング領域の上に長方形断面を作成します。各フレームのROI領域の積分強度を、画像>スタック>測定スタックを使用して記録します。「分析」または「測定の設定」を使用して、測定タイプを「統合密度」>設定します。「結果」ウィンドウに表示される分析強度データを、「ファイル」または「別名で保存」を選択して、.csvまたは.txt形式のテキストファイルとして保存>ます。
メモ: 図5B は、漂白剤領域の微小管およびGFP-MAP65チャネルについて測定された生の16ビット強度データの例を示しています。 - 画像の全体的な強度は、画像によって引き起こされるフォトブリーチングのためにグローバルに時間の経過とともにフェードアウトするため、このグローバルフォトブリーチングを修正する必要があります。これを行うには、同じROIサイズを使用し(ステップ8.2.4)、微小管またはMAP65が見えない画像の背景の領域に移動します。ステップ 8.2.4 の説明に従って、スタックの積分強度を測定します。結果を 2 番目のテキスト ファイルとして保存します。
メモ: 図5B は、バックグラウンド領域の微小管およびGFP-MAP65チャネルについて測定された生の16ビット強度データの例を示しています。 - 背景のフェードを補正するには、タクタイドの信号強度を同じ時点の背景強度で除算します。ここで、IS(t)(信号)は漂白領域で行われた測定であり、BG(t)(
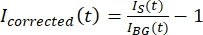 ノイズ)はバックグラウンド領域で行われた測定です(図5C)。これにより、各フレームの信号対雑音比が計算され、ノイズも減算されます。
ノイズ)はバックグラウンド領域で行われた測定です(図5C)。これにより、各フレームの信号対雑音比が計算され、ノイズも減算されます。 - 次に、 を使用して
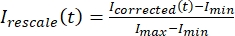 データを 0 から 1 の範囲に再スケーリングします。ここで、I minとImaxは、それぞれ、全時間にわたって補正されたデータのグローバルな最小値と最大値を示します(図5C)。
データを 0 から 1 の範囲に再スケーリングします。ここで、I minとImaxは、それぞれ、全時間にわたって補正されたデータのグローバルな最小値と最大値を示します(図5C)。 - このデータを、次の形式の減衰指数に当てはめます。
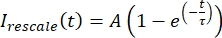 ここで、 A は回復の振幅、τ は回復のタイムスケールです (図 5C)。
ここで、 A は回復の振幅、τ は回復のタイムスケールです (図 5C)。
少数の成分、チューブリン二量体、および微小管架橋剤のみで、微小管タクトイドを形成することができる(図2A)。このプロトコルは、インキュベーター内で微小管を核形成および成長させるためのインキュベーションを記述しているが、核生成および成長は顕微鏡下で直接観察することができる(30分以内に完了する)(図2B)。チューブリンの濃度は13.6μmで一定に保たれ、MAP65-MT結合は10%である。
図 4 は、成功したデータを表しています。タクトイドは、チューブリンチャネルの561nmレーザーとMAP65チャンネルの488nmの両方のレーザーで見えるはずです(図4A)。この系の謎の1つは、微小管の長さ、MAP65濃度、およびクラウディング剤(図4B)22,28の変更を含む、さまざまな実験的変化の下でタクトイドの幅が変化しないように見えることである。長さははるかに可変であり、微小管の長さとMAP65濃度の両方に依存する(図4B)22,28。
FRAPを実行すると、MAP65信号は回復するが、微小管信号は回復しないことが観察されている(図5)。FRAPの回復は、ラベル付けされた物体および光漂白された物体の移動性および移動性によるものである。MAP65の場合、暗くなった分子は解離して微小管から遠ざかり、新しい分子が領域に移動します(図5)。MAP65結合は平衡状態にあるため、結合と非結合の速度は等しくなります(1秒あたりの分子数で測定)。微小管については、微小管がタクトイドを離れることができないことを示唆する回復は見られなかった(図5A、Bi、C)。さらに、黒ずんだ領域の広がりは見られず、微小管は局所的に不動であり、タクチイド形状内の流体ではないことが示唆された。
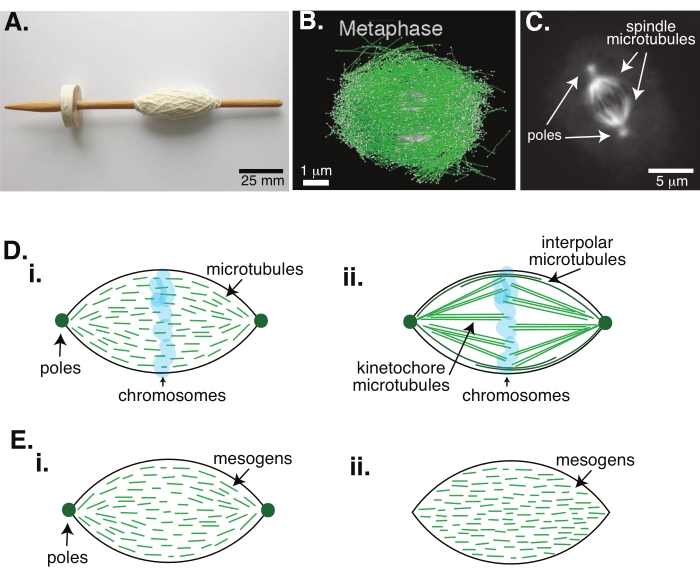
図1:スピンドル形成のさまざまなモデル。 有糸分裂紡錘体は、微小管とそれに関連するタンパク質と酵素で作られた機械で、細胞分裂中に染色体を整列させ、2つの新しい娘細胞に分離します。(A)オランダの細い糸を持つ中世初期のドロップスピンドルレプリカの画像。この図は、Peter van der Sluijs30によるウィキメディアの画像から修正されています。(B)野生型減数分裂の異なる段階における微小管の三次元再構成II.微小管は緑色で示され、染色体は灰色で示されている。スケールバー = 1 μm。この図はLantzschら31から修正されている。(c)分裂したSf9細胞の有糸分裂紡錘体における微小管の顕微鏡観察像。紡錘体の極および微小管は、緑色蛍光タンパク質で標識されている。スケールバー = 5 μm。この図はAdvani et al.32から修正されています。(D)有糸分裂および減数分裂紡錘体微小管がどのように組織化されるかの異なるモデル。(i)アフリカツメガエルの卵抽出物から作成された減数分裂紡錘体について以前に観察された微小管(緑色)は、紡錘体全体にわたって短くて動的であると推定された。これは、液晶内部のバイポーラタクトイド組織に似ています。(ii)有糸分裂紡錘体内部の微小管組織の正準モデルには、染色体の周りの中間帯で架橋する極間または極性微小管(濃い緑色)と、染色体を押したり引いたりするために極からキネトコレに束ねられて引き伸ばされたキネトコリア微小管(薄緑色)の2種類の微小管がある。すべての画像において、染色体は透明な青色で示され、紡錘体極は濃い緑色で表される。(E)(i)バイポーラおよび(ii)均質なタクトイドに対する液晶タクトイド中のメソゲン(緑色の線)の回路図。双極性タクトイドはタクトイドの端に2つの極を持ち、メソゲンはそれらの極を指すように向きを変える。均質なタクトイドは無限大に極を持ち、メソゲンはタクトイドの長さに沿って向きを変えません。 この図の拡大版を表示するには、ここをクリックしてください。
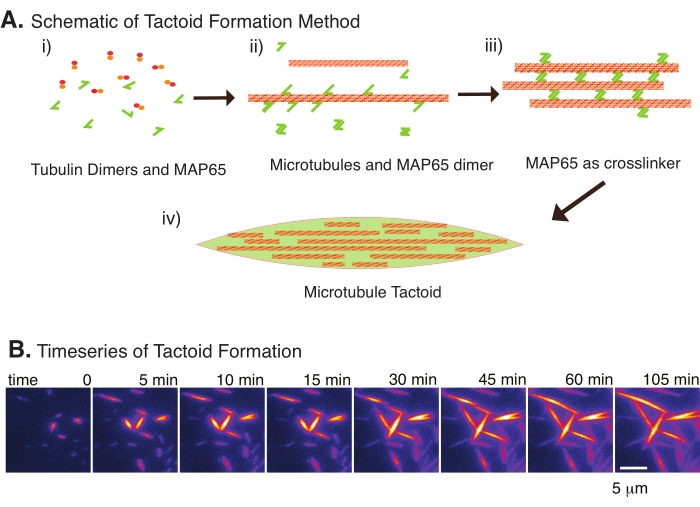
(A)微小管は、イオン種、クラウディング剤によって引き起こされる枯渇力、およびMAP65などの特定の微小管架橋剤を含む様々な方法によって束ねられ、架橋され得る。(i)チューブリン二量体とMAP65タンパク質が混合され、微小管を核形成および成長させる。(ii)微小管は、チューブリンから核形成して成長し、MAP65は直ちに微小管、別のMAP65モノマー、またはその両方に結合し、バンドルを引き起こす。(iii)架橋束中の微小管が核形成して成長する。(iv)最終的な構成は、紡錘体に似た微小管タクトイドである。(b)105分にわたって核形成および成長する微小管タクトイドの時系列。スケールバー = 5 μm。図はEdozie et al.22から翻案された。この図の拡大版を表示するには、ここをクリックしてください。
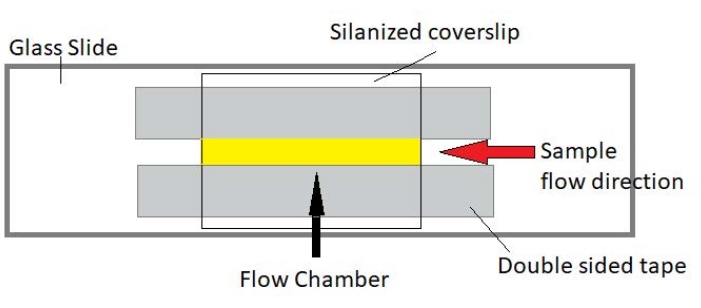
図3:フローチャンバアセンブリ フローチャンバは、スライドガラス、シラン処理カバーガラス、および永久両面テープを使用して作られています。黄色で強調表示された領域は、サンプルが流れて観察される流路です。フローチャンバの容積は約20μLで、エポキシを使用してチャンバの端部を密封し、数時間にわたる長期イメージング中にサンプルが蒸発するのを防ぎました。 この図の拡大版を表示するには、ここをクリックしてください。
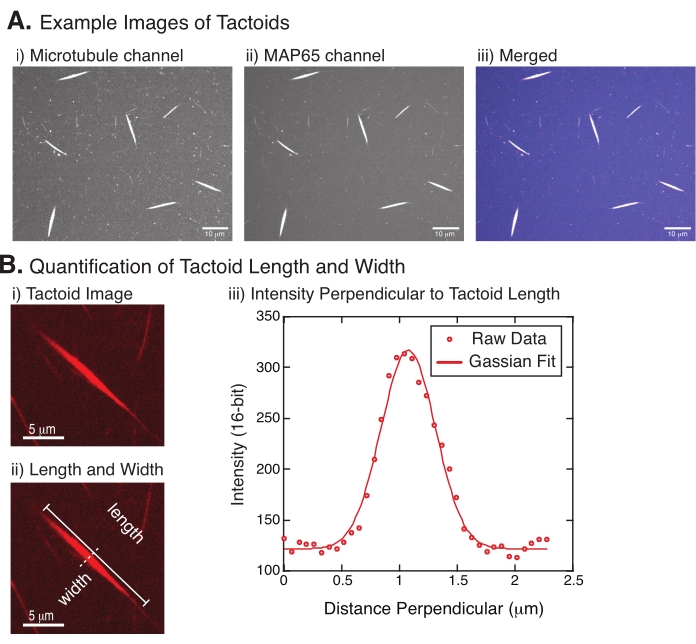
図4:タクトイド画像と長さと幅の分析。 (a)記載されたように形成され、スピニングディスク共焦点を用いて画像化されたタクトイドの例データは、(i)561nmレーザーを用いたローダミン標識チューブリンの微小管チャネルイメージング、(ii)488nmレーザーを用いたGFPのGFP−MAP65チャネルイメージング、および(iii)微小管チャネル(マゼンタ)およびGFP−MAP65チャネル(シアン)からのマージオーバーレイ画像を示す。オーバーラップ領域は白色で表示され、微小管とMAP65が正確に共局在することを示す。スケールバー = (A)のすべての画像について10μm。(B)タクトイドの長さと幅の定量化。(i)標識なしで分析されるタクトイドの画像。スケールバー = 5 μm. (ii) (i) と同じ画像で、長さ (ラインキャップ付きの実線) と幅 (破線) の測定値を示します。(iii)幅は、(ii)で示された垂直な二等分線(破線)でタクトイドを横切る強度プロファイルをとることによって測定した。強度プロファイルは、タクトイドの振幅と幅を明らかにするためにガウス関数に適合した。 この図の拡大版を表示するには、ここをクリックしてください。
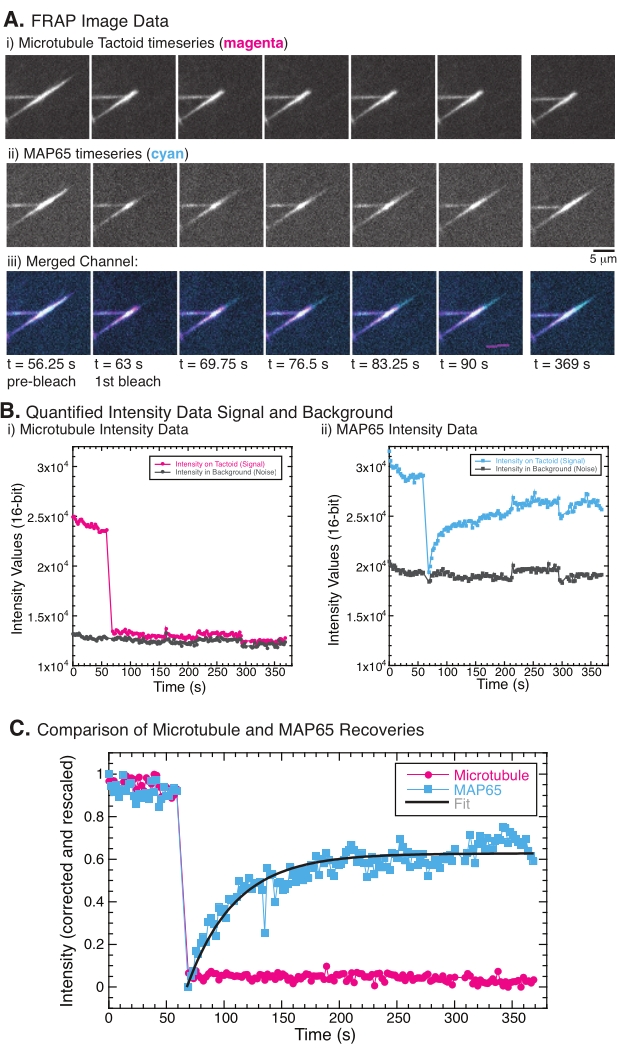
(A)微小管タクトイドおよび(ii)GFP-MAP65の顕微鏡時系列データ、および(iii)マゼンタおよびシアン中のGFP-MAP65の微小管を有する両チャネルのオーバーレイ画像((B)時間63秒でフォトブリーチされ、さらに5分間観察された(i漂白領域(マゼンタ円)および背景(濃い灰色の円)における微小管チャネル(ii)漂白領域におけるGFP−MAP65チャネル(シアンの正方形)および背景(濃い灰色の正方形)。(C)データはバックグラウンドノイズに対して補正され、微小管チャネル(マゼンタ円)およびGFP-MAP65チャネル(シアンの四角)に対して再スケーリングされた。微小管は回復しませんが、GFP-MAP65は上昇する指数関数的減衰にフィットし(濃い灰色の線)、振幅と回復の時間スケールを見つけることができます。この図の拡大版を表示するには、ここをクリックしてください。
ここで説明する方法は、微小管タクトイド(図2)22、28を作成するためにいくつかの論文で使用されている。これらの実験は、ほとんどの細胞型において有糸分裂または減数分裂紡錘体の形状および安定性を制御する組織原理を明らかにするのに役立つ生物学的に関連する。さらに、微小管は、液晶が等方性相からネマチック相を核形成および成長させる方法の詳細を学ぶのに役立つモデル液晶メソゲンである。
ここで概説する手順は、微小管の自己組織化を探索するためのいくつかの利点を有する。第一に、それは再現性が高く、研究室で始める前に、高校生を含む多くの学生によって、予知や訓練がほとんどなく、実験室で行われています。タクトイドは複屈折性22であり、蛍光顕微鏡に加えて透過光で見ることができ、この方法は多くの研究室でアクセス可能であり、この実験手順はハイエンドの研究に加えて教育目的に適応可能である。最後に、このプロセスは、取り除かれた還元主義的なアプローチで生物学的システムを理解し、プローブし続けるための道を開き、各追加の状態、タンパク質、または添加剤がタクトイドの自己組織化をどのように変えるか、そしておそらく最終的にはスピンドルを理解することができます。より良いバイオミミクリーのためのターゲットには、活性、流動性、およびフィラメント極性ソーティングが含まれる。
実験に影響を与えるいくつかの要因が予期しない結果をもたらす可能性があります。例えば、タクトイドが形成されない(図2)が、ファン様のパターンが観察される場合、MAP65は、微小管22、28に存在しないか、または結合していない可能性が高い。これは、GFP-MAP65が微小管に結合しないため、MAP65蛍光チャネルでも明らかであるはずです。
タクトイドが形成されず、背景がガラス上に斑点として表示される場合、これは表面コーティングが原因である可能性があります。一度実行されると、シラナイゼーションはカバースリップでわずか1ヶ月間持続します。それが磨耗すると、チューブリンは露出した表面に非特異的に結合することができる。このバインドは、奇妙なパターンで発生します。
タクトイドが形成されず、チューブリンが様々な形状およびサイズの凝集体で観察される場合、これは質の悪いチューブリンが原因である可能性がある。チューブリンを遠心分離して、微小管重合の代わりにこのオフパスウェイ凝集を駆動できる初期凝集体を除去することができる。表面がチューブリンに結合している場合、溶液中のチューブリンを枯渇させることもできる。低濃度のチューブリンは、微小管を重合するための臨界濃度を下回り、凝集体を生じ得る。
FRAP実験において、MAP65チャネルが何の回復も示さない場合(図5)、フォトブリーチングが微小管を光損傷した可能性がある。光損傷は、フィラメントの局所的な破壊を引き起こす。これは、送信チャネルでの検査によって確認することができます。微小管タクトイドは、周囲の水との不一致の高い屈折率を介して透過チャネルに見える。光誘発性光損傷は、フォトブリーチングを受けたROIの位置における透過光イメージングにおいて、バーンマークまたはコントラストの損失として現れるであろう。これが発生した場合、タンパク質の光損傷を阻害するためにレーザーまたは光パワーを低下させなければならない。
この手順とアプローチでは、いくつかの課題に直面しました。1つの問題は、長さ測定が現在、画像をクリックして手作業で行われていることです。この方法は単純ですが、不確実性が高くなる可能性があります。断面とガウス分布へのフィッティングを使用する幅測定は、サイズを定量化するためのより良い方法です。長さについても同様の方法を採用することができる。2番目の問題は、タクトイドが非常に長くて薄いため、時々、タクトイドが曲がる可能性があることです。これにより、長さの定量化がより困難になります。輪郭の長さはセグメント化された線を使用して定量化できますが、セグメントが追加されるたびに不確実性が増します。
科学的な観点からは、このアプローチは、液晶またはスピンドルのモデルとして使用するために、いくつかの他の課題を有する。最初の課題は、微小管が作り出すタクトイドの長くて薄い形状でした(図3と図4)。先行刊行物22に記されているように、微小管タクトイドは均質なタクトイドであり、双極性ではない。これは、形状を構成する微小管が構造の先端を指すように向きを変えないことを意味する。代わりに、すべての微小管は長軸に平行であり、「極」は無限大に位置する。これは、分子液晶や、液晶メソゲンとしても作用するアクチンやDNAについて観察されるタクトイドとは大きく異なります。これらの他の系では、タクトイドは双極性であり、交差偏光子で見ると、ロッドの再配向の明白な兆候を示す。
このシステムにおける第2の大きな課題は、微小管がタクトイドの内部で不動であることです。これは、微小管の回収率が非常に低いため、FRAP実験および分析から明らかである。その固体状の性質により、微小管タクトイドは大規模な液晶類似体としての価値が低くなります。液晶のネマチック相は、液体(流体)と結晶(組織化)の両方の特性を有するべきである。形状はスピンドルに適しているように見えますが、不動性により、モデルは有糸分裂スピンドルとして刺激的ではありません。一方、この問題は、システムに流動性を高めるために実験をどのように変更できるかを調査する機会を提供します。
これらの科学的課題は、システムに関する新しい知識を可能にするエキサイティングな機会を提供します。微小管タクトイドをより双極性にするために、より短い微小管を使用することができる。しかし、微小管にはアクチンのように長さを制御するためのよく特徴付けられたキャッピングタンパク質があまりないため、追加の課題があります。核生成および成長の使用は、短い微小管を作るために非常に高い濃度のチューブリンおよびGMPCPPの使用を必要とする。チューブリン濃度が高いと、系内のフィラメントの数が多くなり、タクトイドを互いに分離することがより困難になります。DARPin33などの新しい微小管キャッパーの追加は、この状況に役立つ可能性があります。微小管が不動であるという第2の問題は、有糸分裂に使用されるモーターの四量体であるキネシン-534などのモータータンパク質を添加することによって緩和することができる。あるいは、二量体キネシン−1の人工二量体、15を使用することができる。
流動性を高めるもう1つの方法は、微小管が動的不安定性、微小管の成長と縮小を実行できるようにすることです。現在、安定したGMPCPPフィラメントを播種し、その後動的不安定性を受ける微小管は、スピンドルまたはタクトイドを形成するために望まれるよりもはるかに長く、ファンまたはバンドルのような非常に長い組織をもたらすであろう。したがって、微小管の動的不安定性を加えることは、タクチイド形状を維持するために慎重に行う必要がある。長さを制御できる関連タンパク質および酵素の添加は、この問題を緩和する可能性がある。例えば、キネシン-1335などの解重合キネシンや、カタニン36などの切断酵素が必要になる可能性が高い。これらの実験は複雑で困難ですが、結果が何を明らかにしようとも非常に洞察力に富んでいます。将来の実験がどの方向を向いても、微小管タクトイドを作成するためにここで開発されたプラットフォームは、微小管組織の物理的基盤に関する新しい情報を公開することができます。
著者らは、競合する金銭的利益はないと宣言している。
著者らは、2021年夏のロスラボのメンバー、特にK. Alice Lindsayの助けに感謝したいと思います。この研究は、S. Sahu、N. Goodbee、H.B. Lee、J.L. Rossを支援するNSF BIO-2134215からの助成金によって支援されました。KECK Foundation(Rae Anderson, USD, lead PI)からの助成金は、R. BranchとP. Chauhanを部分的に支援した。
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
Tags
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved