
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
Selvmontering av mikrotubule taktoider
Denne artikkelen presenterer en protokoll for dannelsen av mikrotubule samlinger i form av taktoider ved hjelp av MAP65, en plantebasert mikrotubule crosslinker, og PEG som et trengselmiddel.
Cytoskjelettet er ansvarlig for stor intern organisering og omorganisering i cellen, alt uten leder for å lede endringene. Dette er spesielt tilfelle under mitose eller meiosis, hvor mikrotubulene danner spindelen under celledeling. Spindelen er maskineriet som brukes til å skille genetisk materiale under celledeling. Mot å lage selvorganiserte spindler in vitro, utviklet vi nylig en teknikk for å rekonstituere mikrotubuler i spindellignende forsamlinger med et minimalt sett med mikrotubule-assosierte proteiner og trengselmidler. Spesielt ble MAP65 brukt, som er en antiparallel mikrotubule crosslinker fra planter, en homolog av Ase1 fra gjær og PRC1 fra pattedyrorganismer. Denne crosslinkeren organiserer selv mikrotubuler i lange, tynne, spindellignende mikrotubule selvorganiserte forsamlinger. Disse samlingene ligner også flytende krystall taktoider, og mikrotubuler kan brukes som mesoskala mesogener. Her presenteres protokoller for å skape disse mikrotubule taktoidene, samt for å karakterisere formen på samlingene ved hjelp av fluorescensmikroskopi og bestanddelenes mobilitet ved hjelp av fluorescensgjenvinning etter fotobleking.
Celledeling via mitose er en av de viktigste biologiske prosessene for å opprettholde livet. Mikrotubulfilamentene, som består av tubulin dimers, er essensielle strukturelle elementer i denne prosessen. Det forbigående maskineriet som opprettes i metafase når kromosomene justeres i cellesenteret, kalles den mitotiske spindelen på grunn av formen, som er som en spindel av en vevstol dekket av tråder (figur 1A). Det er veletablert på tvers av mange organismer at mikrotubuler brukes i metafase for å presse og trekke kondenserte kromosomer inn i midten av cellen, justere dem og koble dem opp til mikrotubuler som vil trekke dem fra hverandre i anafase (figur 1B, C). Spindelen dannes i både meiose (figur 1B) og mitose (figur 1C), laget av mange overlappende mikrotubuler som ikke er pakket rundt den sentrale aksen som tråd, men kjører parallelt med grensesnittet. Å lage disse mikrotubule-baserte strukturene krever tilknyttede proteiner som krysskobling og tilhørende enzymer som kan fungere som motorer for å bidra til å presse og trekke kromosomene1.
Studier av meiotiske spindler har vist at mikrotubulene er korte, dynamiske og overlappende i krysskoblede matriser 2,3,4,5,6 (figur 1Di). På grunn av den fysiske organiseringen av disse korte mikrotubulene, ligner den meiotiske spindelen en flytende krystall taktoid (figur 1E). Faktisk har spindler vist seg å samle seg og fusjonere, som man kan forvente av flytende krystall taktoider5.
Mange studier som dateres tilbake til 1960-tallet har brukt fiksering, serielle seksjoner og elektronmikroskopi for å fastslå at det er to typer mikrotubuler inne i den mitotiske spindelen 7,8,9,10. Den første typen kalles kinetochore mikrotubules, som forbinder spindelstangen til kinetochore. Den andre typen kalles interpolare eller polare mikrotubuler, som vokser forbi kromosomene og overlapper ved midtsonen (figur 1Dii) 8,9,10. En tredje type kalles astral mikrotubules, som er utenfor spindelen og forbinder polene til cellekanten; Disse mikrotubule organisasjonene er utenfor omfanget av den aktuelle diskusjonen. Det har nylig vært studier på samspillet mellom augmin6 og gamma-tubulin ring kompleks som påvirker nukleasjonssentre for mikrotubuler, noe som resulterer i en mititotisk spindel med kortere mikrotubuler som i figur 1D.
Siden mikrotubuler er lengre enn de er brede, med et høyt størrelsesforhold og høy stivhet, er de som oppskalerte versjoner av flytende krystallmolekyler. I myk materiefysikk har atomer og molekyler blitt tilnærmet ved hjelp av minimale interaksjoner for å utlede de fysiske mekanismene for faseoverganger, inkludert kjernedannelse og smelting av krystaller11. På samme måte er mikrotubuler mesoskala objekter som er oppskalerte versjoner av flytende krystallmolekyler, noe som gir innsikt i fysikken til flytende krystalldynamikk, inkludert kjernen og veksten av de nematiske fasene fra isotrope. Videre, som diskutert ovenfor, viser den meiotiske spindelen egenskaper som en flytende krystall taktoid, en nematisk tilstand som nukleerer og vokser fra isotropisk tilstand av flytende krystallmolekyler 3,4,5. For taktoider er kjernedannelse og vekst som for andre krystaller (dvs. krever en relativt høy konsentrasjon av mesogener [molekylene som danner flytende krystaller]). Den unike "spindel" formen på taktoiden kommer fra den lokale justeringen av flytende krystallmetogener som justerer seg inn i den nematiske fasen (figur 1E). De kan ikke danne en avrundet krystall fordi molekylene er svært asymmetriske. Gitt mikrotubulenes natur er det kanskje ikke overraskende at det mititotiske spindelmaskineriet laget av en høy lokal konsentrasjon av mikrotubuler også er av samme form, enten det kalles taktoid eller spindel. Taktoider kan være bipolare, med poler i de koniske endene (figur 1Ei), eller homogen, med poler effektivt ved uendelig (figur 1Eii).
Gitt betydningen av spindeldannelse, har det pågått innsats mot selvorganisert spindeldannelse in vitro ved å demonstrere mikrotubulkondensasjon i bunter via ioniske arter12,13, trengselmidler som skaper uttømmingsinteraksjoner14,15, og spesifikke mikrotubule krysskoblingsproteiner 13,16,17,18,19, 21. Overraskende, selv om disse midlene alle jobber for å øke den lokale konsentrasjonen av mikrotubuler, resulterer de ofte i lange mikrotubulebunter, men ikke taktoider. En grunn til at disse buntene er lange, kan være at mikrotubulene som består av dem også er lange. Nylig arbeid med kortere mikrotubuler rapporterte også lengre bunter som ikke er avsmalnet på slutten15; I dette tilfellet holdes buntene sammen med motorproteiner som forårsaker forlengelse av buntene og dermed gjør dem lengre. Korte mikrotubuler med ikke-ekstensile krysskoblinger er nødvendig for koniske, spindellignende forsamlinger, som beskrevet her.
Nylig har vi utviklet en teknikk for å muliggjøre opprettelse av mikrotubule taktoider ved hjelp av antiparallel crosslinker, MAP65, i nærvær av nukleerende stabile mikrotubuler22. Mikrotubulene måtte være korte, men få kjente regulatorer av mikrotubullengde kan dekke mikrotubuler mot dynamisk ustabilitet eller ende-til-ende annealing. I stedet ble GMPCPP brukt til å nukleere og stabilisere filamentene etter vekst. Dette tillot å skape en høy tetthet av korte mikrotubuler som selv kunne organisere seg i taktoider. Disse taktoidene var homogene når de ble sett under birefringence. I tillegg til korte mikrotubuler ble en spesifikk antiparallel crosslinker, MAP65, brukt til å danne taktoidene (figur 2). MAP65 er et plantemikrotubul-assosiert protein i PRC1/Ase1-familien av mititotiske krysskoblinger23. MAP65 eksisterer som en dimer, med en sterk affinitet til å binde seg til seg selv så vel som mikrotubulene24. I motsetning til den meiotiske spindelen og taktoidene som observeres med aktinfilamenter 25,26,27, som er bipolare og har de væskelignende egenskapene til flytende krystaller, har mikrotubule taktoider blitt observert å være faste 22,28.
Her presenteres protokoller for å skape mikrotubule taktoider og karakterisere formen på samlingene og mobiliteten til bestanddelene ved hjelp av fluorescensbaserte teknikker.
MERK: Med mindre annet er oppgitt, kan deler av eksperimentet utføres på en labbenk mens du bruker passende verneutstyr (hansker).
1. Coverslip silanisering
MERK: Deksler må silaniseres for å kunne brukes med polymerbørstebelegget som brukes i disse eksperimentene. Dette er en hydrofob silaniseringsbehandling som gjør det mulig for en blokkkopolymer med en hydrofob sentral blokk å binde og lage en polymerbørste. Følgende trinn bør utføres i en avtrekkshette for å forhindre eksponering for giftige damper mens du bruker hansker. Dimethyldichlorolsilane er svært giftig og må håndteres med største forsiktighet.
- Skyll dekslene med ddH2O, 70% etanol og ddH2O i rekkefølge. Tørk dem med lofrie laboratorieservietter mellom hver skylling. Dette fjerner støv og vannløselige eller organiske partikler fra overflaten før behandling.
- Plasser dekslene i et metalldeksleslip-holdestativ og overfør stativet til en UV-ozonmaskin (UVO). Bestråle dekslene med UVO i 20 min for å fjerne bakgrunnsfluorescens. Et plasmakammer kan brukes i stedet for UVO.
- Bruk pinsett, overfør dekslene fra metallstativet som brukes til UVO-behandling til et annet metallstativ som brukes til silanisering. Ikke bruk de samme stativene til begge, da det vil føre til høye oksidasjonsnivåer når UVO påføres. Forrens stativene med vann og etanol slik at ingen gjenværende kjemikalier forblir fra tidligere bruksområder.
- Senk stativet med dekslene i en beholder med 100 % aceton i 1 time. Skyll beholderen 3x med vann fra springen og deretter 3x med ddH2O for å fjerne all aceton.
- Senk stativet ned med dekslene i 100 % etanol i 10 minutter. Skyll beholderen 3x med vann fra springen og deretter 3x med ddH2O for å fjerne all etanol.
- Senk stativet ned med dekslene 3x i ddH2O i 5 minutter hver.
- Senk stativet med dekslene i 0,1 M KOH (50 ml 1 M KOH i 450 ml ddH2O) i 15 minutter. Skyll beholderen 3x med vann fra springen og deretter 3x med ddH2O for å fjerne all KOH.
- Senk stativet med deksler 3x i ddH2O i 5 minutter hver.
- Lufttørk stativet med dekslene over natten i en avtrekkshette eller laminær strømningshette.
- Etter helt tørking av stativet og dekslene, senk dem i 5 min i 2% dimetyldichlorosilane (DDS) tatt i en annen beholder som brukes spesielt til silan. Ikke la noe som ikke er tørt komme i kontakt med silan.
- Senk stativet og dekslene 2x ned i en beholder med 100 % etanol i 5 minutter. Skyll beholderen 3x med vann fra springen og deretter 3x med ddH2O.
- Senk stativet og dekslene 3x ned i ddH2O i 5 minutter hver.
- Lufttørk stativet med dekslene over natten i en avtrekkshette eller laminær strømningshette.
- Etter dette siste tørketrinnet, overfør dekslene tilbake til deksler med pinsett. Disse dekslene kan brukes i løpet av de neste 1-2 månedene. Gamle deksler vil begynne å miste belegget og bør kastes.
2. Tubulin forberedelse
MERK: Den kjøpte tubulinen kommer som et lyofilisert pulver som enten er umerket eller merket med fluoroforer. Lyofilisert tubulin lagres i en -80 °C fryser. Følgende prosedyre brukes til å blande umerket tubulin med merket tubulin i et forhold som er bra for visualisering.
- Fjern fra -80 °C fryser en aliquot av umerket tubulin som inneholder 1 mg lyofilisert tubulinpulver og hold det på is. Tilsett 200 μL kald PEM-80 til røret for å bringe tubulinkonsentrasjonen til 5 mg / ml. Hold den på is i 10 minutter for å oppløse all lyofilat.
- Fjern fra -80 °C fryseren ett aliquot av rhodamin-merket tubulin som inneholder 20 μg lyofilisert tubulinpulver og hold det på is. Tilsett 4 μL kald PEM-80 i røret for å bringe tubulinkonsentrasjonen til 5 mg /ml. Hold den på is i 10 minutter for å oppløse lyofilaten helt.
- Når den er oppløst, tilsett 100 μL umerket resuspendert tubulinoppløsning til 4 μL-løsningen av rhodaminmerket tubulin. Pipette 6x-7x veldig sakte å blande. Hvis aggregater er synlige, sentrifugerer du den solubiliserte tubulinen i 10 minutter ved 90 000 x g for å fjerne aggregater ved å kaste pelletsen og beholde supernatanten. Denne tubulinblandingen vil resultere i ~ 4% merket tubulin.
- Drop-freeze de resterende 100 μL umerket tubulin i flytende nitrogen (LN2) og oppbevar den ved −80 °C som skal brukes til ytterligere tubulinblandinger.
- Ta den merkede tubulinblandingen og aliquot i syv rør med 15 μL hver. Hver aliquot kan brukes til et enkelt eksperimentelt kammer. Drop-freeze de resterende aliquots og lagre dem ved −80 °C for fremtidige eksperimenter.
3. MAP65 rensing
MERK: MAP65 er ikke kommersielt tilgjengelig, og må derfor renses for dette arbeidet. Protokollen er utarbeidet tidligere i flere publikasjoner23,29.
- Forvandle MAP65 plasmid og GFP-MAP65 plasmid til BL21 bakteriestamme for proteinuttrykk.
- Vokse BL21 bakterier til en optisk tetthet på 0,6-1 ved 600 nm. Induser proteinproduksjon ved hjelp av laktoperatøren og dyrk bakteriene over natten.
- Pellet kulturene og lyse bakteriene.
- Samle lysate etter sentrifugation og inkuber den med perler som har en nikkel ion tilgjengelig for å binde 6x-histidin tag.
- Elute proteinet ved hjelp av imidazol og avsalt det.
- Drop-freeze proteinet med flytende nitrogen og oppbevar det ved −80 °C for bruk innen 1 år.
4. Montering av strømningskamre
MERK: Eksperimenter utføres i strømningskamre laget av en glasssklie og silanisert dekselglass (figur 3).
- Ta en glasssklie og rengjør den med ddH2O, etanol og ddH2O i rekkefølge. Tørk med en lofri laboratorieserviett mellom hver skylling.
- Bruk en del av dobbeltsidig tape til å opprette en flytbane. Bruk hanskede hender til å kutte tapen til ~25-30 mm i lengde. Del båndet på langs for å lage to tynnere strimler. Plasser de to båndstrimlene på lysbildet med ca. 5-8 mm mellom dem.
MERK: Ettersom tykkelsen på båndet er standardisert til ca. 80-100 μm, vil bredden på banen mellom tapestykkene bestemme volumet i kammeret. - Plasser de silaniserte dekslene på toppen av strømningsbanen. Forsegle gliden og dekslene til de dobbeltsidige båndstrimlene ved å trykke forsiktig på båndområdet med baksiden av en penn. Sørg for å få en god segl over hele området; båndet skal dreie fra gjennomskinnelig til klart når forseglingen er gjort godt.
- Fjern det ekstra båndet på kantene, og la bare 1 mm fra strømningskammerets inngang ved å kutte båndet med et barberblad.
- Merk kammeret med informasjon om de eksperimentelle parametrene etter behov.
5. Taktoid eksperimenter
MERK: Når alle reagensene og forsyningene er generert, kan de brukes til å nukleere og polymerisere mikrotubule taktoider i strømningskammeret.
- Samle alle reagensene som skal brukes. Tine dem på is og oppbevar dem på is mens du arbeider. Lag flere strømningskamre for forsøkene.
- Bruk ett strømningskammer for hvert eksperiment. Belegge strømningskammeroverflatene med en polymerbørste ved å strømme i 20 μL 5% ikke-ionisk blokk kopolymer overflateaktivt middel (Materialbord) oppløst i PEM-80, med små dråper i begge ender av kammeret for å forhindre dannelse av luftbobler inni. Oppbevar denne i et fuktig kammer (dvs. Petri-tallerken med en våt lofri laboratorieserviett) til den er klar til bruk (minst 5-7 min).
- I et sterilt rør blander du følgende for å lage Tubulin-MAP-blandingen: 9,5 μL PEM-80; 4 μL 10 mM GMPCPP; 4 μL av 5% Pluronic-F127; 1 μL 1M DTT; 1 μL glukose; 2 μL polyetylenglykol (PEG); 12 μL 5 mg/ml tubulinblanding (13,6 μM sluttkonsentrasjon) fra trinn 2.5.; og 5,5 μL arbeidslager av MAP65 der 10% er GFP-MAP65 for visualisering. Hold deg på is mens du blander.
MERK: Det anbefales å bruke en pipette med positiv forskyvning til håndtering av den viskøse PEG-oppløsningen. Vanlige pipetter kan brukes etter kutting av spissen for å gjøre åpningen større; Denne metoden er imidlertid mindre nøyaktig. - Bland 5x-6x ved pipettering.
- Like før du legger til kammeret, tilsett 1 μL av en ferdigblandet oppløsning av glukoseoksidase (0,5 mg / ml) og katalering (0,15 mg / ml) (deoksy) i Tubulin-MAP-blandingen og bland 7x-8x. Del det totale volumet av løsningen (40 μL) i to deler som skal brukes i separate kamre.
- Bland Tubulin-MAP-blandingen i kamre. Siden kamrene allerede har ikke-ionisk blokk-kopolymer overflateaktivt middel i dem, kan mer væske ikke tilsetts uten å fjerne den gamle væsken. For å gjøre dette, bruk et stykke filterpapir eller en lofri laboratorieserviett i den andre enden av kammeret for å fjerne væske via kapillær virkning.
- Når prøven er helt inne i kammeret, forsegler du de to endene av kammeret ved hjelp av 5-min epoksy og holder den ved 37 °C i ca. 30 minutter for å nukleere og dyrke mikrotubule taktoider.
6. Fluorescensmikroskopi
- Bruk et fluorescensmikroskop for å avbilde taktoidene.
MERK: Total intern refleksjon fluorescensmikroskopi eller spinnende plate konfektmikroskopi er gode å fjerne bakgrunnsfluorescens fra fri tubulin, men taktoider er også synlige ved vanlig epi-fluorescens og til og med overført lysmikroskopi når de er dannet, noe som gjør denne prosedyren tilgjengelig uten spesialisert utstyr. - Bruk et numerisk blenderåpningsmål (NA) på 1,2 NA eller høyere med en forstørrelse på 60x eller høyere for å samle nok lys i fluorescens. Disse målene krever ofte nedsenking i enten ddH2O eller olje.
- Ta opp bilder med CMOS- eller CCD-kamera. Bruk en effektiv pikselstørrelse på kameraet på 108 nm.
MERK: Pikselstørrelsen avhenger av kameraet og forstørrelsen som ble brukt, som i dette tilfellet var 60x eller 100x med høy numerisk blenderåpning (1,2 eller 1,49 NA). Flere bildeutvisere kan brukes før kameraet for å oppnå den nødvendige pikselstørrelsen. - Oppbevar prøven ved 37 °C med et miljøkammer innstilt på denne temperaturen. Alternativt kan du bruke andre scenevarmere, inkludert varmluftsvarmere og objektive temperaturkontrollerte krager med sirkulerende varmt vann.
- Bruk eksitasjonskilder som er riktige for fluorescensen som trengs. For rhodamin tubulin, bruk en 561 nm laser med minst 1 mW strøm ved prøven, og for GFP-MAP65, bruk en 488 nm laser med minst 1 mW strøm ved prøven.
MERK: Hvis du bruker bredfelts epifluorescensmikroskopi, bruk en rhodaminfilterkube med eksitasjon: 540 ± 12,5 nm, dichroic: 545 nm ± 12,5 nm cut-off, og utslipp: 575 nm langpasning, og en GFP-filterkube med eksitasjon: 480 ± 15 nm, dichroic: 505 nm ± 15 nm cut-off, og utslipp: 515 nm lang pass. - Ta minst 10 bilder av forskjellige områder for å bilde over 100 taktoider. Ta bilder i både de røde og grønne kanalene og lagre dem som 16-biters tiff-bilder for analyse. Kontroller at belysningseffekten og eksponeringstidene er slik at intensitetsskalaen for kameraet ikke er mettet.
7. Fluorescensgjenoppretting etter fotobleking (FRAP)
MERK: For å studere mobiliteten til de indre bestanddelene i taktoidene, ble FRAP brukt. FRAP fungerer ved å fotobleaching en utvalgt del av rhodamin-merket tubulin og GFP-merket MAP65 taktoid og deretter observere utvinning av fluorescens med tiden i den regionen. Utvinningsgraden avhenger av omsetningen av arten som fotobleached. Denne omsetningsraten kan avhenge av diffusjon og bindende reaksjoner. For MAP65-binding til taktoidene kan bindende valutakurser estimeres. FRAP utføres ved hjelp av et ekstra 405 nm lasersystem som kan skanne laseren i hvilken som helst form. Det er mange muligheter for å utføre FRAP, inkludert bruk av den overførte lampen og blenderåpningen for å fotobleach et lokalt område14.
- Velg en isolert taktoid i kammeret for å skape en region av interesse (ROI) som dekker deler av taktoidene og den omkringliggende løsningen.
- Bruk et mikroskop med en ekstra 405 nm laser for FRAP for å fotobleach både tubulin og MAP65 samtidig. Alternativt kan en lys lampe brukes gjennom feltstoppet til iris14. Juster den spesifikke intensiteten til fotoblekingssystemene empirisk for å unngå å skade proteinene under bleking.
- Ta opp taktoiden som en tidsseriefilm i 30-60 s før fotobleaching for å få informasjon om intensiteten før blekemiddelet. Ta opp både de røde og grønne kanalene.
- Photobleach taktoiden ved å utsette avkastningen med enten lasere eller lampen så lenge det er nødvendig for å fotobleach uten å skade taktoiden. Bestem intensiteten og tiden empirisk.
- Fortsett å spille inn filmen i begge fargekanalene i 5-10 min post photobleaching eller til utvinningen ser ut til å ha nådd likevekt.
- Undersøk GFP-MAP65-kanalen visuelt for gjenoppretting.
8. Dataanalyse
MERK: Kvantitativ analyse av bildene av taktoider ble utført for å lære om effekten av miljøendringer pålagt via forskjellige crowding agenter, ioniske forhold, og tillegg av andre faktorer.
- Karakterisering av taktoid figur
- Kvantifisere lengden og bredden på taktoidene fra de røde og grønne bildene tatt med konfikal mikroskopi.
- Åpne bildene ved hjelp av FIJI/ImageJ.
- Hvis rådata tas i 16-biters, justerer du lysstyrken og kontrasten om nødvendig. Velg Bilde > Juster lysstyrken og kontrasten > for å justere bildet slik at det tydelig kan se taktoiden. Juster lysstyrken og kontrasten uten å bruke innstillingen for ikke å endre intensitetsdataene ved et uhell.
- Når taktoidene er godt synlige, velger du gode taktoider som skal måles (figur 4Bi). Forsikre deg om at taktoidene er tydelig synlige uten overlapping med andre taktoider eller aggregater og ikke er buede eller bøyd for å kunne bruke rettlinjede måleverktøy.
- Kontroller deretter at riktig pikselstørrelse er angitt for bildene. Mikroskopbildene kommer med metadata om pikselstørrelsen. Når du bruker et annet kamera som ikke har metadata eller eksterne bildeutvidelsessystemer som kan endre forventet effektiv pikselstørrelse, justerer du pikselstørrelsen manuelt. I FIJI/ImageJ går du til Analyser > Angi skala for å angi riktig pikselkonvertering.
- Bruk rett linje-verktøyet fra verktøylinjen i FIJI/ImageJ, klikk på den ene enden av taktoiden og dra markøren til den andre enden av taktoiden (figur 4Bii). Når linje-AVKASTNINGEN er valgt, velger du Analyser > mål for å måle lengden. Hvis lengden ikke måles som standard, må du passe på å angi at målingen skal inkludere lengde i dialogboksen Analyser > Angi mål .
MERK: Når du måler ved hjelp av rett linje-verktøyet , vil det vanligvis gi lengden og vinkelen på linjen som tegnes. Som et eksempel viser Figur 4Bii en rett linje trukket til siden av taktoiden for å gjøre sistnevnte synlig, men gjør målingen direkte på taktoiden. - Når du har foretok målingen, bruker du tekstverktøyet på verktøylinjen til å merke taktoiden. Opprett en tekstboks, legg til en nummeretikett, og velg Rediger > Tegn for å rette etiketten inn i bildet. Lagre bildet som en separat ROI-fil.
MERK: Merking og lagring av denne filen gjør det mulig for undersøkeren å vite hvilken måling som tilsvarer hvilken taktoid fra rådataene. Sørg for å måle hver taktoid en gang. - Når taktoidene for hele bildet er målt, lagrer du dataene i resultatvinduet i en komma- eller tabulatordelt tekstfil (ved hjelp av Fil > Lagre som) og åpner dataene i et regnearkprogram for å analysere dataene i tall. Samle alle dataene sammen (rå bildedata, ROI-bilde og tekstfil med resultater) i en mappe med en passende navnekonvensjon for å holde alt organisert.
MERK: Selv om taktoid lengdemålinger utføres for hånd, gitt at taktoidbreddene er smale, er det bedre å bruke en annen metode for å måle taktoid bredde (se nedenfor) for å redusere målefeil. - Bruk ImageJ/FIJI til å tegne et linjeområde ved hjelp av rett linje-verktøyet . Tegn linjen som en vinkelrett bisektor til den taktoide lange aksen (figur 4Bii).
- Velg Analyser > Plottprofil for å opprette intensitetsprofilen til den lineære bisektoren (figur 4Biii). Et plott vises. Hvis du vil hente og lagre dataene fra plottet, velger du Liste-knappen nederst til venstre. Dette genererer tekstfillisten over intensitetsdataene langs lengden på linjen som tegnes. Lagre tekstfilen som en .csv- eller .txt fil.
- Åpne tekstfilen i et passende program, for eksempel MatLab, Python (sciPy) eller andre programmer. Tilpass intensitetsdataene med en variabel funksjon i skjemaet:
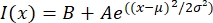 , der I(x) er gråtoneverdien langs lengden, x; B er bakgrunnsnivået. A er gaussianerens amplitude; μ er gjennomsnittet eller sentrum av gaussianeren; og σ er standardavviket til gaussianeren.
, der I(x) er gråtoneverdien langs lengden, x; B er bakgrunnsnivået. A er gaussianerens amplitude; μ er gjennomsnittet eller sentrum av gaussianeren; og σ er standardavviket til gaussianeren. - Rapport 2σ som bredden på taktoiden. Beregn intensiteten av mikrotubulene i taktoiden ved å beregne området under gaussianeren (ikke inkludert bakgrunnen).
MERK: Hvis bildene er innenfor kameraets lineære intensitetsområde og tas med samme eksponeringstid og eksitasjonsintensitet, kan integrerte intensiteter sammenlignes med å estimere det relative antallet mikrotubuler i taktoiden.
- FRAP-analyse
MERK: Eksperimenter for å teste mobiliteten til mikrotubulene og MAP65 brukte FRAP til å registrere spesifikk fotobleaching og gjenoppretting av intensitet på grunn av molekylær bevegelse (figur 5A). Dataene ble kvantifisert fra bildetidsseriedataene ved hjelp av ImageJ/FIJI.- Bruk ImageJ/FIJI til å åpne filmdataene.
- Registrer stakkene (tidsseriedata) over tid for å fjerne drift. Bruk StackReg plugin sammen med den hjelpende TurboReg plugin; For instruksjoner om bruk av plugins, se weblinkene som er gitt i materialtabellen. Velg oversettelse for å forskyve plasseringen av rammene og dermed registrere bildene.
- Når bildene er registrert for å fjerne drift, roterer du bildet slik at taktoiden enten er vertikal eller vannrett i rammen ved å velge Bilde > Transformer > Roter. Velg vinkelen du vil rotere, og bruk Forhåndsvisning til å finne ut om taktoiden er rotert nok. Når forhåndsvisningen viser at taktoiden er enten loddrett eller vannrett, velger du OK for å rotere alle bildene i filmen.
- Bruk markeringsverktøyet Rektangel på verktøylinjen til å opprette en rektangulær inndeling over det fotoblekkede området i taktoiden. Registrer den integrerte intensiteten i AVKASTNING-området for hvert delbilde ved hjelp av bilde- > stakker >-målestakken. Sett målingstypen til Integrert tetthet ved hjelp av Analyser > Angi mål. Lagre de analyserte intensitetsdataene som vises i resultatvinduet , som en tekstfil i .csv- eller .txt-format ved å velge Fil > Lagre som.
MERK: Figur 5B viser et eksempel på rå 16-biters intensitetsdata målt for mikrotubul- og GFP-MAP65-kanalene i blekemiddelområdet. - Ettersom den totale intensiteten til bildene vil falme over tid globalt på grunn av fotobleaching forårsaket av avbildning, må denne globale fotobleaching korrigeres. Hvis du vil gjøre dette, bruker du samme avkastningsstørrelse (trinn 8.2.4.), og flytter den til et område i bakgrunnen av bildet der ingen mikrotubuler eller MAP65 er synlige. Mål den integrerte intensiteten til stakken som beskrevet i trinn 8.2.4. Lagre resultatene som en annen tekstfil.
MERK: Figur 5B viser et eksempel på rå 16-biters intensitetsdata målt for mikrotubul- og GFP-MAP65-kanalene i bakgrunnsområdet. - For å korrigere bakgrunnsfadingen, del signalintensiteten på taktoiden med bakgrunnsintensiteten for samme tidspunkt. Beregn at jegkorrigerte (t) som:
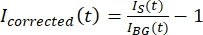 , der IS (t) (signalet) er målingen som er gjort på det blekede området, og IBG (t) (støyen) er målingen som er gjort på bakgrunnsområdet (figur 5C). Dette beregner signal-til-støy-forholdet for hver ramme og trekker også fra støyen.
, der IS (t) (signalet) er målingen som er gjort på det blekede området, og IBG (t) (støyen) er målingen som er gjort på bakgrunnsområdet (figur 5C). Dette beregner signal-til-støy-forholdet for hver ramme og trekker også fra støyen. - Deretter skalerer du dataene på nytt til område mellom null og ett ved hjelp
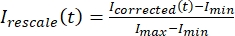 av , der jegmin . og jegmaks angir henholdsvis det globale minimumet og maksimumet for dekorrigerte dataene for jeg gjennom hele tiden (figur 5C).
av , der jegmin . og jegmaks angir henholdsvis det globale minimumet og maksimumet for dekorrigerte dataene for jeg gjennom hele tiden (figur 5C). - Tilpass disse dataene til en forfallende eksponentiell for skjemaet:
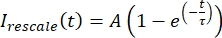 , der A er amplituden for gjenopprettingen og τ er tidsskalaen for gjenopprettingen (figur 5C).
, der A er amplituden for gjenopprettingen og τ er tidsskalaen for gjenopprettingen (figur 5C).
Med bare et lite antall komponenter, tubulin dimmere og mikrotubule krysskoblinger, kan mikrotubule taktoider dannes (figur 2A). Selv om denne protokollen beskriver inkubasjon for å nukleere og dyrke mikrotubuler i en inkubator, kan kjernen og veksten observeres direkte under mikroskopet (som er fullført innen 30 min) (figur 2B). Konsentrasjonen av tubulin holdes konstant på 13,6 μm og MAP65-MT binding ved 10%.
Figur 4 representerer vellykkede data. Taktoidene skal være synlige med både en 561 nm laser i tubulinkanalen og 488 nm i MAP65-kanalen, som perfekt overlapper med hverandre (figur 4A). Et mysterium i systemet har vært at bredden på taktoidene ikke ser ut til å variere under en rekke eksperimentelle endringer, inkludert endring av mikrotubullengdene, MAP65-konsentrasjonen og trengsemidlene (figur 4B) 22,28. Lengden er mye mer variabel og avhenger av både mikrotubullengdene og MAP65-konsentrasjonen (figur 4B) 22,28.
Når du utfører FRAP, har det blitt observert at MAP65-signalet gjenoppretter, men mikrotubulsignalet gjenoppretter ikke (figur 5). Gjenopprettingen i FRAP skyldes mobiliteten og bevegelsen til de merkede og fotobleachede objektene. Når det gjelder MAP65, dissosierer de mørke molekylene og beveger seg bort fra mikrotubulen og nye beveger seg inn i regionen (figur 5). MAP65 bindingen er i likevekt, så bindingshastigheten og ubindingen er lik (målt i molekyler per sekund). For mikrotubulene ble det ikke sett noen gjenoppretting som antyder at mikrotubulene ikke er i stand til å forlate taktoiden (figur 5A, Bi, C). Videre ble det ikke sett noen spredning av den mørke regionen, noe som tyder på at mikrotubulene er lokalt immobile og ikke en væske i taktoidformen.
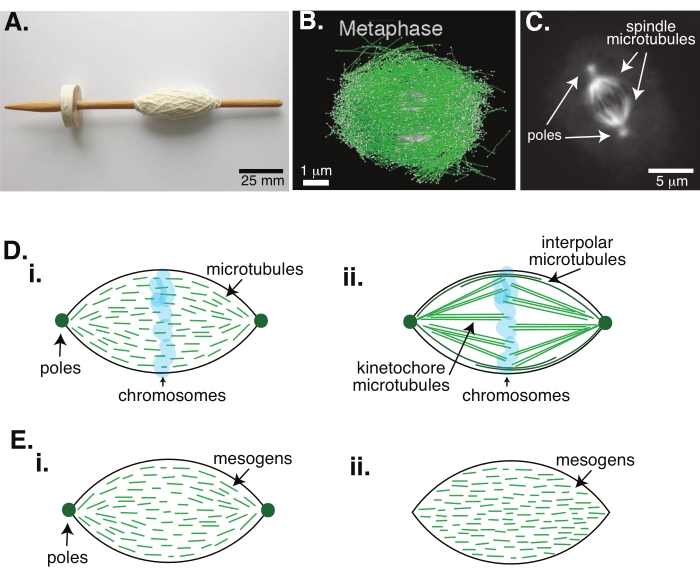
Figur 1: Ulike modeller for spindeldannelse. En mititotisk spindel er en maskin laget av mikrotubuler og deres tilknyttede proteiner og enzymer som justerer og skiller kromosomene inn i de to nye dattercellene under celledeling. (A) Bilde av en tidlig middelalder slippe spindel kopi med fint garn fra Nederland. Denne figuren er endret fra et Wikimedia-bilde av Peter van der Sluijs30. (B) Tredimensjonal rekonstruksjon av mikrotubuler på ulike stadier av villtype meiosis II. Mikrotubuler vises i grønt, og kromosomer vises i grått. Skalastang = 1 μm. Denne figuren er endret fra Lantzsch et al.31. (C) Mikroskopi bilde av mikrotubulene i en mititotisk spindel av en deling Sf9 celle. Spindelstengene og mikrotubulene på spindelen er merket med et grønt fluorescerende protein. Skalastang = 5 μm. Denne figuren er endret fra Advani et al.32. (D) Ulike modeller for hvordan mitotiske og meiotiske spindelmikrotubuler er organisert. (i) Tidligere observert for meiotiske spindler laget av Xenopus egg ekstrakter, ble mikrotubulene (grønne) utledet til å være korte og dynamiske gjennom spindelen. Dette ligner en bipolar taktoid organisasjon inne i en flytende krystall. (ii) Den kanoniske modellen for mikrotubul organisasjon inne i en mititotisk spindel har to typer mikrotubuler: interpolariske eller polare mikrotubuler (mørkegrønn) som krysslinker i midtsonen rundt kromosomene og kinetochore mikrotubulene (lysegrønne) som er buntet og strukket fra polen til kinetochore for å presse og trekke kromosomene. I alle bilder vises kromosomer i gjennomsiktig blå, og spindelstenger er representert i mørkegrønn. (E) Skjematikk av mesogener (grønne linjer) i en flytende krystall taktoid for (i) bipolare og (ii) homogene taktoider. Bipolare taktoider har to poler på slutten av taktoiden, og mesogenene orienterer seg om å peke på disse polene. Homogene taktoider har poler ved uendelig, og mesogenene endrer ikke orientering langs tactoidens lengde. Klikk her for å se en større versjon av denne figuren.
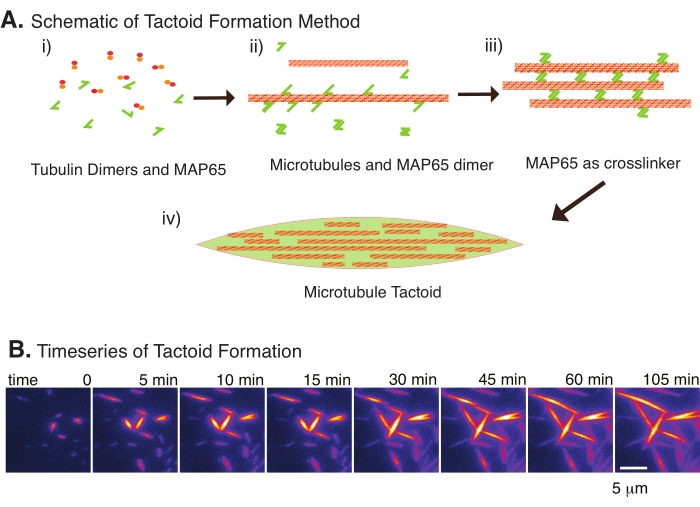
Figur 2: Mikrotubulkondensasjon. (A) Mikrotubuler kan buntes og krysskobles av en rekke metoder, inkludert ioniske arter, uttømmingskrefter forårsaket av trengselsmidler og spesifikke mikrotubule crosslinkers, for eksempel MAP65. (i) Tubulin dimers og MAP65 proteiner blandes for å nukleere og vokse mikrotubuler. (ii) Mikrotubuler nukleerer og vokser fra tubulin, og MAP65 binder seg umiddelbart til mikrotubuler, en annen MAP65 monomer, eller begge deler og forårsaker bunting. (iii) Mikrotubuler i de krysskoblede buntene nukleerer og vokser. (iv) Den endelige konfigurasjonen er en mikrotubul taktoid som ligner en spindel. (B) Tidsserier av mikrotubule taktoider nukleerer og vokser over 105 min. Skalastang = 5 μm. Figur tilpasset fra Edozie et al.22. Klikk her for å se en større versjon av denne figuren.
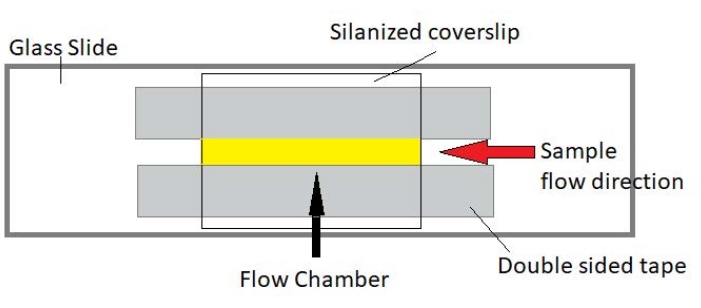
Figur 3: Montering av strømningskammer. Strømningskammeret er laget ved hjelp av en glasssklie, silanisert dekselglass og permanent dobbeltsidig tape. Det gule uthevede området er strømningsbanen der prøven flyter og observeres. Volumet av strømningskammeret er ~ 20 μL. Epoxy ble brukt til å forsegle endene av kammeret for å forhindre at prøven fordamper under langvarig avbildning over flere timer. Klikk her for å se en større versjon av denne figuren.
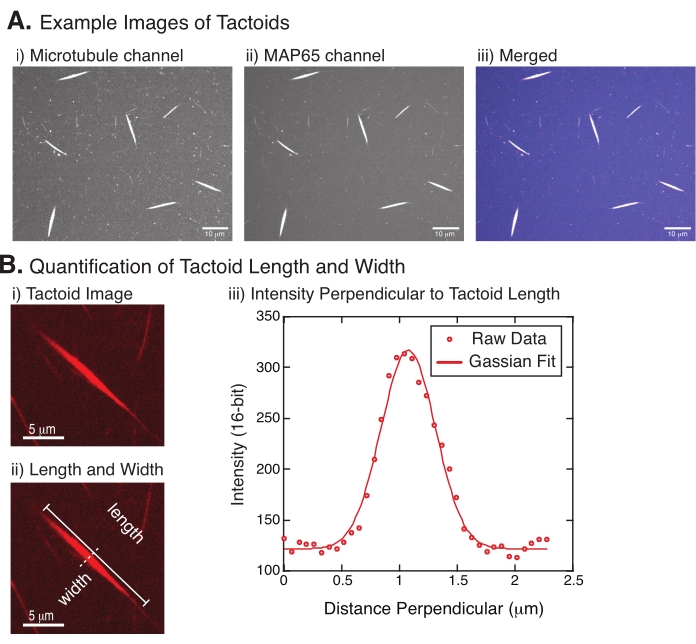
Figur 4: Taktoidbilder og lengde- og breddeanalyse. (A) Eksempeldata for taktoider dannet som beskrevet og avbildet ved hjelp av roterende plate confocal viser (i) mikrotubule kanalavbildning rhodamin-merket tubulin ved hjelp av en 561 nm laser, (ii) GFP-MAP65 kanal bildebehandling GFP ved hjelp av en 488 nm laser, og (iii) fusjonert overleggsbilde fra mikrotubulekanalen (magenta) og GFP-MAP65 kanal (cyan). Overlappingsområder vises som hvite og viser at mikrotubulene og MAP65 colokaliseres nøyaktig. Skalalinje = 10 μm for alle bilder i (A). (B) Kvantifisering av taktoid lengde og bredde. (i) Bilde av en taktoid som skal analyseres uten etiketter. Skalalinje = 5 μm. (ii) Samme bilde som i (i), der lengden (heltrukket linje med linjeendepunkter) og breddemål (stiplet linje) er merket. Skalastang = 5 μm. (iii) Bredden ble målt ved å ta intensitetsprofilen over taktoiden ved vinkelrett komektor (stiplet linje) betegnet i (ii). Intensitetsprofilen var egnet til en gaussisk funksjon for å avsløre amplituden og bredden på taktoiden. Klikk her for å se en større versjon av denne figuren.
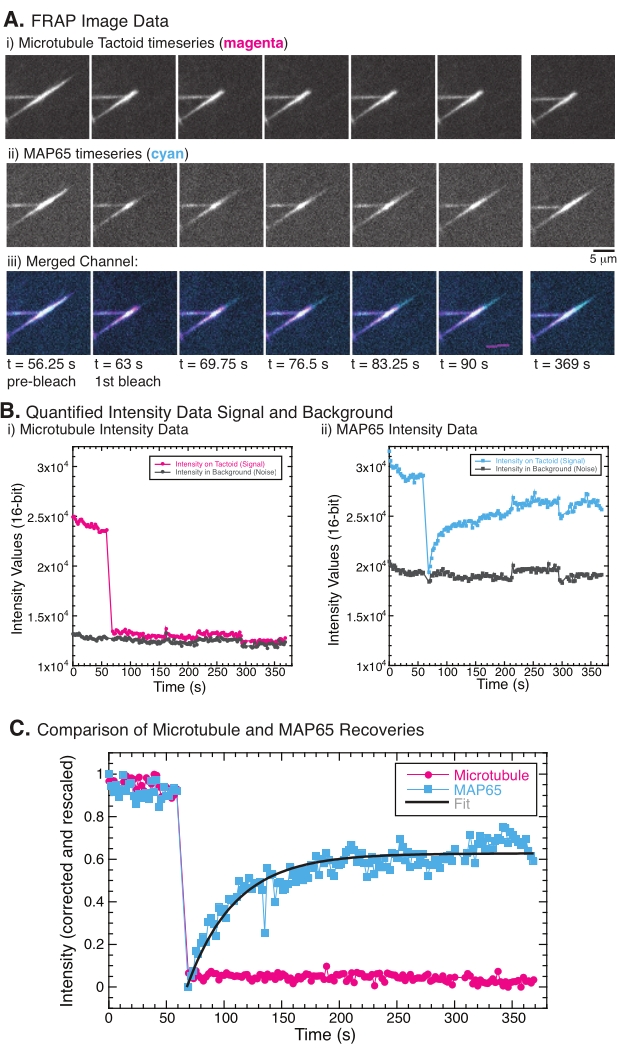
Figur 5: Representative FRAP-data og -analyser. (A) Mikroskopitidsseriedata av (i) mikrotubul tactoid og (ii) GFP-MAP65, og (iii) overleggsbilde av begge kanaler med mikrotubuler i magenta og GFP-MAP65 i cyan som ble fotobleket på tidspunktet 63 s og observert i ytterligere 5 min (B) Kvantifisert intensitet av (i ) mikrotubulkanal i bleket område (magenta sirkler) og bakgrunnen (mørkegrå sirkler) og (ii) GFP-MAP65 kanal i bleket område (cyan firkanter) og bakgrunnen (mørkegrå firkanter). (C) Dataene ble korrigert for bakgrunnsstøyen og skalert på nytt for mikrotubulkanalen (magenta sirkler) og GFP-MAP65-kanalen (cyan firkanter). Mikrotubulene gjenoppretter ikke, men GFP-MAP65 gjør og kan passe (mørkegrå linje) til et stigende eksponentielt forfall for å finne amplituden og tidsskalaen for utvinning. Klikk her for å se en større versjon av denne figuren.
Metodene beskrevet her har blitt brukt i flere artikler for å lage mikrotubule taktoider (figur 2)22,28. Disse eksperimentene er biologisk relevante for å bidra til å avdekke de organisatoriske prinsippene som styrer formen og stabiliteten til den mitotiske eller meiotiske spindelen i de fleste celletyper. I tillegg er mikrotubuler modell flytende krystallmetogener som kan hjelpe til med å lære mer om hvordan flytende krystaller nukleerer og vokser nematiske faser fra isotrope faser.
Prosedyren som er skissert her har flere fordeler for å utforske mikrotubul selvorganisering. For det første er det svært reproduserbart, etter å ha blitt utført i laboratoriet av mange studenter, inkludert elever på videregående skole, med liten forkunnskap eller trening før de begynte i laboratoriet. Taktoidene er birefringent22, slik at de kan sees i overført lys i tillegg til fluorescensmikroskopi, noe som gjør denne metoden tilgjengelig for mange laboratorier og denne eksperimentelle prosedyren som kan tilpasses pedagogiske formål, i tillegg til avansert forskning. Til slutt åpner denne prosessen veier for å fortsette å forstå og sondere biologiske systemer i en nedstrippet, reduksjonistisk tilnærming, slik at man kan forstå hvordan hver ekstra tilstand, protein eller additiv kan endre selvorganisering av taktoider og kanskje til slutt spindelen. Mål for bedre biomimicry inkluderer aktivitet, fluiditet og filament polaritet sortering.
Det kan være flere faktorer som påvirker eksperimentet og gir uventede resultater. Hvis taktoidene for eksempel ikke dannes (figur 2), men viftelignende mønstre observeres, er MAP65 sannsynligvis ikke til stede eller ikke bindende for mikrotubulene22,28. Dette bør også være åpenbart i MAP65 fluorescenskanalen fordi GFP-MAP65 ikke vil være bundet til mikrotubulene.
Hvis taktoidene ikke dannes og bakgrunnen vises som flekker på glasset, kan dette skyldes overflatebelegget. Når den er utført, varer silaniseringen bare 1 måned på coverlips. Når den slites av, vil tubulinen kunne binde seg til den eksponerte overflaten ikke-spesifikt. Denne bindingen vil forekomme i rare mønstre.
Hvis taktoidene ikke dannes og tubulin observeres i aggregater av forskjellige former og størrelser, kan dette skyldes tubulin av dårlig kvalitet. Tubulin kan sentrifugeres for å fjerne innledende aggregater som kan drive denne off-pathway aggregasjonen i stedet for mikrotubul polymerisering. Hvis overflaten er bindende til tubulinen, kan den også tømme tubulinen i løsningen. Lave konsentrasjoner av tubulin, under den kritiske konsentrasjonen for polymerisering av mikrotubuler, kan resultere i aggregater.
I FRAP-eksperimenter, hvis MAP65-kanalen ikke viser noen gjenoppretting (figur 5), er det mulig at fotobleaching fotodamaging mikrotubules. Fotodamage forårsaker lokalisert ødeleggelse av filamentene. Dette kan kontrolleres ved undersøkelse i den overførte kanalen. Mikrotubule taktoider er synlige i den overførte kanalen gjennom en høy indeks av brytningskonflikt med det omkringliggende vannet. Lysindusert fotodamage vil vises som et brennmerke eller tap av kontrast i overført lysavbildning på stedet der avkastningen utsettes for fotobleking. Hvis dette skjer, må laseren eller lyseffekten reduseres for å hemme proteinenes fotodamage.
Det var flere utfordringer i denne prosedyren og tilnærmingen. Et problem er at lengdemålene for øyeblikket utføres for hånd ved å klikke på bildet. Denne metoden, selv om den er grei, kan føre til stor usikkerhet. Breddemålet som bruker tverrsnittet og tilpasningen til en gaussianer, er en bedre metode for å kvantifisere størrelsen. En lignende metode kan brukes for lengden. Et annet problem er at noen ganger kan taktoidene, fordi de er så lange og tynne, bøye seg. Dette gjør kvantifisering av lengden vanskeligere. Konturlengden kan kvantifiseres ved hjelp av en segmentert linje, men det er ekstra usikkerhet hver gang et segment legges til.
Fra et vitenskapelig perspektiv har denne tilnærmingen noen andre utfordringer for bruken som modell for flytende krystaller eller spindler. Den første utfordringen har vært den lange, tynne formen på taktoidene som mikrotubuler skaper (figur 3 og figur 4). Som nevnt i tidligere publikasjoner22, er mikrotubule taktoider homogene taktoider, ikke bipolare. Dette betyr at mikrotubulene som utgjør formen ikke reorienterer for å peke mot spissene på strukturen. I stedet er alle mikrotubulene parallelle med den lange aksen, og "polene" ligger ved uendelig. Dette er svært forskjellig fra taktoidene som observeres for molekylære flytende krystaller eller til og med for aktin eller DNA som også kan fungere som flytende krystallmetogener. I disse andre systemene er taktoidene bipolare, og når de ses i kryssede polarisatorer, viser de de fortellende tegn på reorientering av stengene.
En annen stor utfordring i dette systemet er at mikrotubulene er immobile inne i taktoiden. Dette er klart fra FRAP-eksperimenter og analyse, da utvinningen av mikrotubulene er svært lav. Deres solide natur gjør mikrotubule taktoider mindre verdifulle som store flytende krystallanaloger. Den nematiske fasen av en flytende krystall skal ha både flytende (væske) og krystall (organisert) egenskaper. Selv om formen virker riktig for spindelen, gjør immobiliteten systemet mindre spennende som en modellgeitotisk spindel. På den annen side gir dette problemet muligheter til å undersøke hvordan man kan endre forsøkene for å skape mer flyt i systemet.
Disse vitenskapelige utfordringene gir spennende muligheter som vil gi ny kunnskap om systemet. For å gjøre mikrotubule taktoidene mer bipolare, kan man bruke kortere mikrotubuler. Det er imidlertid en ekstra utfordring, siden mikrotubuler ikke har mange godt karakteriserte cappingproteiner for å kontrollere lengden som aktin gjør. Bruk av kjernedannelse og vekst krever bruk av svært høye konsentrasjoner av tubulin og GMPCPP for å lage korte mikrotubuler. Den høye tubulinkonsentrasjonen resulterer i et større antall filamenter i systemet, noe som gjør det vanskeligere å skille taktoider fra hverandre. Tillegg av nye mikrotubule cappers, for eksempel DARPin33, kan hjelpe med denne situasjonen. Det andre problemet med at mikrotubulene er immobile, kan reduseres ved tilsetning av motorproteiner, som kinesin-534, som er tetramerer av motorer som brukes i mitose. Alternativt kan kunstige dimmere av dimeriske kinesin-1 brukes15.
En annen måte å legge til mer flyt på ville være å la mikrotubulene utføre sin dynamiske ustabilitet, vekst og krymping av mikrotubuler. For tiden er mikrotubuler som er sådd med stabile GMPCPP-filamenter og deretter gjennomgår dynamisk ustabilitet langt lengre enn ønsket for å danne en spindel eller taktoid, noe som vil resultere i svært lange organisasjoner som fans eller bunter. Så å legge til mikrotubul dynamisk ustabilitet må gjøres nøye for å bevare taktoidformen. Tilsetningen av tilknyttede proteiner og enzymer som kan kontrollere lengden, kan redusere dette problemet. For eksempel vil depolymeriserende kinesiner, som kinesin-1335, eller kutte enzymer, som katanin36, sannsynligvis være nødvendig. Disse eksperimentene er komplekse og vanskelige, selv om de ville være veldig innsiktsfulle uansett hva resultatene avslører. Uansett hvilken retning fremtidige eksperimenter tar, kan plattformen som er utviklet her for å lage mikrotubule taktoider, avsløre ny informasjon på fysisk grunnlag av mikrotubulorganisasjon.
Forfatterne erklærer at de ikke har konkurrerende økonomiske interesser.
Forfatterne vil takke alle Ross Lab-medlemmene sommeren 2021, spesielt K. Alice Lindsay, for deres hjelp. Dette arbeidet ble støttet av et stipend fra NSF BIO-2134215 som støttet S. Sahu, N. Goodbee, H.B. Lee og J.L. Ross. Et stipend fra KECK Foundation (Rae Anderson, USD, lead PI) støttet delvis R. Branch og P. Chauhan
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved