
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
Auto-montagem de Tactóides microtúbulos
Este artigo apresenta um protocolo para a formação de conjuntos de microtúbulos na forma de tatoides usando MAP65, um interlinker de microtúbulos à base de plantas, e PEG como um agente de aglomeração.
O citoesqueleto é responsável pela grande organização interna e reorganização dentro da célula, tudo sem um gerente para direcionar as mudanças. Este é especialmente o caso durante a mitose ou meiose, onde os microtúbulos formam o fuso durante a divisão celular. O eixo é o maquinário usado para segregar material genético durante a divisão celular. Para criar fusos auto-organizados in vitro, recentemente desenvolvemos uma técnica para reconstituir microtúbulos em conjuntos semelhantes a fusíveis com um conjunto mínimo de proteínas associadas a microtúbulos e agentes de aglomeração. Especificamente, map65 foi usado, que é um interlinker antiparalha de microtúbulos de plantas, um homólogo de Ase1 de levedura e PRC1 de organismos mamíferos. Este crosslinker auto-organiza microtúbulos em conjuntos auto-organizados de microtúbulos longos, finos e semelhantes a fusíveis. Estes conjuntos também são semelhantes aos tatoides de cristal líquido, e microtúbulos poderiam ser usados como mesogênicos mesoescala. Aqui, são apresentados protocolos para a criação desses tatoides microtúbulos, bem como para caracterizar a forma dos conjuntos utilizando microscopia de fluorescência e a mobilidade dos constituintes utilizando recuperação de fluorescência após fotobleachamento.
A divisão celular via mitose é um dos processos biológicos mais importantes para sustentar a vida. Os filamentos microtúbulos, compostos por dimers tubulin, são elementos estruturais essenciais desse processo. A maquinaria transitória criada em metafase quando os cromossomos estão se alinhando no centro celular é chamada de fuso mitótico devido à sua forma, que é como um fuso de um tear coberto de fios (Figura 1A). É bem estabelecido em muitos organismos que microtúbulos são usados em metafase para empurrar e puxar cromossomos condensados para o centro da célula, alinhando-os e conectando-os a microtúbulos que os separarão em anaphase (Figura 1B,C). O fuso se forma tanto em meiose (Figura 1B) quanto em mitose (Figura 1C), criado a partir de muitos microtúbulos sobrepostos não envoltos ao redor do eixo central como o fio, mas correndo paralelamente à interface. A criação dessas estruturas baseadas em microtúbulos requer proteínas associadas que cruzam e enzimas associadas que podem atuar como motores para ajudar a empurrar e puxar os cromossomos1.
Estudos de fusos meioticos mostraram que os microtúbulos são curtos, dinâmicos e sobrepostos em matrizes interligadas 2,3,4,5,6 (Figura 1Di). Devido à organização física desses microtúbulos curtos, o fuso meiotic é semelhante a um tatoide de cristal líquido (Figura 1E). De fato, os fusos têm sido mostrados para coalesce e fusão, como seria de esperar dos tatoides de cristal líquido5.
Muitos estudos que datam da década de 1960 usaram fixação, seções seriais e microscopia eletrônica para determinar que existem dois tipos de microtúbulos dentro do fuso mitotístico 7,8,9,10. O primeiro tipo é chamado de microtúbulos kinetochore, que conectam o polo do eixo ao kinetochore. O segundo tipo é chamado de microtúbulos interpolares ou polares, que crescem além dos cromossomos e se sobrepõem à zona média (Figura 1Dii)8,9,10. Um terceiro tipo é chamado de microtúbulos astrais, que estão fora do eixo e conectam os polos à borda celular; essas organizações de microtúbulos estão fora do escopo da discussão atual. Tem havido estudos recentes sobre a interação entre o complexo de anel augmin6 e gama-tubulina influenciando os centros de nucleação para microtúbulos, resultando em um fuso mitotístico com microtúbulos mais curtos como na Figura 1D.
Como os microtúbulos são mais longos do que são largos, com uma alta proporção e alta rigidez, eles são como versões dimensionadas de moléculas de cristal líquido. Na física da matéria macia, átomos e moléculas foram aproximados usando interações mínimas para deduzir os mecanismos físicos das transições de fase, incluindo a nucleação e o derretimento dos cristais11. Da mesma forma, microtúbulos são objetos mesoescalados que são versões dimensionadas de moléculas de cristal líquido, dando insights sobre a física da dinâmica do cristal líquido, incluindo a nucleação e o crescimento das fases nemáticas das isotrópicas. Além disso, como discutido acima, o fuso meiotic exibe propriedades como as de um tatoide de cristal líquido, um estado nemático que nuclea e cresce a partir do estado isotrófico de moléculas de cristal líquido 3,4,5. Para tatoides, nucleação e crescimento são como os de outros cristais (ou seja, exigindo uma concentração relativamente alta de mesógenos [as moléculas que formam cristais líquidos]). A forma única de "fuso" do tatoide vem do alinhamento local dos mesógenos de cristal líquido que se alinham à fase nemática (Figura 1E). Eles não podem formar um cristal arredondado porque as moléculas são altamente assimétricas. Dada a natureza dos microtúbulos, talvez não seja surpreendente que o fuso-máquinas mitóticos feitos a partir de uma alta concentração local de microtúbulos também seja da mesma forma, seja ele chamado de tatoide ou fuso. Os tatoides podem ser bipolares, com polos nas extremidades afiladas (Figura 1Ei), ou homogêneas, com polos efetivamente no infinito (Figura 1Eii).
Dada a importância da formação do fuso, esforços têm sido em andamento para a formação de fusos auto-organizados in vitro, demonstrando condensação de microtúbulos em feixes através de espécies iônicas12,13, agentes de aglomeração que criam interações de esgotamento14,15, e proteínas de microtúbulo específicos 13,16,17,18,19, 21. Surpreendentemente, embora todos esses agentes trabalhem para aumentar a concentração local de microtúbulos, eles muitas vezes resultam em longos feixes de microtúbulos, mas não tatoides. Uma das razões pelas quais esses pacotes são longos pode ser que os microtúbulos que os compõem também são longos. Trabalhos recentes usando microtúbulos mais curtos também relataram feixes mais longos que não são afunilados no finaldos 15 anos; Neste caso, os feixes são mantidos em conjunto com proteínas motoras que causam a extensão dos feixes e, assim, torná-los mais longos. Microtúbulos curtos com crosslinkers não extensíveis são necessários para conjuntos afilados, semelhantes a fusíveis, conforme descrito aqui.
Recentemente, desenvolvemos uma técnica para permitir a criação de tatoides microtúbulos utilizando o crosslinker antiparallel, MAP65, na presença de microtúbulos estáveisnucleantes 22. Os microtúbulos precisavam ser curtos, mas poucos reguladores conhecidos de comprimento de microtúbulos podem limitar microtúbulos contra instabilidade dinâmica ou ressarcer de ponta a ponta. Em vez disso, o GMPCPP foi usado para nuclear e estabilizar os filamentos após o crescimento. Isso permitiu criar uma alta densidade de microtúbulos curtos que poderiam se auto-organizar em tatoides. Estes tatoides eram homogêneos quando vistos sob birefringência. Além de microtúbulos curtos, um crosslinker antiparalha específico, MAP65, foi empregado para formar os tatoides (Figura 2). MAP65 é uma proteína associada a microtúbulos vegetais na família PRC1/Ase1 de cruzlinkers mitotéticos23. MAP65 existe como um dimer, com uma forte afinidade para se ligar a si mesmo, bem como aos microtúbulos24. Ao contrário do fuso meiotico e dos tactóides observados com filamentos de actina 25,26,27, que são bipolares e possuem as propriedades líquidas de cristais líquidos, os tatoides microtúbulos têm sido observados comosólidos 22,28.
Aqui, são apresentados protocolos para a criação dos tactóides microtúbulos e caracterização da forma dos conjuntos e da mobilidade dos constituintes utilizando técnicas baseadas em fluorescência.
NOTA: Salvo afirmou o contrário, partes do experimento podem ser realizadas em um banco de laboratório enquanto usam equipamentos de proteção apropriados (luvas).
1. Silanização de deslizamento de cobertura
NOTA: As manchas devem ser silanizadas para serem utilizadas com o revestimento de pincel de polímero empregado nestes experimentos. Trata-se de um tratamento de silanização hidrofóbica que permite que um copolímero de bloco com um bloco central hidrofóbico se ligue e crie uma escova de polímero. As etapas a seguir devem ser realizadas em um capô de fumaça para evitar a exposição a vapores tóxicos durante o uso de luvas. Dimetildcloolsilane é altamente tóxico e deve ser tratado com o maior cuidado.
- Enxágüe as tampas com ddH2O, 70% etanol e ddH2O em ordem. Seque-os com lenços de laboratório sem fiapos entre cada enxágue. Isso remove poeira e partículas solúveis em água ou orgânicas da superfície antes do tratamento.
- Coloque as tampas em um rack de retenção de tampas metálicas e transfira o rack para uma máquina UV-Ozone (UVO). Irradiar as tampas com UVO por 20 min para remover qualquer fluorescência de fundo. Uma câmara de plasma pode ser usada em vez de UVO.
- Utilizando pinças, transfira as tampas do rack de metal usado para o tratamento UVO para um rack de metal diferente usado para silanização. Não utilize os mesmos racks para ambos, pois causará altos níveis de oxidação quando o UVO for aplicado. Limpe previamente os racks com água e etanol para que não haja produtos químicos residuais de usos anteriores.
- Mergulhe o rack com as tampas em um recipiente com 100% de acetona por 1 h. Enxágüe o recipiente 3x com água da torneira e depois 3x com ddH2O para remover toda a acetona.
- Mergulhe o rack com as tampas em 100% etanol por 10 minutos. Enxágüe o recipiente 3x com água da torneira e depois 3x com ddH2O para remover todo o etanol.
- Mergulhe o rack com as tampas 3x em ddH2O por 5 min cada.
- Mergulhe o rack com as tampas em 0,1 M KOH (50 mL de 1 M KOH em 450 mL de ddH2O) por 15 min. Enxágüe o recipiente 3x com água da torneira e depois 3x com ddH2O para remover todo o KOH.
- Mergulhe o rack com tampas 3x em ddH2O por 5 min cada.
- Seque o rack com as tampas durante a noite em um capô de fumaça ou capô de fluxo laminar.
- Depois de secar completamente o rack e as tampas, mergulhe-os por 5 min em 2% de dimetildichlorosilano (DDS) tomado em um recipiente diferente que é usado especificamente para silano. Não deixe que nada que não esteja seco entre em contato com o silano.
- Mergulhe o rack e cubra 2x em um recipiente com 100% de etanol por 5 min. Enxágüe o recipiente 3x com água da torneira e depois 3x com ddH2O.
- Mergulhe o rack e cubra 3x em ddH2O por 5 min cada.
- Seque o rack com as tampas durante a noite em um capô de fumaça ou capô de fluxo laminar.
- Após esta etapa final de secagem, transfira as tampas de volta para caixas de deslizamento de cobertura usando pinças. Estas tampas podem ser usadas nos próximos 1-2 meses. As tampas antigas começarão a perder seu revestimento e devem ser descartadas.
2. Preparação de tubulina
NOTA: A tubulina comprada vem como um pó liofilizado que não tem rótulo ou rótulo com fluoroforos. A tubulina liofilizada é armazenada em um congelador de −80 °C. O procedimento a seguir é usado para misturar tubulina sem rótulo com tubulina rotulada em uma proporção que é boa para visualização.
- Retire do congelador de −80 °C uma alíquota de tubulina sem rótulo contendo 1 mg de pó de tubulina liophilizada e mantenha-a no gelo. Adicione 200 μL de PEM-80 frio ao tubo para levar a concentração de tubulina a 5 mg/mL. Mantenha-o no gelo por 10 minutos para dissolver todo o liofilato.
- Retire do congelador de −80 °C uma alíquota de tubulina rotulada por rhodamina contendo 20 μg de pó de tubulina liofilizada e mantenha-a no gelo. Adicione 4 μL de PEM-80 frio ao tubo para levar a concentração de tubulina a 5 mg/mL. Mantenha-o no gelo por 10 minutos para dissolver totalmente o liofilato.
- Uma vez dissolvida, adicione 100 μL de solução de tubulina resuspended não rotulada à solução de 4 μL de tubulina com rótulo de rhodamina. Pipeta 6x-7x muito lentamente para misturar. Se os agregados forem visíveis, centrifugar a tubulina solubilizada por 10 min a 90.000 x g para remover agregados descartando a pelota e retendo o sobrenatante. Esta mistura de tubulina resultará em ~4% de tubulina rotulada.
- Congele os 100 μL restantes de tubulina sem rótulo em nitrogênio líquido (LN2) e armazene-o a −80 °C para ser usado para misturas adicionais de tubulina.
- Leve a mistura de tubulina rotulada e aliquot em sete tubos com 15 μL cada. Cada alíquota pode ser usada para uma única câmara experimental. Congele as alíquotas restantes e armazene-as a −80 °C para experimentos futuros.
3. Purificação MAP65
NOTA: O MAP65 não está disponível comercialmente e, portanto, precisa ser purificado para este trabalho. O protocolo já foi elaborado anteriormente em diversas publicações 23,29.
- Transforme o plasmídeo MAP65 e o plasmídeo GFP-MAP65 em cepa de bactérias BL21 para expressão proteica.
- Cresça bactérias BL21 a uma densidade óptica de 0,6-1 a 600 nm. Induzir a produção de proteínas usando o operador de lac e cultivar as bactérias durante a noite.
- Pelotar as culturas e lise as bactérias.
- Colete o lysate após a centrifugação e incuba-o com contas que têm um íon de níquel disponível para amarrar a etiqueta 6x-histidina.
- Elute a proteína usando imidazol e deságua..
- Congele a proteína com nitrogênio líquido e armazene-a a -80 °C para uso dentro de 1 ano.
4. Montagem de câmaras de fluxo
NOTA: Os experimentos são realizados em câmaras de fluxo feitas a partir de um escorregador de vidro e vidro de cobertura silanizado (Figura 3).
- Pegue um slide de vidro e limpe-o usando ddH2O, etanol e ddH2O em ordem. Seque com um lenço de laboratório sem fiapos entre cada enxágue.
- Use um pedaço de fita dupla face para criar um caminho de fluxo. Usando as mãos enluvadas, corte a fita para ~25-30 mm de comprimento. Divida a fita longitudinalmente para criar duas tiras mais finas. Coloque as duas tiras de fita no slide com cerca de 5-8 mm entre elas.
NOTA: Como a espessura da fita é padronizada para cerca de 80-100 μm, a largura do caminho entre as peças de fita determinará o volume na câmara. - Coloque as tampas silanizadas em cima do caminho de fluxo. Sele o slide e a tampa nas tiras de fita de duas faces pressionando suavemente a região da fita com a parte de trás de uma caneta. Certifique-se de obter um bom selo sobre toda a área; a fita deve passar de translúcida para clara quando o selo é bem feito.
- Remova a fita extra nas bordas, deixando apenas 1 mm da entrada da câmara de fluxo cortando a fita com uma lâmina de barbear.
- Rotule a câmara com informações sobre os parâmetros experimentais, conforme necessário.
5. Experimentos de tatoide
NOTA : Uma vez gerados todos os reagentes e suprimentos, eles podem ser usados para nuclear e politizar tactóides de microtúbulos na câmara de fluxo.
- Colete todos os reagentes a serem usados. Descongele-os no gelo e guarde-os no gelo enquanto trabalha. Faça várias câmaras de fluxo para os experimentos.
- Use uma câmara de fluxo para cada experimento. Cubra as superfícies da câmara de fluxo com uma escova de polímero fluindo em 20 μL de 5% de surfactante de bloco não iônico (Tabela de Materiais) dissolvido em PEM-80, com pequenas gotas nas duas extremidades da câmara para evitar a formação de bolhas de ar no interior. Mantenha isso em uma câmara úmida (ou seja, placa de Petri com um lenço de laboratório sem fiapos molhados) até estar pronto para usar (pelo menos 5-7 min).
- Em um tubo estéril, misture o seguinte para criar a mistura Tubulin-MAP: 9,5 μL de PEM-80; 4 μL de 10 mM GMPCPP; 4 μL de 5% Pluronic-F127; 1 μL de 1M DTT; 1 μL de glicose; 2 μL de polietilenoglicol (PEG); 12 μL de 5 mg/mL de mix de tubulina (concentração final de 13,6 μM) da Etapa 2.5.; e 5,5 μL de estoque de trabalho da MAP65, onde 10% é GFP-MAP65 para visualização. Mantenha o gelo enquanto mistura.
NOTA: Recomenda-se o uso de uma pipeta de deslocamento positiva para o manuseio da solução PEG viscosa. Pipetas regulares podem ser usadas após cortar a ponta para tornar a abertura maior; no entanto, este método é menos preciso. - Misture 5x-6x por pipetação.
- Pouco antes de adicionar à câmara, adicione 1 μL de uma solução pré-misturada de glicose oxidase (0,5 mg/mL) e catalase (0,15 mg/mL) (Deoxy) na mistura Tubulin-MAP e misture 7x-8x. Divida o volume total da solução (40 μL) em duas porções a serem utilizadas em câmaras separadas.
- Flua a mistura Tubulin-MAP em câmaras. Como as câmaras já possuem um surfactante de bloco não iônico, mais líquido não pode ser adicionado sem remover o líquido antigo. Para isso, use um pedaço de papel filtro ou um lenço de laboratório sem fiapos na outra extremidade da câmara para remover o líquido através de ação capilar.
- Uma vez que a amostra esteja totalmente dentro da câmara, sele as duas extremidades da câmara usando epóxi de 5 minutos e mantenha-a a 37 °C por ~30 min para nuclear e cultivar tatoides microtúbulos.
6. Microscopia de fluorescência
- Use um microscópio de fluorescência para fotografar os tactóides.
NOTA: A microscopia de fluorescência de reflexão interna total ou microscopia confocal do disco giratório são boas para remover a fluorescência de fundo da tubulina livre, mas os táctidos também são visíveis por epi-fluorescência regular e até mesmo microscopia de luz transmitida uma vez formada, tornando este procedimento acessível sem equipamento especializado. - Use um objetivo de abertura numérica (NA) de 1,2 NA ou superior com uma ampliação de 60x ou mais para coletar luz suficiente em fluorescência. Esses objetivos muitas vezes requerem imersão em ddH2O ou óleo.
- Grave imagens com CMOS ou câmera CCD. Use um tamanho de pixel eficaz na câmera de 108 nm.
NOTA: O tamanho do pixel depende da câmera e da ampliação utilizada, que neste caso foi de 60x ou 100x com uma alta abertura numérica (1,2 ou 1,49 NA). Expansores adicionais de imagem podem ser usados antes da câmera para alcançar o tamanho necessário do pixel. - Mantenha a amostra a 37 °C usando uma câmara ambiental definida para esta temperatura. Alternativamente, empregue outros aquecedores de estágio, incluindo aquecedores de palco de ar quente e colares controlados pela temperatura objetiva com água quente circulante.
- Use fontes de excitação corretas para a fluorescência necessária. Para a tubulina de rhodamina, use um laser de 561 nm com pelo menos 1 mW de potência na amostra, e para GFP-MAP65, use um laser de 488 nm com pelo menos 1 mW de potência na amostra.
NOTA: Se usar microscopia de epi-fluorescência de campo largo, use um cubo de filtro de rhodamina com excitação: 540 ± 12,5 nm, dicroico: 545 nm ± corte de 12,5 nm, e emissão: 575 nm de passagem longa, e um cubo de filtro GFP com excitação: 480 ± 15 nm,dicroico: 505 nm ± corte de 15 nm e emissão: 515 nm de passagem longa. - Tire pelo menos 10 imagens de diferentes áreas para imagem de mais de 100 tatoides. Tire imagens nos canais vermelho e verde e salve-as como imagens de tiff de 16 bits para análise. Certifique-se de que o poder de iluminação e os tempos de exposição são tais que a escala de intensidade para a câmera não esteja saturada.
7. Recuperação da fluorescência após fotobleaching (FRAP)
NOTA: Para estudar a mobilidade dos constituintes internos dos tatoides, foi utilizado o FRAP. O FRAP funciona fotobleaching uma parte selecionada de tubulina com rótulo de rhodamina e táctóide MAP65 rotulado gfp e, em seguida, observando a recuperação da fluorescência com o tempo naquela região. A taxa de recuperação depende do volume de negócios das espécies que estão sendo fotobleachadas. Essa taxa de rotatividade pode depender de difusão e reações vinculantes. Para a vinculação MAP65 aos tactóides, podem ser estimadas taxas de câmbio vinculantes. FRAP é realizado usando um sistema laser adicional de 405 nm que pode escanear o laser em qualquer forma. Existem muitas possibilidades para realizar FRAP, incluindo o uso da lâmpada transmitida e abertura para fotobleach uma área local14.
- Selecione um tatoide isolado na câmara para criar uma região de interesse (ROI) cobrindo partes dos tatoides e da solução circundante.
- Use um microscópio com um laser adicional de 405 nm para FRAP para fotobleach tanto a tubulina quanto o MAP65 simultaneamente. Alternativamente, uma lâmpada brilhante pode ser usada através da parada de campo da íris14. Ajuste a intensidade específica dos sistemas de fotobleachamento empiricamente para evitar danificar as proteínas durante o branqueamento.
- Grave o tatoide como um filme de séries temporadas para 30-60 s antes de fotobleaching para obter informações sobre a intensidade antes do alvejante. Grave os canais vermelho e verde.
- Fotobleach o tatoide expondo o ROI com os lasers ou a lâmpada pelo tempo necessário para fotobleach sem danificar o tatoide. Determine a intensidade e o tempo empiricamente.
- Continue gravando o filme em ambos os canais de cores por 5-10 minutos postando fotobleaching ou até que a recuperação parece ter atingido o equilíbrio.
- Inspecione visualmente o canal GFP-MAP65 para recuperação.
8. Análise de dados
NOTA: A análise quantitativa das imagens dos tatoides foi realizada para conhecer os efeitos das mudanças ambientais impostas por meio de diferentes agentes de aglomeração, condições iônicas e a adição de outros fatores.
- Caracterização da forma tactóide
- Quantifique o comprimento e largura dos tatoides das imagens vermelha e verde tiradas com microscopia confocal.
- Abra as imagens usando FIJI/ImageJ.
- Se os dados brutos forem tomados em 16 bits, ajuste o brilho e o contraste, se necessário. Selecione > de imagem Ajuste > brilho e contraste para ajustar a imagem para poder ver o tatoide claramente. Ajuste o brilho e o contraste sem aplicar a configuração para não alterar acidentalmente os dados de intensidade.
- Uma vez que os tatoides estejam claramente visíveis, selecione bons tatoides para medir (Figura 4Bi). Certifique-se de que os tatoides são claramente visíveis sem sobreposição com qualquer outro tatoide ou agregados e não são curvados ou dobrados para poder usar ferramentas de medição em linha reta.
- Em seguida, verifique se o tamanho correto do pixel está definido para as imagens. As imagens do microscópio vêm com metadados sobre o tamanho do pixel. Ao usar uma câmera diferente que não tenha metadados ou sistemas externos de expansão de imagem que possam alterar o tamanho do pixel efetivo esperado, ajuste o tamanho do pixel manualmente. Em FIJI/ImageJ, vá para a Escala de conjunto de > analisar para definir a conversão correta de pixels.
- Usando a ferramenta Linha Reta da barra de ferramentas em FIJI/ImageJ, clique em uma extremidade do tatoide e arraste o cursor para a outra extremidade do tatoide (Figura 4Bii). Uma vez que o ROI de linha seja selecionado, selecione Analisar > Medida para medir o comprimento. Se o comprimento não for medido por padrão, certifique-se de definir a medida para incluir comprimento na caixa de diálogo Analisar > definir medidas .
NOTA: Normalmente, ao medir usando a ferramenta Linha Reta , ele dará o comprimento e o ângulo da linha desenhada. Como exemplo, a Figura 4Bii mostra uma linha reta desenhada para o lado do tatoide para tornar o último visível, mas tornar a medição diretamente no tatoide. - Após fazer a medição, use a ferramenta Texto na barra de ferramentas para rotular o tatoide. Crie uma caixa de texto, adicione um rótulo numéde e selecione Editar > Desenhar para corrigir o rótulo na imagem. Salve a imagem como um arquivo ROI separado.
NOTA: Rotular e salvar este arquivo permite que o investigador saiba qual medida corresponde a qual tatoide dos dados brutos. Certifique-se de medir cada tatoide uma vez. - Depois que os tatoides de toda a imagem forem medidos, salve os dados na janela Resultados para um arquivo de texto delimitado por vírgula ou guia (usando o Arquivo > Salve As) e abra os dados em um programa de planilha para analisar os dados em números. Colete todos os dados em conjunto (dados de imagem bruta, imagem roi e arquivo de texto dos resultados) em uma pasta com uma convenção de nomeação apropriada para manter tudo organizado.
NOTA: Embora as medidas de comprimento tatoide sejam realizadas manualmente, dado que as larguras tatoides são estreitas, é melhor empregar um método diferente para medir a largura tatoide (veja abaixo) para reduzir o erro de medição. - Usando ImageJ/FIJI, desenhe uma região de linha usando a ferramenta Linha Reta . Desenhe a linha como um bisetor perpendicular para o eixo longo tatoide (Figura 4Bii).
- Selecione Analisar > Perfil de Enredo para criar o perfil de intensidade do bisetor linear (Figura 4Biii). Um enredo aparecerá. Para recuperar e salvar os dados da trama, selecione o botão Listar no inferior esquerdo; isso gera a lista de arquivos de texto dos dados de intensidade ao longo do comprimento da linha desenhada. Salve o arquivo de texto como um arquivo .csv ou .txt.
- Abra o arquivo de texto em um programa de montagem, como MatLab, Python (sciPy) ou outros programas. Ajuste os dados de intensidade com uma função gaussiana da forma:
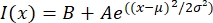 , onde I(x) é o valor em escala de cinza ao longo do comprimento, x; B é o nível de fundo; A é a amplitude do gaussiano; μ é a média ou o centro do gaussiano; e σ é o desvio padrão do gaussiano.
, onde I(x) é o valor em escala de cinza ao longo do comprimento, x; B é o nível de fundo; A é a amplitude do gaussiano; μ é a média ou o centro do gaussiano; e σ é o desvio padrão do gaussiano. - Relatório 2σ como a largura do tatoide. Estime a intensidade dos microtúbulos no tatoide calculando a área sob o gaussiano (sem incluir o fundo).
NOTA: Se as imagens estiverem dentro da faixa de intensidade linear da câmera e forem tiradas com o mesmo tempo de exposição e intensidade de excitação, intensidades integradas podem ser comparadas para estimar o número relativo de microtúbulos no tatoide.
- Análise FRAP
NOTA: Experimentos para testar a mobilidade dos microtúbulos e do MAP65 utilizaram FRAP para registrar o fotobleaching específico e a recuperação da intensidade devido ao movimento molecular (Figura 5A). Os dados foram quantificados a partir dos dados da série de tempo de imagem usando ImageJ/FIJI.- Use ImageJ/FIJI para abrir os dados do filme.
- Registre as pilhas (dados da série temporal) ao longo do tempo para remover a deriva. Use o plugin StackReg junto com o plugin Auxiliar TurboReg ; para obter instruções sobre o uso dos plugins, consulte os weblinks dados na Tabela de Materiais. Selecione a tradução para mudar a posição dos quadros e, assim, registre as imagens.
- Uma vez que as imagens sejam registradas para remover a deriva, gire a imagem para que o tatoide seja vertical ou horizontal no quadro, selecionando imagem > Transformar > Girar. Selecione o ângulo para girar e use o Preview para determinar se o tatoide está girado o suficiente. Quando a visualização mostrar que o tatoide é vertical ou horizontal, selecione OK para girar todas as imagens do filme.
- Use a ferramenta de seleção retângulo na barra de ferramentas para criar uma seção retangular sobre a região fotobleachada do tatoide. Regissuça a intensidade integrada da área do ROI para cada quadro usando Image > Stacks > Measure Stack. Defina o tipo de medição como densidade integrada usando medidas de > de medição. Salve os dados de intensidade analisados mostrados na janela Resultados como um arquivo de texto em formato .csv ou .txt, selecionando Arquivo > Salvar As.
NOTA: A Figura 5B mostra um exemplo dos dados de intensidade bruta de 16 bits medidos para os canais microtúbulo e GFP-MAP65 na região alvejante. - Como a intensidade geral das imagens desaparecerá ao longo do tempo globalmente devido ao fotobleaching causado por imagens, este fotobleaching global deve ser corrigido. Para isso, use o mesmo tamanho do ROI (Passo 8.2.4.), e mova-o para uma região no fundo da imagem onde não há microtúbulos ou MAP65 visíveis. Meça a intensidade integrada da pilha conforme descrito na Etapa 8.2.4. Salve os resultados como um segundo arquivo de texto.
NOTA: A Figura 5B mostra um exemplo dos dados de intensidade brutos de 16 bits medidos para os canais microtúbulo e GFP-MAP65 na região de fundo. - Para corrigir o desbotamento do fundo, divida a intensidade do sinal no tatoide pela intensidade de fundo para o mesmo ponto de tempo. Calculo quecorrigi (t) como:
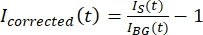 , onde IS (t) (o sinal) é a medição feita na região branqueada e IBG (t) (o ruído) é a medição feita na região de fundo (Figura 5C). Isso calcula a relação sinal-ruído para cada quadro e subtrai o ruído também.
, onde IS (t) (o sinal) é a medição feita na região branqueada e IBG (t) (o ruído) é a medição feita na região de fundo (Figura 5C). Isso calcula a relação sinal-ruído para cada quadro e subtrai o ruído também. - Em seguida, redimensione os dados para variar entre zero e um utilizando
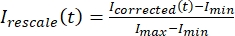 , onde eumin e eumax denotar, respectivamente, o mínimo global e o máximo dos dados corrigidos ao longo de todo o tempo (Figura 5C).
, onde eumin e eumax denotar, respectivamente, o mínimo global e o máximo dos dados corrigidos ao longo de todo o tempo (Figura 5C). - Encaixe esses dados em um exponencial decadente da forma:
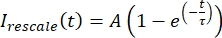 , onde A é a amplitude da recuperação e τ é a escala de tempo da recuperação (Figura 5C).
, onde A é a amplitude da recuperação e τ é a escala de tempo da recuperação (Figura 5C).
Com apenas um pequeno número de componentes, escurecedores de tubulina e interlinkers de microtúbulos, tatoides de microtúbulos podem se formar (Figura 2A). Embora este protocolo descreva a incubação de microtúbulos nucleados e de cultivo em uma incubadora, a nucleação e o crescimento podem ser observados diretamente sob o microscópio (que estão completos dentro de 30 minutos) (Figura 2B). A concentração de tubulina é mantida constante em 13,6 μm e a ligação MAP65-MT em 10%.
A Figura 4 representa dados bem sucedidos. Os tatoides devem ser visíveis com um laser de 561 nm no canal tubulina e 488 nm no canal MAP65, que se sobrepõem perfeitamente entre si (Figura 4A). Um mistério do sistema é que a largura dos tatoides não parece variar sob uma variedade de alterações experimentais, incluindo a alteração dos comprimentos do microtúbulo, a concentração MAP65 e os agentes de aglomeração (Figura 4B)22,28. O comprimento é muito mais variável e depende tanto dos comprimentos do microtúbulo quanto da concentração MAP65 (Figura 4B)22,28.
Ao realizar o FRAP, observou-se que o sinal MAP65 se recupera, mas o sinal de microtúbulo não se recupera (Figura 5). A recuperação no FRAP deve-se à mobilidade e movimentação dos objetos rotulados e fotobleachados. No caso do MAP65, as moléculas escurecidas se dissociam e se afastam do microtúbulo e novas se movem para a região (Figura 5). A ligação MAP65 está em equilíbrio, de modo que a taxa de ligação e desvinculação é igual (medida em moléculas por segundo). Para os microtúbulos, não foi observada recuperação implicando que os microtúbulos não são capazes de sair do tatoide (Figura 5A, Bi, C). Além disso, não foi observada propagação da região escura, sugerindo que os microtúbulos são localmente imóveis e não um fluido dentro da forma tatoide.
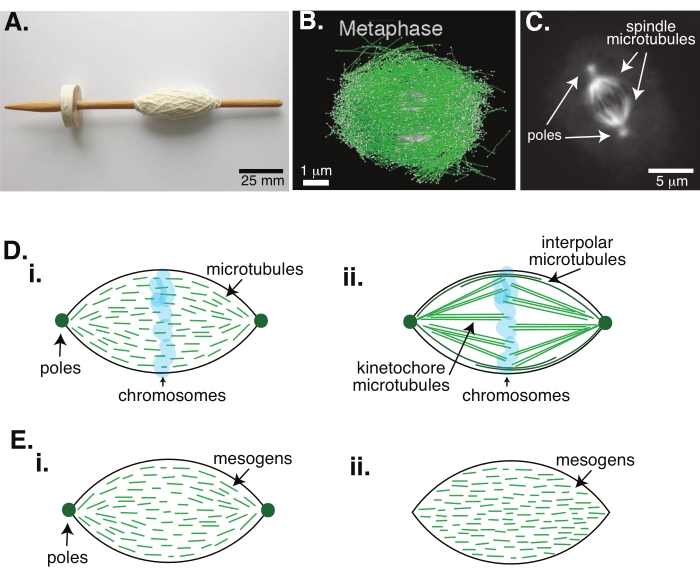
Figura 1: Diferentes modelos de formação de fuso. Um fuso mitotístico é uma máquina feita de microtúbulos e suas proteínas e enzimas associadas que alinha e separa os cromossomos nas duas novas células filhas durante a divisão celular. (A) Imagem de uma réplica de fuso de queda da Idade Média com fios finos da Holanda. Esta figura foi modificada a partir de uma imagem wikimedia por Peter van der Sluijs30. (B) Reconstrução tridimensional de microtúbulos em diferentes estágios da meiose selvagem II. Microtúbulos são mostrados em verde, e cromossomos são mostrados em cinza. Barra de escala = 1 μm. Este número foi modificado de Lantzsch et al.31. (C) Imagem de microscopia dos microtúbulos em um fuso mitotístico de uma célula Sf9 divisória. Os polos de fuso e os microtúbulos do fuso são rotulados com uma proteína fluorescente verde. Barra de escala = 5 μm. Este número foi modificado a partir de Advani et al.32. (D) Modelos diferentes de como microtúbulos de fuso místico e meiotic são organizados. (i) Previamente observados para fusos meioticos criados a partir de extratos de ovos xenopus, os microtúbulos (verde) foram deduzidos como curtos e dinâmicos em todo o eixo. Isto é semelhante a uma organização bipolar de tatoide dentro de um cristal líquido. (ii) O modelo canônico para organização de microtúbulos dentro de um fuso mitótico tem dois tipos de microtúbulos: microtúbulos interpolares ou polares (verde escuro) que se cruzam na zona média ao redor dos cromossomos e microtúbulos kinetochore (verde claro) que são agrupados e esticados do polo até a kinetochore para empurrar e puxar os cromossomos. Em todas as imagens, os cromossomos são mostrados em azul transparente, e os polos de fuso são representados em verde escuro. (E) Esquemas de mesógenos (linhas verdes) em um tatoide de cristal líquido para (i) tatoides bipolares e (ii) homogêneos. Tatoides bipolares têm dois polos no final do tatoide, e os mesógenos re-orientam para apontar para esses polos. Tatoides homogêneos têm polos no infinito, e os mesógenos não mudam a orientação ao longo do comprimento do tatoide. Clique aqui para ver uma versão maior desta figura.
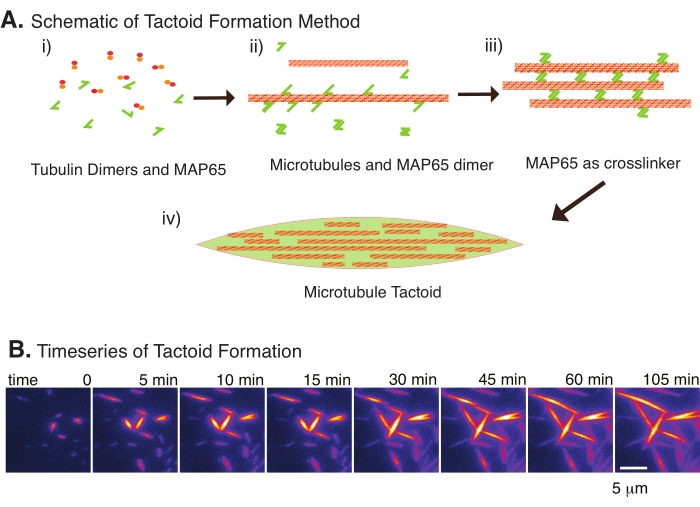
Figura 2: Condensação de microtubulos. (A) Microtubulos podem ser agrupados e interligados por uma variedade de métodos, incluindo espécies iônicas, forças de esgotamento causadas por agentes de aglomeração e interlinkers de microtúbulos específicos, como MAP65. i Os dimers tubulin e as proteínas MAP65 são misturados a nucleados e microtúbulos de cultivo. (ii) Os microtúbulos nucleam e crescem a partir da tubulina, e o MAP65 se liga imediatamente a microtúbulos, outro monômero MAP65, ou ambos e causa agrupamento. (iii) Microtúbulos nos feixes transligados nucleares e crescer. (iv) A configuração final é um tactóide de microtúbulo semelhante a um fuso. (B) Série temporal de tatoides microtúbulos nucleando e crescendo mais de 105 min. Barra de escala = 5 μm. Figura adaptada de Edozie et al.22. Clique aqui para ver uma versão maior desta figura.
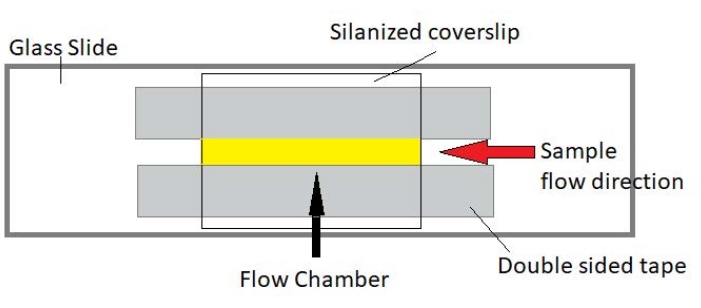
Figura 3: Montagem da câmara de fluxo. A câmara de fluxo é feita usando um escorregador de vidro, vidro de cobertura silanizado e fita permanente de dupla face. A região destacada amarela é o caminho de fluxo onde a amostra é escoada e observada. O volume da câmara de fluxo é ~20 μL. Epoxy foi usado para selar as extremidades da câmara para evitar que a amostra evaporasse durante a imagem de longo prazo ao longo de várias horas. Clique aqui para ver uma versão maior desta figura.
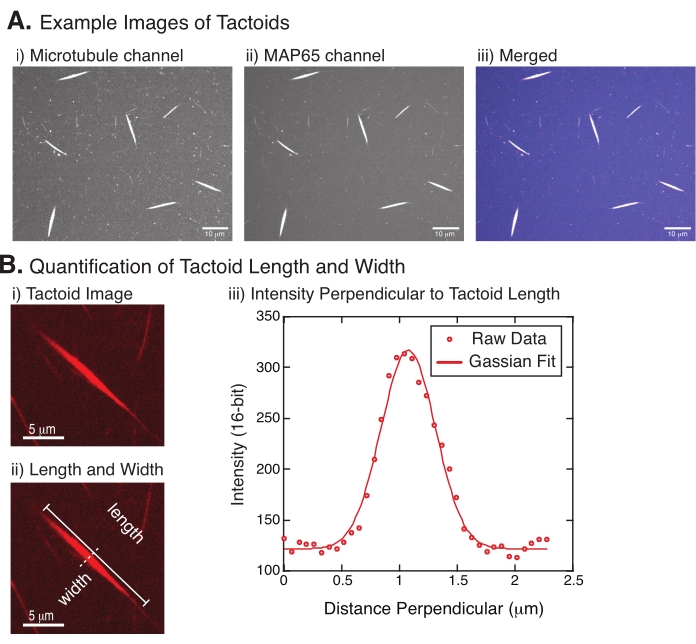
Figura 4: Imagens tactóides e análise de comprimento e largura. (A) Exemplos de dados de tatoides formados como descritos e imagens usando o disco giratório confocal mostrando (i) tubulina de canal microtúbulo com imagem de rodízio rotulado por rhodamina usando um laser de 561 nm, (ii) GFP-MAP65 canal de imagem do GFP usando um laser de 488 nm, e (iii) imagem de sobreposição mesclada do canal microtúbulo (magenta) e GFP-MAP65 (cyan). As regiões de sobreposição são exibidas como brancas e demonstram que os microtúbulos e MAP65 se colocam exatamente. Barra de escala = 10 μm para todas as imagens em (A). (B) Quantificação do comprimento e largura do tactóide. i Imagem de um tatoide a ser analisado sem rótulos. Barra de escala = 5 μm. (ii) Mesma imagem que em (i), onde as medidas de comprimento (linha sólida com tampas de linha) e largura (linha tracejada) são denotadas. Barra de escala = 5 μm. (iii) A largura foi medida tomando o perfil de intensidade através do tatoide no bisetor perpendicular (linha tracejada) denotado (ii). O perfil de intensidade foi adequado a uma função gaussiana para revelar a amplitude e largura do tatoide. Clique aqui para ver uma versão maior desta figura.
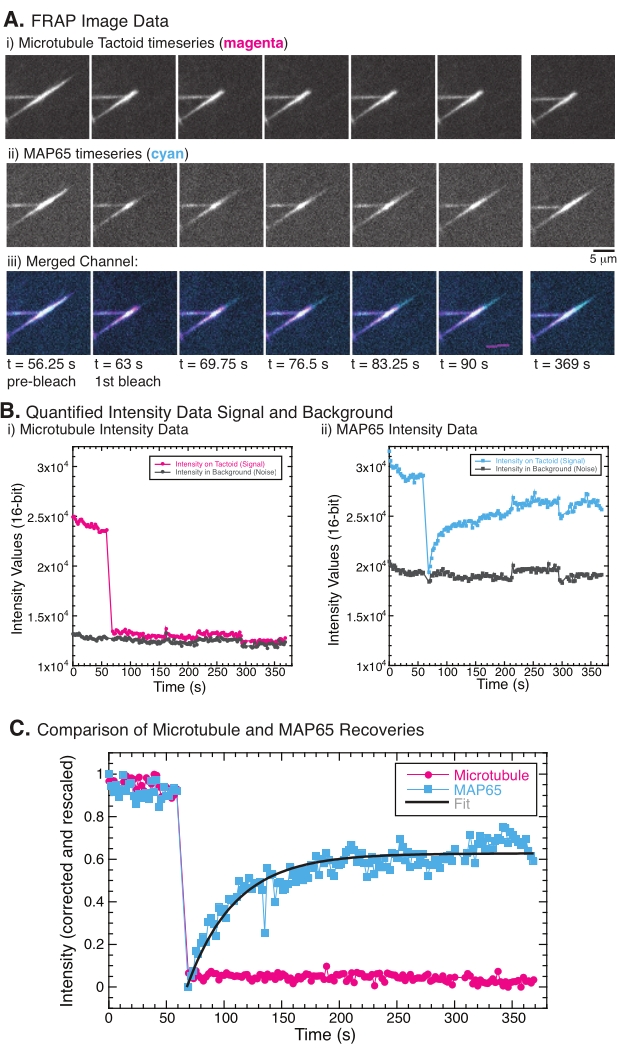
Figura 5: Dados e análises representativas frap. (A) Dados da série de tempo de microscopia de (i) tactóide de microtúbulo e (ii) GFP-MAP65, e (iii) imagem sobreposição de ambos os canais com microtúbulos em magenta e GFP-MAP65 em ciano que foram fotobleachados no momento 63 s e observados por uma intensidade quantificada adicional de 5 minutos (B) da intensidade quantificada (i i ) canal de microtúbulo na região branqueada (círculos magenta) e fundo (círculos cinza escuro) e (ii) canal GFP-MAP65 na região branqueada (quadrados de ciano) e no fundo (quadrados cinza escuro). (C) Os dados foram corrigidos para o ruído de fundo e redimensionados para o canal microtúbulo (círculos magenta) e o canal GFP-MAP65 (quadrados de ciano). Os microtúbulos não se recuperam, mas o GFP-MAP65 faz e pode caber (linha cinza escura) a uma crescente decadência exponencial para encontrar a amplitude e a escala de tempo de recuperação. Clique aqui para ver uma versão maior desta figura.
Os métodos aqui descritos têm sido utilizados em vários artigos para criar tatoides microtúbulos (Figura 2)22,28. Esses experimentos são biologicamente relevantes para ajudar a descobrir os princípios organizacionais que controlam a forma e estabilidade do fuso mitotístico ou meiotic na maioria dos tipos de células. Além disso, os microtúbulos são mesogens de cristal líquido modelo que podem ajudar a aprender mais sobre como cristais líquidos nucleam e crescem fases nemáticas a partir de fases isotrópicas.
O procedimento aqui descrito tem várias vantagens para explorar a auto-organização do microtúbulo. Primeiro, é altamente reproduzível, tendo sido realizado em laboratório por muitos alunos, incluindo estudantes do ensino médio, com pouco conhecimento ou treinamento antes de começar no laboratório. Os tatoides são birefringentes22, permitindo que sejam vistos em luz transmitida, além da microscopia de fluorescência, tornando este método acessível a muitos laboratórios e a este procedimento experimental adaptável a propósitos educacionais, além de pesquisas de ponta. Finalmente, esse processo abre caminhos para continuar a compreender e sondar sistemas biológicos em uma abordagem despojada e reducionista, permitindo entender como cada condição adicional, proteína ou aditivo pode alterar a auto-organização de tatoides e, talvez, em última instância, o fuso. Os alvos para melhor biomimimicmento incluem atividade, fluidez e classificação de polaridade de filamentos.
Pode haver vários fatores afetando o experimento dando resultados inesperados. Por exemplo, se os tatoides não se formam (Figura 2), mas padrões semelhantes aos ventiladores são observados, o MAP65 provavelmente não está presente ou não se liga aos microtúbulos22,28. Isso deve ser óbvio no canal de fluorescência MAP65 também porque o GFP-MAP65 não estará vinculado aos microtúbulos.
Se os tatoides não se formarem e o fundo aparecer como manchas no vidro, isso pode ser devido ao revestimento superficial. Uma vez realizada, a silanização dura apenas 1 mês em deslizamentos. Quando passar, a tubulina será capaz de se ligar à superfície exposta não especificamente. Esta ligação ocorrerá em padrões estranhos.
Se os tatoides não se formarem e a tubulina for observada em agregados de várias formas e tamanhos, isso pode ser devido à tubulina de má qualidade. A tubulina pode ser centrifugada para remover agregados iniciais que podem impulsionar essa agregação fora da via em vez de polimerização de microtúbulos. Se a superfície estiver ligando a tubulina, também pode esgotar a tubulina na solução. Baixas concentrações de tubulina, abaixo da concentração crítica para microtúbulos polimerizadores, podem resultar em agregados.
Nos experimentos FRAP, se o canal MAP65 não mostrar qualquer recuperação (Figura 5), é possível que o fotobleaching tenha sido fotodambulando os microtúbulos. Fotodamage causa a destruição localizada dos filamentos. Isso pode ser verificado por exame no canal transmitido. Tatoides microtúbulos são visíveis no canal transmitido através de um alto índice de incompatibilidade de refração com a água circundante. A fotodamagem induzida pela luz aparecerá como uma marca de queimadura ou perda de contraste em imagens de luz transmitidas na localização do ROI submetida a fotobleaching. Se isso ocorrer, o laser ou a energia da luz devem ser reduzidos para inibir a fotodamagem das proteínas.
Foram encontrados diversos desafios nesse procedimento e abordagem. Uma questão é que as medidas de comprimento são atualmente executadas manualmente clicando na imagem. Este método, embora simples, pode resultar em alta incerteza. A medição de largura que usa a seção transversal e a montagem de um gaussiano é um método melhor para quantificar o tamanho. Um método semelhante poderia ser empregado para o comprimento. Uma segunda questão é que, às vezes, os tatoides, porque são tão longos e finos, podem dobrar. Isso dificulta a quantificação do comprimento. O comprimento do contorno pode ser quantificado usando uma linha segmentada, mas há mais incerteza cada vez que um segmento é adicionado.
Do ponto de vista científico, essa abordagem tem alguns outros desafios para seu uso como modelo para cristais líquidos ou fusos. O primeiro desafio tem sido a forma longa e fina dos tatoides que os microtúbulos criam (Figura 3 e Figura 4). Como observado em publicações anteriores22, os tatoides microtúbulos são tatoides homogêneos, não bipolares. Isso significa que os microtúbulos que compõem a forma não reorientam para apontar para as pontas da estrutura. Em vez disso, todos os microtúbulos são paralelos ao eixo longo e os "polos" estão localizados no infinito. Isso é muito diferente dos tatoides observados para cristais líquidos moleculares ou mesmo para actina ou DNA que também podem atuar como mesógenos de cristal líquido. Nesses outros sistemas, os tatoides são bipolares e, quando vistos em polarizadores cruzados, mostram os sinais de reorientação das hastes.
Um segundo grande desafio neste sistema é que os microtúbulos estão imóveis dentro do tatoide. Isso é claro a partir de experimentos e análises FRAP, pois a recuperação dos microtúbulos é muito baixa. Sua natureza sólida torna os tatoides microtúbulos menos valiosos como análogos de cristal líquido em larga escala. A fase nemática de um cristal líquido deve ter propriedades líquidas (fluidas) e cristal (organizadas). Embora a forma pareça certa para o fuso, a imobilidade torna o sistema menos excitante como um modelo de fuso mitotístico. Por outro lado, esta questão oferece oportunidades para investigar como se pode modificar os experimentos para criar mais fluidez no sistema.
Esses desafios científicos oferecem oportunidades emocionantes que permitirão novos conhecimentos sobre o sistema. Para tornar os tatoides microtúbulos mais bipolares, pode-se usar microtúbulos mais curtos. Há um desafio adicional, no entanto, uma vez que os microtúbulos não têm muitas proteínas de tampa bem caracterizadas para controlar o comprimento como o actin faz. O uso de nucleação e crescimento requer o uso de concentrações muito altas de tubulina e GMPCPP para fazer microtúbulos curtos. A alta concentração de tubulina resulta em um número maior de filamentos no sistema, o que torna mais difícil separar tatoides uns dos outros. A adição de novos cappers de microtúbulos, como o DARPin33, pode ajudar nessa situação. A segunda questão dos microtúbulos imóveis poderia ser atenuada pela adição de proteínas motoras, como a quinase-534, que são tetramers de motores utilizados na mitose. Alternativamente, dimers artificiais de cinesina dimeric-1 poderiam ser usados15.
Outra forma de adicionar mais fluidez seria permitir que os microtúbulos realizassem sua instabilidade dinâmica, o crescimento e o encolhimento dos microtúbulos. Atualmente, microtúbulos que são semeados com filamentos GMPCPP estáveis e, em seguida, sofrem instabilidade dinâmica são muito mais longos do que o desejado para formar um fuso ou tatoide, o que resultaria em organizações muito longas como ventiladores ou pacotes. Assim, adicionar instabilidade dinâmica de microtúbulo precisaria ser feito cuidadosamente para preservar a forma tatoide. A adição de proteínas e enzimas associadas que possam controlar o comprimento pode mitigar esse problema. Por exemplo, cinesinas despomerizantes, como a cinesina-1335, ou enzimas de corte, como katanin36, provavelmente seriam necessárias. Esses experimentos são complexos e difíceis, embora sejam muito perspicazes, não importa o que os resultados revelem. Seja qual for a direção que os experimentos futuros tomem, a plataforma desenvolvida aqui para a criação de tatoides microtúbulos pode expor novas informações com base física na organização de microtúbulos.
Os autores declaram que não têm interesses financeiros concorrentes.
Os autores gostariam de agradecer a todos os membros do Ross Lab de verão de 2021, especialmente K. Alice Lindsay, por sua ajuda. Este trabalho foi apoiado por uma subvenção da NSF BIO-2134215 que apoiou S. Sahu, N. Goodbee, H.B. Lee e J.L. Ross. Uma bolsa da Fundação KECK (Rae Anderson, USD, chumbo PI) apoiou parcialmente R. Branch e P. Chauhan
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved