
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
Самостоятельная сборка тактоидов микротрубочек
В данной статье представлен протокол формирования сборок микротрубочек в форме тактоидов с использованием MAP65, сшивающего микротрубочек на растительной основе, и ПЭГ в качестве краудингового агента.
Цитоскелет отвечает за крупную внутреннюю организацию и реорганизацию внутри клетки, и все это без менеджера, который будет руководить изменениями. Это особенно актуально во время митоза или мейоза, когда микротрубочки образуют веретено во время деления клеток. Веретено - это механизм, используемый для разделения генетического материала во время деления клеток. Для создания самоорганизованных веретен in vitro мы недавно разработали технику воссоздания микротрубочек в веретенообразные сборки с минимальным набором белков, связанных с микротрубочками, и агентов скученности. В частности, был использован MAP65, который является антипараллельным сшивателем микротрубочек из растений, гомологом Ase1 из дрожжей и PRC1 из организмов млекопитающих. Этот кросслинкер самоорганизует микротрубочки в длинные, тонкие, веретенообразные микротрубочки самоорганизованные сборки. Эти сборки также похожи на жидкокристаллические тактоиды, а микротрубочки могут быть использованы в качестве мезомасштабных мезогенов. Здесь представлены протоколы создания этих микротрубочек тактоидов, а также для характеристики формы сборок с помощью флуоресцентной микроскопии и подвижности составляющих с помощью флуоресцентного восстановления после фотоотбеливания.
Деление клеток с помощью митоза является одним из наиболее важных биологических процессов для поддержания жизни. Нити микротрубочек, состоящие из тубулиновых димеров, являются важными структурными элементами этого процесса. Переходный механизм, созданный в метафазе, когда хромосомы выравниваются в центре клетки, называется митотическим веретеном из-за его формы, которая похожа на веретено ткацкого станка, покрытого нитями (рисунок 1A). Во многих организмах хорошо известно, что микротрубочки используются в метафазе для проталкивания и вытягивания конденсированных хромосом в центр клетки, выравнивания их и подключения к микротрубочкам, которые будут раздвигать их в анафазе (рисунок 1B, C). Веретено образуется как в мейозе (рисунок 1B), так и в митозе (рисунок 1C), созданном из множества перекрывающихся микротрубочек, не обернутых вокруг центральной оси, как нить, но идущих параллельно интерфейсу. Создание этих структур на основе микротрубочек требует ассоциированных белков, которые сшиваются, и связанных ферментов, которые могут действовать как двигатели, помогающие толкать и тянуть хромосомы1.
Исследования мейотических веретен показали, что микротрубочки короткие, динамичные и перекрываются в сшитых массивах 2,3,4,5,6 (рисунок 1Di). Из-за физической организации этих коротких микротрубочек мейотическое веретено похоже на жидкокристаллический тактоид (рисунок 1Е). Действительно, было показано, что шпиндели объединяются и сливаются, как и следовало ожидать от жидкокристаллических тактоидов5.
Многие исследования, относящиеся к 1960-м годам, использовали фиксацию, последовательные сечения и электронную микроскопию, чтобы определить, что внутри митотического веретенаесть два типа микротрубочек 7,8,9,10. Первый тип называется кинетохорными микротрубочками, которые соединяют веретенообразный полюс с кинетохорой. Второй тип называется межполярными или полярными микротрубочками, которые прорастают мимо хромосом и перекрываются в средней зоне (рисунок 1Dii)8,9,10. Третий тип называется астральными микротрубочками, которые находятся вне веретена и соединяют полюса с краем клетки; эти организации, занимающиеся микротрубочками, выходят за рамки нынешнего обсуждения. Недавно были проведены исследования взаимодействия между аугмином6 и гамма-тубулиновым кольцевым комплексом, влияющим на центры нуклеации микротрубочек, в результате чего получилось митотическое веретено с более короткими микротрубочками, как на рисунке 1D.
Поскольку микротрубочки длиннее, чем они широкие, с высоким соотношением сторон и высокой жесткостью, они похожи на увеличенные версии жидкокристаллических молекул. В физике мягкой материи атомы и молекулы были аппроксимированы с использованием минимальных взаимодействий для вывода физических механизмов фазовых переходов, включая зарождение и плавление кристаллов11. Аналогичным образом, микротрубочки являются мезомасштабными объектами, которые являются увеличенными версиями молекул жидких кристаллов, что дает представление о физике динамики жидких кристаллов, включая зарождение и рост нематических фаз из изотропных. Далее, как обсуждалось выше, мейотическое веретено проявляет свойства, подобные свойствам жидкокристаллического тактоида, нематического состояния, которое нуклеативно и растет из изотропного состояния жидкокристаллических молекул 3,4,5. Для тактоидов нуклеация и рост аналогичны зарождению и росту других кристаллов (т. е. требуют относительно высокой концентрации мезогенов [молекул, образующих жидкие кристаллы]). Уникальная «веретенообразная» форма тактоида происходит от локального выравнивания жидкокристаллических мезогенов, которые выравниваются в нематическую фазу (рисунок 1E). Они не могут образовать округлый кристалл, потому что молекулы очень асимметричны. Учитывая природу микротрубочек, возможно, неудивительно, что митотический веретенообразный механизм, изготовленный из высокой локальной концентрации микротрубочек, также имеет одинаковую форму, независимо от того, называется ли он тактоидом или веретеном. Тактоиды могут быть биполярными, с полюсами на конических концах (рисунок 1Ei), или однородными, с полюсами, эффективно находящимися в бесконечности (рисунок 1Eii).
Учитывая важность формирования веретена, предпринимаются усилия по самоорганизующемуся образованию веретена in vitro путем демонстрации конденсации микротрубочек в пучки через ионные виды12,13, скученные агенты, которые создают истощение взаимодействий14,15, и специфические белки сшивания микротрубочек 13,16,17,18,19, 21. Удивительно, но, хотя все эти агенты работают для увеличения местной концентрации микротрубочек, они часто приводят к длинным пучкам микротрубочек, но не к тактоидам. Одна из причин, почему эти пучки длинные, может заключаться в том, что микротрубочки, составляющие их, также длинные. Недавняя работа с использованием более коротких микротрубочек также сообщила о более длинных пучках, которые не сужаются в конце15; в этом случае пучки удерживаются вместе с моторными белками, которые вызывают расширение пучков и, тем самым, делают их длиннее. Короткие микротрубочки с неэкстензионными сшивающимися соединениями необходимы для конических, шпиндельно-подобных сборок, как описано здесь.
Недавно мы разработали методику, позволяющую создавать тактоиды микротрубочек с использованием антипараллельного сшивки MAP65 в присутствии нуклеирующих стабильных микротрубочек22. Микротрубочки должны были быть короткими, но немногие известные регуляторы длины микротрубочек могут закрывать микротрубочки против динамической нестабильности или сквозного отжига. Вместо этого GMPCPP использовался для нуклеатизации и стабилизации нитей после роста. Это позволило создать высокую плотность коротких микротрубочек, которые могли бы самоорганизоваться в тактоиды. Эти тактоиды были однородными при просмотре под двулучепреломлением. В дополнение к коротким микротрубочкам для формирования тактоидов использовался специфический антипараллельный сшиватель MAP65 (рисунок 2). MAP65 — растительный микротрубочки-ассоциированный белок из семейства митотических сшивающих сшивателей PRC1/Ase11. MAP65 существует в виде димера, с сильным сродством к связыванию с самим собой, а также с микротрубочками24. В отличие от мейотических веретен и тактоидов, наблюдаемых с актиновыми нитями 25,26,27, которые являются биполярными и обладают жидкоподобными свойствами жидких кристаллов, тактоиды микротрубочек были твердоподобными22,28.
Здесь представлены протоколы создания тактоидов микротрубочек и характеристики формы сборок и подвижности составляющих с использованием флуоресцентных методов.
ПРИМЕЧАНИЕ: Если не указано иное, части эксперимента могут быть выполнены на лабораторной скамье в соответствующем защитном снаряжении (перчатках).
1. Силанизация покрова
ПРИМЕЧАНИЕ: Покровные листы должны быть силанизированы для использования с полимерным щеточным покрытием, используемым в этих экспериментах. Это гидрофобная силанизация, которая позволяет блок-сополимеру с гидрофобным центральным блоком связывать и создавать полимерную щетку. Следующие шаги следует выполнять в вытяжном капюшоне, чтобы предотвратить воздействие токсичных паров во время ношения перчаток. Диметилдихлорсилан очень токсичен и должен обрабатываться с максимальной осторожностью.
- Промойте крышки ddH2O, 70% этанолом и ddH2O по порядку. Высушите их безворсовыми лабораторными салфетками между каждым ополаскивателем. Это удаляет пыль и водорастворимые или органические частицы с поверхности перед обработкой.
- Поместите крышки в металлическую стойку для чехлов и перенесите стойку на машину с УФ-озоном (UVO). Облучите крышки UVO в течение 20 минут, чтобы удалить фоновую флуоресценцию. Вместо УВО можно использовать плазменную камеру.
- Используя пинцет, перенесите крышки с металлической стойки, используемой для обработки UVO, на другую металлическую стойку, используемую для силанизации. Не используйте одни и те же стойки для обоих, так как это вызовет высокий уровень окисления при применении UVO. Предварительно очистите стойки водой и этанолом, чтобы от предыдущих применений не осталось остаточных химических веществ.
- Погрузите стойку с крышками в емкость со 100% ацетоном на 1 ч. Промойте контейнер 3x водопроводной водой, а затем 3x с ddH2O, чтобы удалить весь ацетон.
- Погрузите стойку с крышками в 100% этанол на 10 мин. Промойте контейнер 3x водопроводной водой, а затем 3x с ddH2O, чтобы удалить весь этанол.
- Погружайте стойку с крышками 3x в ddH2O на 5 минут каждая.
- Погружайте стойку с крышками в 0,1 М КОН (50 мл 1 М КОН в 450 мл ddH2O) в течение 15 мин. Промойте контейнер 3x водопроводной водой, а затем 3x с ddH2O, чтобы удалить весь KOH.
- Погружайте стойку с чехлами 3x в ddH2O на 5 мин каждый.
- Высушите стойку с помощью крышек на ночь в вытяжном шкафу или ламинарной вытяжке.
- После полного высыхания стеллажа и обшивки погружают их на 5 мин в 2% диметилдихлорсилан (DDS), взятый в другой контейнер, который используется специально для силана. Не позволяйте ничему, что не является сухим, вступать в контакт с силаном.
- Погружайте стойку и крышки в 2 раза в емкость со 100% этанолом на 5 мин. Промойте контейнер 3x водопроводной водой, а затем 3x с ddH2O.
- Погружайте стойку и крышки в 3 раза в ddH2O на 5 минут каждая.
- Высушите стойку с помощью крышек на ночь в вытяжном шкафу или ламинарной вытяжке.
- После этого заключительного этапа сушки переложите крышки обратно в коробки для чехлов с помощью пинцета. Эти чехлы можно использовать в ближайшие 1-2 месяца. Старые крышки начнут терять свое покрытие и должны быть выброшены.
2. Препарат тубулина
ПРИМЕЧАНИЕ: Купленный тубулин поставляется в виде лиофилизированного порошка, который либо не маркирован, либо помечен флуорофорами. Лиофилизированный тубулин хранится в морозильной камере при температуре −80 °C. Следующая процедура используется для смешивания немаркированного тубулина с меченым тубулином в соотношении, которое хорошо подходит для визуализации.
- Извлеките из морозильной камеры −80 °C одну аликвоту немаркированного тубулина, содержащего 1 мг лиофилизированного порошка тубулина, и держите его на льду. Добавьте 200 мкл холодного PEM-80 в пробирку, чтобы довести концентрацию тубулина до 5 мг/мл. Держите его на льду в течение 10 минут, чтобы растворить весь лиофилат.
- Извлеките из морозильной камеры при температуре −80 °C одну аликвоту меченого родамином тубулина, содержащего 20 мкг лиофилизированного порошка тубулина, и держите его на льду. Добавьте 4 мкл холодного PEM-80 в пробирку, чтобы довести концентрацию тубулина до 5 мг/мл. Держите его на льду в течение 10 минут, чтобы полностью растворить лиофилат.
- После растворения добавьте 100 мкл немаркированного повторно суспендированного раствора тубулина к 4 мкл раствора тубулина, меченного родамином. Пипетка 6х-7х очень медленно перемешивается. Если видны агрегаты, центрифугируют солюбилизированный тубулин в течение 10 мин при 90 000 х г , чтобы удалить агрегаты, выбросив гранулу и сохранив супернатант. Эта смесь тубулина приведет к образованию ~ 4% меченого тубулина.
- Заморозьте оставшиеся 100 мкл немаркированного тубулина в жидкий азот (LN2) и храните его при -80 °C для использования для дополнительных тубулиновых смесей.
- Возьмите меченую смесь тубулина и аликвоту в семь пробирок по 15 мкл в каждой. Каждая аликвота может быть использована для одной экспериментальной камеры. Заморозьте оставшиеся аликвоты и храните их при −80 °C для будущих экспериментов.
3. Очистка MAP65
ПРИМЕЧАНИЕ: MAP65 не является коммерчески доступным и, следовательно, нуждается в очистке для этой работы. Протокол был разработан ранее в нескольких публикациях23,29.
- Преобразование плазмиды MAP65 и плазмиды GFP-MAP65 в штамм бактерий BL21 для экспрессии белка.
- Вырастите бактерии BL21 до оптической плотности 0,6-1 при 600 нм. Индуцируйте выработку белка с помощью оператора lac и выращивайте бактерии в течение ночи.
- Гранулируйте культуры и лизируйте бактерии.
- Соберите лизат после центрифугирования и инкубируйте его с шариками, которые имеют ион никеля, доступный для связывания 6x-гистидоновой метки.
- Элюируют белок с помощью имидазола и обессоливают его.
- Заморозьте белок жидким азотом и храните его при температуре −80 °C для использования в течение 1 года.
4. Сборка проточных камер
ПРИМЕЧАНИЕ: Эксперименты проводятся в проточных камерах, изготовленных из стеклянной горки и силанизированного покровного стекла (рисунок 3).
- Возьмите стеклянный слайд и очистите его, используя ddH2O, этанол и ddH2O по порядку. Высушите безворсовой лабораторной салфеткой между каждым ополаскивателем.
- Используйте кусок двусторонней ленты, чтобы создать путь потока. Руками в перчатках разрежьте ленту до ~25-30 мм в длину. Разделите ленту вдоль, чтобы создать две более тонкие полоски. Поместите две ленточные полоски на слайд с расстоянием около 5-8 мм между ними.
ПРИМЕЧАНИЕ: Поскольку толщина ленты стандартизирована примерно до 80-100 мкм, ширина пути между кусками ленты будет определять объем в камере. - Поместите силанизированные крышки поверх пути потока. Запечатайте слайд и крышку на двухсторонних ленточных полосках, осторожно надавливая на область ленты задней частью ручки. Убедитесь, что вы получили хорошее уплотнение по всей площади; лента должна превратиться из полупрозрачной в прозрачную, когда уплотнение сделано хорошо.
- Снимите лишнюю ленту по краям, оставив всего 1 мм от входа в проточную камеру, разрезав ленту лезвием бритвы.
- Маркировка камеры с информацией об экспериментальных параметрах, по мере необходимости.
5. Тактоидные эксперименты
ПРИМЕЧАНИЕ: После того, как все реагенты и материалы сгенерированы, их можно использовать для нуклеатизации и полимеризации тактоидов микротрубочек в проточной камере.
- Соберите все реагенты, которые будут использоваться. Оттаивайте их на льду и храните на льду во время работы. Сделайте несколько проточных камер для экспериментов.
- Используйте одну проточную камеру для каждого эксперимента. Покрытие поверхностей проточной камеры полимерной щеткой путем перетекания в 20 мкл 5% неионного блока сополимерного поверхностно-активного вещества (Таблица материалов), растворенного в ПЭМ-80, с небольшими каплями на обоих концах камеры для предотвращения образования пузырьков воздуха внутри. Храните его во влажной камере (т.е. чашке Петри с влажной безворсовой лабораторной салфеткой) до готовности к использованию (не менее 5-7 мин).
- В стерильной пробирке смешайте для создания смеси Тубулин-MAP следующее: 9,5 мкл PEM-80; 4 мкл 10 мМ GMPCPP; 4 мкл 5% плуроника-F127; 1 мкл 1M DTT; 1 мкл глюкозы; 2 мкл полиэтиленгликоля (ПЭГ); 12 мкл смеси тубулина 5 мг/мл (конечная концентрация 13,6 мкМ) со стадии 2.5.; и 5,5 мкл рабочего фонда MAP65, где 10% составляет GFP-MAP65 для визуализации. Держите на льду во время перемешивания.
ПРИМЕЧАНИЕ: Для обработки вязкого раствора ПЭГ рекомендуется использовать пипетку с положительным смещением. Обычные пипетки можно использовать после резки наконечника, чтобы сделать отверстие больше; однако этот метод менее точен. - Перемешать 5х-6х путем пипетки.
- Непосредственно перед добавлением в камеру добавляют 1 мкл предварительно смешанного раствора глюкозооксидазы (0,5 мг/мл) и каталазы (0,15 мг/мл) (Дезокси) в смесь Тубулин-МАП и смешивают 7x-8x. Разделить общий объем раствора (40 мкл) на две порции для использования в отдельных камерах.
- Переведите смесь Тубулин-МАП в камеры. Поскольку камеры уже имеют неионное блок-сополимерное поверхностно-активное вещество, больше жидкости не может быть добавлено без удаления старой жидкости. Для этого используйте лист фильтровальной бумаги или безворсовую лабораторную салфетку на другом конце камеры, чтобы удалить жидкость с помощью капиллярного действия.
- Как только образец полностью окажется внутри камеры, запечатайте два конца камеры с помощью 5-минутной эпоксидной смолы и держите ее при 37 °C в течение ~ 30 минут, чтобы зародить и вырастить тактоиды микротрубочек.
6. Флуоресцентная микроскопия
- Используйте флуоресцентный микроскоп для визуализации тактоидов.
ПРИМЕЧАНИЕ: Флуоресцентная микроскопия полного внутреннего отражения или конфокальная микроскопия вращающегося диска хороши для удаления фоновой флуоресценции из свободного тубулина, но тактоиды также видны при регулярной эпифлуоресценции и даже прозрачной световой микроскопии после их формирования, что делает эту процедуру доступной без специализированного оборудования. - Используйте объектив числовой апертуры (NA) 1,2 NA или выше с увеличением 60x или выше, чтобы собрать достаточно света во флуоресценции. Эти цели часто требуют погружения либо в ddH2O, либо в масло.
- Записывайте изображения с помощью CMOS или ПЗС-камеры. Используйте эффективный размер пикселя на камере 108 нм.
ПРИМЕЧАНИЕ: Размер пикселя зависит от камеры и используемого увеличения, которое в данном случае было 60x или 100x с высокой числовой диафрагмой (1.2 или 1.49 NA). Дополнительные расширители изображения могут быть использованы перед камерой для достижения необходимого размера пикселя. - Держите образец при температуре 37 °C, используя камеру окружающей среды, установленную на эту температуру. В качестве альтернативы можно использовать другие нагреватели ступеней, включая нагреватели ступеней с горячим воздухом и объективную регулируемую температуру воротники с циркулирующей теплой водой.
- Используйте источники возбуждения, которые являются правильными для необходимой флуоресценции. Для тубулина родамина используют лазер 561 нм мощностью не менее 1 мВт в образце, а для GFP-MAP65 используют лазер 488 нм мощностью не менее 1 мВт в образце.
ПРИМЕЧАНИЕ: При использовании широкоугольной эпифлуоресцентной микроскопии используйте куб фильтра родамина с возбуждением: 540 ± 12,5 нм, дихроичность: 545 нм ± 12,5 нм отсечки и излучением: 575 нм длинный проход, и куб фильтра GFP с возбуждением: 480 ± 15 нм, дихроичность: 505 нм ± 15 нм отсечка и излучение: 515 нм длинный проход. - Сделайте не менее 10 изображений различных областей, чтобы изобразить более 100 тактоидов. Делайте снимки как в красном, так и в зеленом каналах и сохраняйте их как 16-битные изображения tiff для анализа. Убедитесь, что мощность освещения и время экспозиции таковы, что шкала интенсивности для камеры не насыщена.
7. Восстановление флуоресценции после фотоотбеливания (FRAP)
ПРИМЕЧАНИЕ: Для изучения подвижности внутренних составляющих тактоидов использовался FRAP. FRAP работает путем фотоотбеливания выбранной части меченого родамином тубулина и тактоида MAP65, меченного GFP, а затем наблюдения за восстановлением флуоресценции со временем в этой области. Скорость восстановления зависит от оборота фотоотбеливаемых видов. Эта скорость оборота может зависеть от реакций диффузии и связывания. Для привязки MAP65 к тактоидам можно оценить связывающие обменные курсы. FRAP выполняется с использованием дополнительной лазерной системы 405 нм, которая может сканировать лазер в любой форме. Существует множество возможностей для выполнения FRAP, включая использование передающейся лампы и диафрагмы для фотоотбеливания локальной области14.
- Выберите изолированный тактоид в камере, чтобы создать интересующую область (ROI), охватывающую части тактоидов и окружающий раствор.
- Используйте микроскоп с дополнительным лазером 405 нм для FRAP для фотоотбеливания как тубулина, так и MAP65 одновременно. Альтернативно, яркая лампа может быть использована через полевой упор диафрагмы14. Отрегулируйте удельную интенсивность систем фотоотбеливания эмпирически, чтобы избежать повреждения белков во время отбеливания.
- Запишите тактоид как фильм временного ряда в течение 30-60 с перед фотоотбеливанием, чтобы получить информацию об интенсивности до отбеливания. Записывайте красный и зеленый каналы.
- Фотоотбеливайте тактоид, подвергая окупаемость инвестиций с помощью лазеров или лампы до тех пор, пока это необходимо для фотоотбеливания, не повреждая тактоид. Определите интенсивность и время эмпирически.
- Продолжайте записывать фильм в обоих цветных каналах в течение 5-10 минут после фотоотбеливания или до тех пор, пока восстановление, кажется, не достигнет равновесия.
- Визуально проверьте канал GFP-MAP65 для восстановления.
8. Анализ данных
ПРИМЕЧАНИЕ: Количественный анализ изображений тактоидов был выполнен, чтобы узнать о последствиях изменений окружающей среды, налагаемых различными агентами скученности, ионными условиями и добавлением других факторов.
- Характеристика тактоидной формы
- Количественно оцените длину и ширину тактоидов по красным и зеленым изображениям, сделанным с помощью конфокальной микроскопии.
- Откройте изображения с помощью FIJI/ImageJ.
- Если необработанные данные взяты в 16-битном формате, при необходимости отрегулируйте яркость и контрастность. Выберите «Изображение > «Настроить яркость и контрастность > », чтобы настроить изображение, чтобы иметь возможность четко видеть тактоид. Отрегулируйте яркость и контрастность, не применяя настройку, чтобы случайно не изменить данные об интенсивности.
- Как только тактоиды будут хорошо видны, выберите хорошие тактоиды для измерения (рисунок 4Bi). Убедитесь, что тактоиды хорошо видны без перекрытия с любыми другими тактоидами или агрегатами и не изогнуты или изогнуты, чтобы иметь возможность использовать инструменты измерения по прямой линии.
- Затем убедитесь, что для изображений установлен правильный размер пикселя. Изображения микроскопа поставляются с метаданными о размере пикселя. При использовании другой камеры, не имеющей метаданных, или внешних систем расширения изображения, которые могут изменять ожидаемый эффективный размер пикселя, отрегулируйте размер пикселя вручную. В РАЗДЕЛЕ FIJI/ImageJ перейдите в раздел Анализ > Задать масштаб , чтобы задать правильное преобразование пикселей.
- Используя инструмент «Прямая линия » с панели инструментов в FIJI/ImageJ, нажмите на один конец тактоида и перетащите курсор на другой конец тактоида (рисунок 4Bii). Выбрав рентабельность инвестиций в линию, выберите «Анализировать > меру», чтобы измерить длину. Если длина не измеряется по умолчанию, убедитесь, что измерение включает длину в диалоговом окне Анализ > Задание измерений .
ПРИМЕЧАНИЕ: Как правило, при измерении с помощью инструмента «Прямая линия » он дает длину и угол проведенной линии. В качестве примера на рисунке 4Bii показана прямая линия, проведенная сбоку от тактоида, чтобы сделать последний видимым, но сделать измерение непосредственно на тактоиде. - После выполнения измерения используйте инструмент «Текст» на панели инструментов, чтобы пометить тактоид. Создайте текстовое поле, добавьте числовую метку и выберите «Изменить > нарисовать», чтобы закрепить метку на изображении. Сохраните изображение как отдельный файл ROI.
ПРИМЕЧАНИЕ: Маркировка и сохранение этого файла позволяет исследователю узнать, какое измерение соответствует какому тактоиду из необработанных данных. Обязательно измерьте каждый тактоид один раз. - После измерения тактоидов для всего изображения сохраните данные в окне «Результаты » в текстовый файл с разделителями-запятыми или табуляцией (с помощью > «Сохранить как») и откройте данные в программе для работы с электронными таблицами, чтобы разобрать данные на числа. Соберите все данные вместе (необработанные данные изображения, изображение ROI и текстовый файл результатов) в папку с соответствующим соглашением об именовании, чтобы все было организовано.
ПРИМЕЧАНИЕ: Хотя измерения длины тактоидов выполняются вручную, учитывая, что ширина тактоидов узкая, лучше использовать другой метод измерения тактоидной ширины (см. Ниже), чтобы уменьшить погрешность измерения. - С помощью ImageJ/FIJI нарисуйте область линии с помощью инструмента «Прямая линия ». Нарисуйте линию в виде перпендикулярного бисектора к тактоидной длинной оси (рисунок 4Bii).
- Выберите Analyze > Plot Profile (Анализ профиля графика ), чтобы создать профиль интенсивности линейного биссектора (рисунок 4Biii). Появится сюжет. Чтобы извлечь и сохранить данные с участка, нажмите кнопку Список в левом нижнем углу; при этом в текстовом файле создается список данных об интенсивности по длине проведенной линии. Сохраните текстовый файл в виде файла .csv или .txt.
- Откройте текстовый файл в подходящей программе, такой как MatLab, Python (sciPy) или других программах. Подогнать данные интенсивности с гауссовской функцией вида:
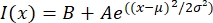 , где I(x) — значение оттенков серого по длине, x; B - фоновый уровень; A — амплитуда Гаусса; μ — среднее или центр гаусса; и σ является стандартным отклонением гаусса.
, где I(x) — значение оттенков серого по длине, x; B - фоновый уровень; A — амплитуда Гаусса; μ — среднее или центр гаусса; и σ является стандартным отклонением гаусса. - Отчет 2σ как ширина тактоида. Оцените интенсивность микротрубочек в тактоиде, вычислив площадь под гауссовской (не включая фон).
ПРИМЕЧАНИЕ: Если изображения находятся в пределах линейного диапазона интенсивности камеры и сделаны с одинаковым временем экспозиции и интенсивностью возбуждения, можно сравнить интегрированные интенсивности для оценки относительного количества микротрубочек в тактоиде.
- Анализ FRAP
ПРИМЕЧАНИЕ: Эксперименты по проверке подвижности микротрубочек и MAP65 использовали FRAP для регистрации специфического фотоотбеливания и восстановления интенсивности за счет молекулярного движения (рисунок 5A). Данные были количественно определены на основе данных временных рядов изображений с использованием ImageJ/FIJI.- Используйте ImageJ/FIJI, чтобы открыть данные фильма.
- Зарегистрируйте стеки (данные временных рядов) с течением времени, чтобы удалить дрейф. Используйте плагин StackReg вместе с вспомогательным плагином TurboReg ; Инструкции по использованию плагинов см. в веб-ссылках, приведенных в Таблице материалов. Выберите перевод, чтобы сместить положение кадров и, таким образом, зарегистрировать изображения.
- После регистрации изображений для удаления дрейфа поверните изображение так, чтобы тактоид был вертикальным или горизонтальным в кадре, выбрав «Изображение > «Преобразовать > Повернуть». Выберите угол поворота и используйте функцию «Просмотр», чтобы определить, достаточно ли повернут тактоид. Когда предварительный просмотр покажет, что тактоид является вертикальным или горизонтальным, нажмите кнопку ОК , чтобы повернуть все изображения в фильме.
- Используйте инструмент « Выделение прямоугольников» на панели инструментов, чтобы создать прямоугольный раздел над фотоотбеленной областью тактоида. Запишите интегрированную интенсивность области окупаемости инвестиций для каждого кадра с помощью стеков > изображений > стека мер. Установите для типа измерения значение Интегрированная плотность с помощью анализа > набора измерений. Сохраните анализируемые данные об интенсивности, отображаемые в окне Результаты , в виде текстового файла в формате .csv или .txt, выбрав Файл > Сохранить как.
ПРИМЕЧАНИЕ: На рисунке 5B показан пример необработанных 16-битных данных об интенсивности, измеренных для каналов микротрубочек и GFP-MAP65 в области отбеливания. - Поскольку общая интенсивность изображений со временем будет уменьшаться во всем мире из-за фотоотбеливания, вызванного изображением, это глобальное фотоотбеливание должно быть исправлено. Для этого используйте тот же размер ROI (шаг 8.2.4.) и переместите его в область на заднем плане изображения, где не видны микротрубочки или MAP65. Измерьте интегрированную интенсивность стека, как описано в шаге 8.2.4. Сохраните результаты в виде второго текстового файла.
ПРИМЕЧАНИЕ: На рисунке 5B показан пример необработанных 16-битных данных об интенсивности, измеренных для каналов микротрубочек и GFP-MAP65 в фоновой области. - Чтобы исправить затухание фона, разделите интенсивность сигнала на тактоиде на интенсивность фона за ту же точку времени. Вычислить iс поправкой (t) как:
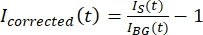 , где IS (t) (сигнал) - измерение, произведенное на обесцвеченной области, а IBG (t) (шум) - измерение, произведенное в фоновой области (рисунок 5C). При этом вычисляется отношение сигнал/шум для каждого кадра, а также вычитается шум.
, где IS (t) (сигнал) - измерение, произведенное на обесцвеченной области, а IBG (t) (шум) - измерение, произведенное в фоновой области (рисунок 5C). При этом вычисляется отношение сигнал/шум для каждого кадра, а также вычитается шум. - Затем перемасштабируйте данные в диапазоне от нуля до единицы, используя
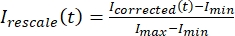 , где Imin и Imax обозначают, соответственно, глобальный минимум и максимум данных icorrected за все время (рисунок 5C).
, где Imin и Imax обозначают, соответственно, глобальный минимум и максимум данных icorrected за все время (рисунок 5C). - Подогнать эти данные к распадающейся экспоненциальной форме:
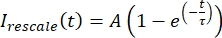 , где A — амплитуда восстановления, а τ — временная шкала восстановления (рисунок 5C).
, где A — амплитуда восстановления, а τ — временная шкала восстановления (рисунок 5C).
С небольшим количеством компонентов, димеров тубулина и сшивающих микротрубочек, могут образовываться тактоиды микротрубочек (рисунок 2A). Хотя этот протокол описывает инкубацию для нуклеации и выращивания микротрубочек в инкубаторе, зарождение и рост можно наблюдать непосредственно под микроскопом (которые завершаются в течение 30 минут) (рисунок 2B). Концентрация тубулина поддерживается постоянной на уровне 13,6 мкм и связыванием MAP65-MT на уровне 10%.
На рисунке 4 показаны успешные данные. Тактоиды должны быть видны как с лазером 561 нм в канале тубулина, так и с 488 нм в канале MAP65, которые идеально перекрываются друг с другом (рисунок 4A). Одна из загадок системы заключалась в том, что ширина тактоидов, по-видимому, не изменяется при различных экспериментальных изменениях, включая изменение длины микротрубочек, концентрации MAP65 и агентов скученности (рисунок 4B)22,28. Длина гораздо более изменчива и зависит как от длины микротрубочек, так и от концентрации MAP65 (рисунок 4B)22,28.
При выполнении FRAP было замечено, что сигнал MAP65 восстанавливается, но сигнал микротрубочки не восстанавливается (рисунок 5). Восстановление FRAP происходит за счет мобильности и перемещения меченых и фотоотбеленных объектов. В случае MAP65 затемненные молекулы диссоциируют и удаляются от микротрубочек, а новые перемещаются в область (рисунок 5). Связывание MAP65 находится в равновесии, поэтому скорость связывания и развязывания равна (измеряется в молекулах в секунду). Для микротрубочек не было замечено никакого восстановления, подразумевающего, что микротрубочки не способны покинуть тактоид (рисунок 5A, Bi, C). Кроме того, не было замечено распространения затемненной области, что говорит о том, что микротрубочки локально неподвижны и не являются жидкостью внутри тактоидной формы.
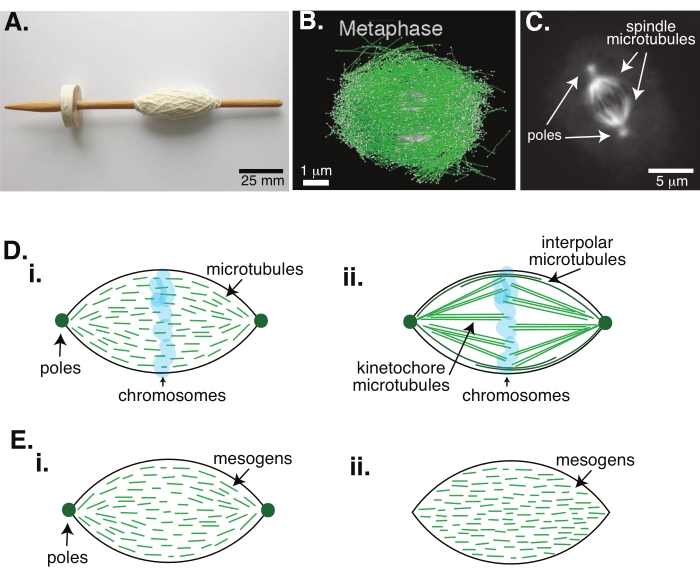
Рисунок 1: Различные модели формирования шпинделя. Митотическое веретено представляет собой машину, состоящую из микротрубочек и связанных с ними белков и ферментов, которая выравнивает и разделяет хромосомы на две новые дочерние клетки во время деления клеток. (A) Изображение реплики веретена раннего средневековья с тонкой пряжей из Нидерландов. Эта цифра была изменена по сравнению с изображением Викимедиа Питера ван дер Слуйса30. (B) Трехмерная реконструкция микротрубочек на различных стадиях мейоза дикого типа II. Микротрубочки показаны зеленым цветом, а хромосомы — серым. Шкала бара = 1 мкм. Эта цифра была изменена по сравнению с Lantzsch et al.31. (C) Микроскопическое изображение микротрубочек в митотическом веретенде делящейся клетки Sf9. Полюса веретена и микротрубочки веретена помечены зеленым флуоресцентным белком. Шкала бара = 5 мкм. Эта цифра была изменена по сравнению с Advani et al.32. (D) Различные модели того, как организованы митотические и мейотические веретенообразные микротрубочки. (i) Ранее наблюдаемые для мейотических веретен, созданных из экстрактов яиц Xenopus, микротрубочки (зеленые) были выведены как короткие и динамичные по всему веретену. Это похоже на биполярную тактоидную организацию внутри жидкого кристалла. (ii) Каноническая модель организации микротрубочек внутри митотического веретена имеет два типа микротрубочек: межполярные или полярные микротрубочки (темно-зеленые), которые сшиваются в средней зоне вокруг хромосом, и кинетохорные микротрубочки (светло-зеленые), которые связываются и растягиваются от полюса до кинетохора, чтобы толкать и тянуть хромосомы. На всех изображениях хромосомы показаны прозрачным синим цветом, а веретенообразные полюса представлены темно-зеленым цветом. (E) Схемы мезогенов (зеленых линий) в жидкокристаллическом тактоиде для (i) биполярных и (ii) однородных тактоидов. Биполярные тактоиды имеют два полюса в конце тактоида, и мезогены переориентируются, чтобы указать на эти полюса. Однородные тактоиды имеют полюса в бесконечности, а мезогены не меняют ориентацию по длине тактоида. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
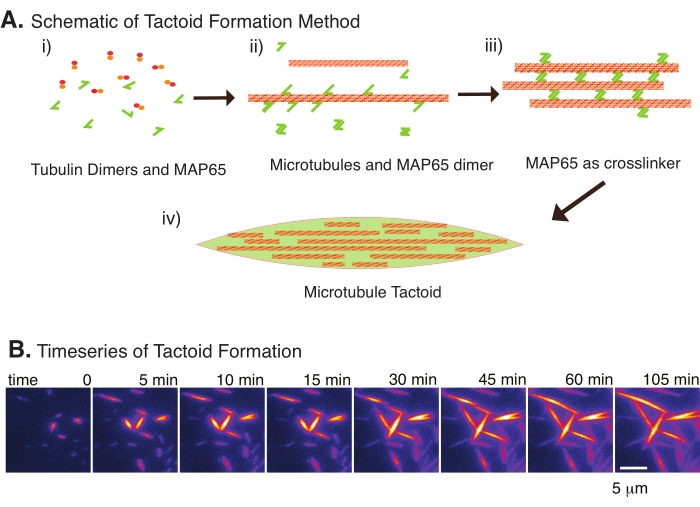
Рисунок 2: Конденсация микротрубочек. (A) Микротрубочки могут быть связаны и сшиты различными методами, включая ионные виды, силы истощения, вызванные агентами скученности, и специфические сшивки микротрубочек, такие как MAP65. (i) Димеры тубулина и белки MAP65 смешиваются с нуклеатом и выращиванием микротрубочек. (ii) Микротрубочки ядра и растут из тубулина, а MAP65 немедленно связывается с микротрубочками, другим мономером MAP65 или и тем, и другим и вызывает объединение. iii) микротрубочки в сшитых пучках зарождаются и растут. iv) Окончательная конфигурация представляет собой тактоид микротрубочки, похожий на шпиндель. (B) Временные ряды тактоидов микротрубочек, зарождающихся и растущих в течение 105 мин. Шкала бара = 5 мкм. Рисунок взят из Edozie et al.22. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
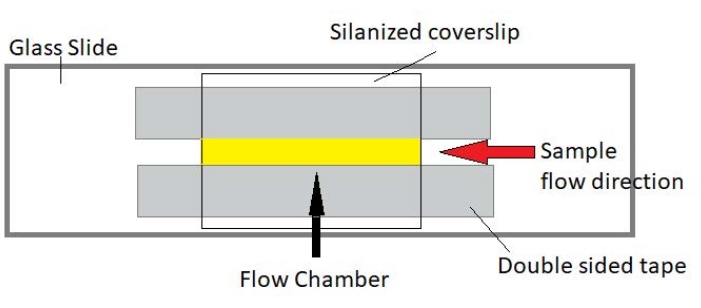
Рисунок 3: Сборка проточной камеры. Проточная камера выполнена с использованием стеклянного затвора, силанизированного покровного стекла и постоянной двусторонней ленты. Выделенная желтым цветом область — это путь потока, по которому проба течет и наблюдается. Объем проточной камеры составляет ~20 мкл. Эпоксидная смола использовалась для герметизации концов камеры, чтобы сохранить образец от испарения во время длительной визуализации в течение нескольких часов. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
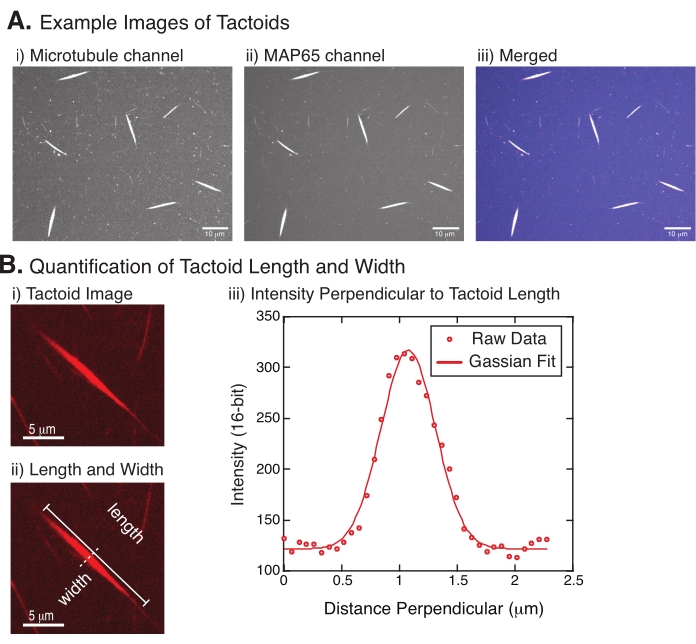
Рисунок 4: Тактоидные изображения и анализ длины и ширины. (A) Примеры данных тактоидов, сформированных в соответствии с описанием и изображенных с использованием конфокального вращающегося диска, показывающих (i) изображение каналов микротрубочек, меченых родамином тубулина с использованием лазера 561 нм, (ii) визуализацию канала GFP-MAP65 с использованием лазера 488 нм и (iii) объединенное наложенное изображение из канала микротрубочек (пурпурный) и канала GFP-MAP65 (голубой). Перекрывающиеся области отображаются белым цветом и демонстрируют, что микротрубочки и MAP65 точно колокализуются. Шкала = 10 мкм для всех изображений в (A). (B) Количественная оценка длины и ширины тактоидов. i) изображение тактоида, подлежащего анализу без меток. Шкала = 5 мкм. (ii) То же изображение, что и в (i), где обозначены размеры длины (сплошная линия с прописными буквами) и ширины (пунктирная линия). Шкала шкалы = 5 мкм. (iii) Ширина измерялась путем взятия профиля интенсивности через тактоид в перпендикулярной биссектрисе (пунктирной линии), обозначенной в (ii). Профиль интенсивности соответствовал функции Гаусса, чтобы выявить амплитуду и ширину тактоида. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
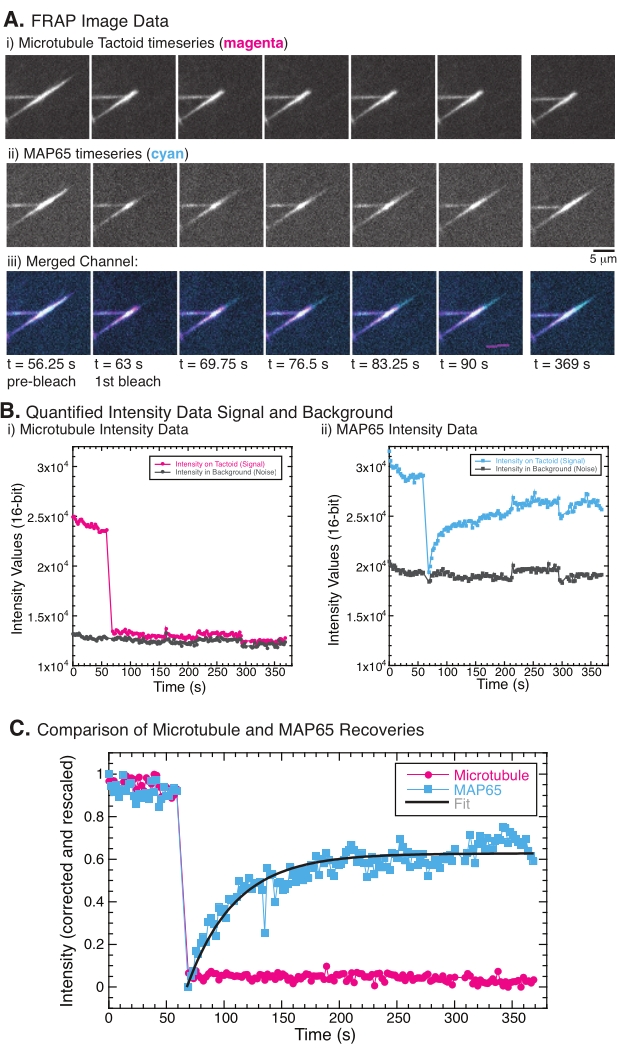
Рисунок 5: Репрезентативные данные и анализ FRAP. (A) Данные временных рядов микроскопии (i) тактоида микротрубочек и (ii) GFP-MAP65 и (iii) наложение изображения обоих каналов с микротрубочками в пурпурном цвете и GFP-MAP65 в голубом, которые были фотоотбелены в момент 63 с и наблюдались в течение дополнительных 5 мин. (B) Количественная интенсивность (i) ) канал микротрубочек в отбеленной области (пурпурные круги) и фон (темно-серые круги) и (ii) канал GFP-MAP65 в отбеленной области (голубые квадраты) и фон (темно-серые квадраты). (C) Данные были скорректированы для фонового шума и перемасштабированы для канала микротрубочек (пурпурные круги) и канала GFP-MAP65 (голубые квадраты). Микротрубочки не восстанавливаются, но GFP-MAP65 подходит и может быть приспособлен (темно-серая линия) к растущему экспоненциальному распаду, чтобы найти амплитуду и временную шкалу восстановления. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
Методы, описанные здесь, были использованы в нескольких работах для создания тактоидов микротрубочек (рисунок 2)22,28. Эти эксперименты биологически значимы, чтобы помочь раскрыть организационные принципы, которые контролируют форму и стабильность митотического или мейотического веретена в большинстве типов клеток. Кроме того, микротрубочки являются модельными жидкокристаллическими мезогенами, которые могут помочь узнать больше о том, как жидкие кристаллы нуклеат и растут нематические фазы из изотропных фаз.
Процедура, описанная здесь, имеет ряд преимуществ для изучения самоорганизации микротрубочек. Во-первых, он очень воспроизводим, будучи выполненным в лаборатории многими студентами, включая старшеклассников, с небольшим предвидением или обучением, прежде чем начать в лаборатории. Тактоиды являются двулучепреломляющими22, что позволяет просматривать их в проходящем свете в дополнение к флуоресцентной микроскопии, что делает этот метод доступным для многих лабораторий, а эту экспериментальную процедуру адаптируемой к образовательным целям, в дополнение к высококлассным исследованиям. Наконец, этот процесс открывает возможности для дальнейшего понимания и исследования биологических систем в урезанном, редукционистском подходе, позволяя понять, как каждое дополнительное условие, белок или добавка могут изменить самоорганизацию тактоидов и, возможно, в конечном счете, веретена. Цели для улучшения биомимикрии включают сортировку активности, текучести и полярности нити.
Может быть несколько факторов, влияющих на эксперимент, дающих неожиданные результаты. Например, если тактоиды не образуются (рисунок 2), но наблюдаются веерообразные паттерны, MAP65, вероятно, не присутствует или не связывается с микротрубочками22,28. Это должно быть очевидно и в канале флуоресценции MAP65, потому что GFP-MAP65 не будет связан с микротрубочками.
Если тактоиды не образуются и фон выглядит в виде пятен на стекле, это может быть связано с покрытием поверхности. После выполнения силанизация длится всего 1 месяц на покровных листах. Когда он изнашивается, тубулин сможет связываться с открытой поверхностью неспецифично. Эта привязка будет происходить в нечетных шаблонах.
Если тактоиды не образуются и тубулин наблюдается в агрегатах различных форм и размеров, это может быть связано с некачественным тубулином. Тубулин может быть центрифугирован для удаления исходных агрегатов, которые могут управлять этой внепутевой агрегацией вместо полимеризации микротрубочек. Если поверхность связывается с тубулином, это также может истощить тубулин в растворе. Низкие концентрации тубулина, ниже критической концентрации для полимеризующих микротрубочек, могут привести к образованию агрегатов.
В экспериментах FRAP, если канал MAP65 не показывает никакого восстановления (рисунок 5), возможно, что фотоотбеливание было фотопоражением микротрубочек. Фотоповреждение вызывает локализованное разрушение нитей. Это можно проверить путем обследования в транслируемом канале. Тактоиды микротрубочек видны в передаваемом канале через высокий показатель несоответствия рефракции окружающей воде. Световой фотоповреждение будет отображаться как след ожога или потеря контраста в изображениях, передаваемых светом, в месте ROI, подвергаемого фотоотбеливанию. Если это происходит, мощность лазера или света должна быть уменьшена, чтобы ингибировать фотоповреждение белков.
В рамках этой процедуры и подхода возникло несколько проблем. Одна из проблем заключается в том, что измерения длины в настоящее время выполняются вручную, нажав на изображение. Этот метод, хотя и прост, может привести к высокой неопределенности. Измерение ширины, которое использует поперечное сечение и подгонку к гауссову, является лучшим методом количественной оценки размера. Аналогичный метод может быть использован для длины. Вторая проблема заключается в том, что иногда тактоиды, поскольку они такие длинные и тонкие, могут изгибаться. Это затрудняет количественную оценку длины. Длина контура может быть количественно определена с помощью сегментированной линии, но при добавлении сегмента добавляется неопределенность.
С научной точки зрения, этот подход имеет некоторые другие проблемы для его использования в качестве модели для жидких кристаллов или веретен. Первой проблемой была длинная, тонкая форма тактоидов, которые создают микротрубочки (рисунок 3 и рисунок 4). Как отмечалось в предыдущих публикациях22, тактоиды микротрубочек являются однородными тактоидами, а не биполярными. Это означает, что микротрубочки, составляющие форму, не переориентируются, чтобы указать на кончики структуры. Вместо этого все микротрубочки параллельны длинной оси, а «полюса» расположены в бесконечности. Это сильно отличается от тактоидов, наблюдаемых для молекулярных жидких кристаллов или даже для актина или ДНК, которые также могут действовать как жидкокристаллические мезогены. В этих других системах тактоиды являются биполярными, и при рассмотрении в скрещенных поляризаторах они показывают явные признаки переориентации стержней.
Вторая серьезная проблема в этой системе заключается в том, что микротрубочки неподвижны внутри тактоида. Это ясно из экспериментов и анализа FRAP, поскольку восстановление микротрубочек очень низкое. Их твердая природа делает тактоиды микротрубочек менее ценными, чем крупномасштабные жидкокристаллические аналоги. Нематическая фаза жидкого кристалла должна обладать как жидкими (жидкими), так и кристаллическими (организованными) свойствами. Хотя форма кажется подходящей для шпинделя, неподвижность делает систему менее захватывающей в качестве модели митотического шпинделя. С другой стороны, этот вопрос дает возможность исследовать, как можно модифицировать эксперименты, чтобы создать больше текучести в системе.
Эти научные проблемы предлагают захватывающие возможности, которые позволят получить новые знания о системе. Чтобы сделать тактоиды микротрубочек более биполярными, можно использовать более короткие микротрубочки. Однако существует дополнительная проблема, поскольку микротрубочки не имеют много хорошо охарактеризованных укупорочных белков для контроля длины, как это делает актин. Использование нуклеации и роста требует использования очень высоких концентраций тубулина и GMPCPP для создания коротких микротрубочек. Высокая концентрация тубулина приводит к большему количеству нитей в системе, что затрудняет отделение тактоидов друг от друга. Добавление новых капперов микротрубочек, таких как DARPin33, может помочь в этой ситуации. Вторая проблема неподвижности микротрубочек может быть смягчена добавлением моторных белков, таких как кинезин-534, которые являются тетрамерами двигателей, используемых в митозе. В качестве альтернативы могут быть использованы искусственные димеры димерного кинезина-15.
Другим способом добавить больше текучести было бы позволить микротрубочкам выполнять свою динамическую нестабильность, рост и сокращение микротрубочек. В настоящее время микротрубочки, которые засеяны стабильными нитями GMPCPP, а затем подвергаются динамической нестабильности, намного длиннее, чем хотелось бы, чтобы сформировать шпиндель или тактоид, что приведет к очень длинным организациям, таким как вентиляторы или пучки. Таким образом, добавление динамической нестабильности микротрубочек должно быть сделано осторожно, чтобы сохранить тактоидную форму. Добавление связанных белков и ферментов, которые могут контролировать длину, может смягчить эту проблему. Например, вероятно, потребуются деполимеризирующие кинезины, такие как кинезин-1335, или разрывные ферменты, такие как катанин36. Эти эксперименты сложны и трудны, хотя они были бы очень проницательными, независимо от того, что показывают результаты. В каком бы направлении ни развивались будущие эксперименты, разработанная здесь платформа для создания тактоидов микротрубочек может выдавать новую информацию на физической основе организации микротрубочек.
Авторы заявляют, что у них нет конкурирующих финансовых интересов.
Авторы хотели бы поблагодарить всех членов Ross Lab летом 2021 года, особенно К. Элис Линдси, за их помощь. Эта работа была поддержана грантом от NSF BIO-2134215, который поддержал С. Саху, Н. Гудби, Х.Б. Ли и Дж.Л. Росса. Грант от Фонда KECK (Rae Anderson, USD, ведущий PI) частично поддержал Р. Бранча и. Чаухана
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
Tags
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved