
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Biochemistry
Autoensamblaje de tactoides de microtúbulos
Este artículo presenta un protocolo para la formación de conjuntos de microtúbulos en forma de tactoides utilizando MAP65, un reticulante de microtúbulos a base de plantas, y PEG como agente de apiñamiento.
El citoesqueleto es responsable de la principal organización interna y reorganización dentro de la célula, todo sin un gerente que dirija los cambios. Este es especialmente el caso durante la mitosis o meiosis, donde los microtúbulos forman el huso durante la división celular. El huso es la maquinaria utilizada para segregar el material genético durante la división celular. Hacia la creación de husos autoorganizados in vitro, recientemente desarrollamos una técnica para reconstituir microtúbulos en ensamblajes similares a husos con un conjunto mínimo de proteínas asociadas a microtúbulos y agentes de apiñamiento. Específicamente, se utilizó MAP65, que es un reticulador de microtúbulos antiparalelos de plantas, un homólogo de Ase1 de levadura y PRC1 de organismos de mamíferos. Este reticulante autoorganiza los microtúbulos en ensamblajes autoorganizados de microtúbulos largos, delgados y similares a husos. Estos ensamblajes también son similares a los tactoides de cristal líquido, y los microtúbulos podrían usarse como mesógenos de mesoescala. Aquí, se presentan protocolos para crear estos tactoides de microtúbulos, así como para caracterizar la forma de los ensamblajes utilizando microscopía de fluorescencia y la movilidad de los constituyentes utilizando la recuperación de fluorescencia después del fotoblanqueo.
La división celular a través de la mitosis es uno de los procesos biológicos más importantes para mantener la vida. Los filamentos de microtúbulos, compuestos por dímeros de tubulina, son elementos estructurales esenciales de este proceso. La maquinaria transitoria creada en metafase cuando los cromosomas se alinean en el centro celular se llama huso mitótico debido a su forma, que es como un huso de un telar cubierto de hilos (Figura 1A). Está bien establecido en muchos organismos que los microtúbulos se utilizan en la metafase para empujar y tirar de los cromosomas condensados en el centro de la célula, alineándolos y conectándolos a microtúbulos que los separarán en anafase (Figura 1B, C). El huso se forma tanto en la meiosis (Figura 1B) como en la mitosis (Figura 1C), creada a partir de muchos microtúbulos superpuestos que no están envueltos alrededor del eje central como hilo, sino que corren paralelos a la interfaz. La creación de estas estructuras basadas en microtúbulos requiere proteínas asociadas que se entrecruzan y enzimas asociadas que pueden actuar como motores para ayudar a empujar y tirar de los cromosomas1.
Los estudios de husos meióticos han demostrado que los microtúbulos son cortos, dinámicos y se superponen en matrices reticuladas 2,3,4,5,6 (Figura 1Di). Debido a la organización física de estos microtúbulos cortos, el huso meiótico es similar a un tactoide de cristal líquido (Figura 1E). De hecho, se ha demostrado que los husos se fusionan y se fusionan, como cabría esperar de los tactoides de cristal líquido5.
Muchos estudios que datan de la década de 1960 han utilizado la fijación, las secciones en serie y la microscopía electrónica para determinar que hay dos tipos de microtúbulos dentro del huso mitótico 7,8,9,10. El primer tipo se llama microtúbulos cinetocoros, que conectan el polo del huso con el cinetocoro. El segundo tipo se denomina microtúbulos interpolares o polares, que crecen más allá de los cromosomas y se superponen en la zona media (Figura 1Dii)8,9,10. Un tercer tipo se llama microtúbulos astrales, que están fuera del huso y conectan los polos con el borde celular; estas organizaciones de microtúbulos están fuera del alcance de la discusión actual. Ha habido estudios recientes sobre la interacción entre augmin6 y el complejo de anillos gamma-tubulina que influyen en los centros de nucleación de los microtúbulos, lo que resulta en un huso mitótico con microtúbulos más cortos como en la Figura 1D.
Dado que los microtúbulos son más largos que anchos, con una alta relación de aspecto y una alta rigidez, son como versiones ampliadas de moléculas de cristal líquido. En física de la materia blanda, los átomos y las moléculas se han aproximado utilizando interacciones mínimas para deducir los mecanismos físicos de las transiciones de fase, incluida la nucleación y la fusión de cristales11. Del mismo modo, los microtúbulos son objetos a mesoescala que son versiones ampliadas de moléculas de cristal líquido, lo que proporciona información sobre la física de la dinámica de los cristales líquidos, incluida la nucleación y el crecimiento de las fases nemáticas a partir de las isotrópicas. Además, como se discutió anteriormente, el huso meiótico muestra propiedades como las de un tactoide de cristal líquido, un estado nemático que nuclea y crece a partir del estado isotrópico de las moléculas de cristal líquido 3,4,5. Para los tactoides, la nucleación y el crecimiento son como los de otros cristales (es decir, requieren una concentración relativamente alta de mesógenos [las moléculas que forman los cristales líquidos]). La forma única de "huso" del tactoide proviene de la alineación local de los mesógenos de cristal líquido que se alinean en la fase nemática (Figura 1E). No pueden formar un cristal redondeado porque las moléculas son altamente asimétricas. Dada la naturaleza de los microtúbulos, tal vez no sea sorprendente que la maquinaria del huso mitótico hecha de una alta concentración local de microtúbulos también tenga la misma forma, ya sea que se llame tactoide o huso. Los tactoides pueden ser bipolares, con polos en los extremos cónicos (Figura 1Ei), u homogéneos, con polos efectivamente en el infinito (Figura 1Eii).
Dada la importancia de la formación del huso, se han realizado esfuerzos hacia la formación de husos autoorganizados in vitro mediante la demostración de condensación de microtúbulos en haces a través de especies iónicas 12,13, agentes de apiñamiento que crean interacciones de agotamiento14,15 y proteínas de reticulación de microtúbulos específicos 13,16,17,18,19, 21. Sorprendentemente, aunque todos estos agentes trabajan para aumentar la concentración local de microtúbulos, a menudo resultan en largos haces de microtúbulos pero no tactoides. Una razón por la que estos haces son largos podría ser que los microtúbulos que los componen también son largos. Trabajos recientes con microtúbulos más cortos también reportaron haces más largos que no están cónicos al final15; en este caso, los haces se mantienen unidos con proteínas motoras que causan la extensión de los haces y, por lo tanto, los hacen más largos. Se necesitan microtúbulos cortos con reticulantes no extensiles para ensamblajes cónicos en forma de husillo, como se describe aquí.
Recientemente, hemos desarrollado una técnica para permitir la creación de tactoides de microtúbulos utilizando el reticulador antiparalelo, MAP65, en presencia de microtúbulos estables nucleantes22. Los microtúbulos debían ser cortos, sin embargo, pocos reguladores conocidos de la longitud de los microtúbulos pueden tapar los microtúbulos contra la inestabilidad dinámica o el recocido de extremo a extremo. En cambio, GMPCPP se utilizó para nuclear y estabilizar los filamentos después del crecimiento. Esto permitió crear una alta densidad de microtúbulos cortos que podrían autoorganizarse en tactoides. Estos tactoides eran homogéneos cuando se veían bajo birrefringencia. Además de los microtúbulos cortos, se empleó un reticulador antiparalelo específico, MAP65, para formar los tactoides (Figura 2). MAP65 es una proteína asociada a microtúbulos vegetales de la familia PRC1/Ase1 de reticulantes mitóticos23. MAP65 existe como un dímero, con una fuerte afinidad para unirse a sí mismo, así como a los microtúbulos24. A diferencia del huso meiótico y los tactoides observados con los filamentosde actina 25,26,27, que son bipolares y tienen las propiedades líquidas de los cristales líquidos, se ha observado que los tactoides de microtúbulos son sólidossimilares a 22,28.
Aquí, se presentan protocolos para crear los tactoides de microtúbulos y caracterizar la forma de los ensamblajes y la movilidad de los constituyentes utilizando técnicas basadas en fluorescencia.
NOTA: A menos que se indique lo contrario, partes del experimento se pueden realizar en un banco de laboratorio mientras se usa el equipo de protección adecuado (guantes).
1. Silanización de coverslip
NOTA: Los coverslips deben estar silanizados para ser utilizados con el recubrimiento de cepillo de polímero empleado en estos experimentos. Este es un tratamiento de silanización hidrofóbico que permite que un copolímero de bloque con un bloque central hidrófobo se una y cree un cepillo de polímero. Los siguientes pasos deben realizarse en una campana de humos para evitar la exposición a vapores tóxicos mientras se usan guantes. El dimetildiclolsilano es altamente tóxico y debe manipularse con el máximo cuidado.
- Enjuague las fundas con ddH2O, 70% de etanol y ddH2O en orden. Séquelos con toallitas de laboratorio sin pelusa entre cada enjuague. Esto elimina el polvo y las partículas solubles en agua u orgánicas de la superficie antes del tratamiento.
- Coloque las cubiertas en un bastidor de sujeción de cubiertas metálicas y transfiera el bastidor a una máquina de ozono UV (UVO). Irradie las cubiertas con UVO durante 20 minutos para eliminar cualquier fluorescencia de fondo. Se puede utilizar una cámara de plasma en lugar de UVO.
- Usando pinzas, transfiera las cubiertas del estante de metal utilizado para el tratamiento de UVO a un estante de metal diferente utilizado para la silanización. No utilice los mismos bastidores para ambos, ya que provocará altos niveles de oxidación cuando se aplique UVO. Limpie previamente los bastidores con agua y etanol para que no queden productos químicos residuales de usos anteriores.
- Sumergir el bastidor con las fundas en un recipiente con 100% acetona durante 1 h. Enjuague el recipiente 3x con agua del grifo y luego 3x con ddH2O para eliminar toda la acetona.
- Sumerja el bastidor con las fundas en etanol al 100% durante 10 min. Enjuague el recipiente 3 veces con agua del grifo y luego 3 veces con ddH2O para eliminar todo el etanol.
- Sumerja el bastidor con las fundas 3x en ddH2O durante 5 min cada una.
- Sumerja el bastidor con las cubiertas en 0,1 M KOH (50 mL de 1 M KOH en 450 mL de ddH2O) durante 15 min. Enjuague el recipiente 3x con agua del grifo y luego 3x con ddH2O para eliminar todo KOH.
- Sumerja el bastidor con fundas 3x en ddH2O durante 5 min cada una.
- Seque al aire el bastidor con las cubiertas durante la noche en una campana de humos o una campana de flujo laminar.
- Después de secar completamente la rejilla y las cubiertas, sumérjalas durante 5 minutos en dimetildiclorosilano al 2% (DDS) tomado en un recipiente diferente que se usa específicamente para silano. No deje que nada que no esté seco entre en contacto con el silano.
- Sumergir el bastidor y las fundas 2x en un recipiente con etanol al 100% durante 5 min. Enjuague el recipiente 3x con agua del grifo y luego 3x con ddH2O.
- Sumerja el bastidor y las fundas 3x en ddH2O durante 5 min cada una.
- Seque al aire el bastidor con las cubiertas durante la noche en una campana de humos o una campana de flujo laminar.
- Después de este paso final de secado, transfiera las cubiertas de nuevo a las cajas de cubiertas con pinzas. Estos cubrehojas se pueden usar en los próximos 1-2 meses. Las cubiertas viejas comenzarán a perder su recubrimiento y deben desecharse.
2. Preparación de tubulina
NOTA: La tubulina comprada viene como un polvo liofilizado que no está etiquetado o etiquetado con fluoróforos. La tubulina liofilizada se almacena en un congelador de -80 °C. El siguiente procedimiento se utiliza para mezclar tubulina sin etiquetar con tubulina etiquetada en una proporción que es buena para la visualización.
- Retire del congelador -80 °C una alícuota de tubulina sin etiquetar que contenga 1 mg de polvo de tubulina liofilizada y manténgala en hielo. Añadir 200 μL de PEM-80 frío al tubo para llevar la concentración de tubulina a 5 mg/ml. Manténgalo en hielo durante 10 minutos para disolver todo el liofilato.
- Retire del congelador -80 °C una alícuota de tubulina marcada con rodamina que contenga 20 μg de polvo de tubulina liofilizada y manténgala en hielo. Añadir 4 μL de PEM-80 frío al tubo para llevar la concentración de tubulina a 5 mg/ml. Manténgalo en hielo durante 10 minutos para disolver completamente el liofilato.
- Una vez disuelto, agregue 100 μL de solución de tubulina resuspendida sin etiquetar a la solución de 4 μL de tubulina marcada con rodamina. Pipeta 6x-7x muy lentamente para mezclar. Si los agregados son visibles, centrifugue la tubulina solubilizada durante 10 min a 90,000 x g para eliminar los agregados desechando el pellet y reteniendo el sobrenadante. Esta mezcla de tubulina dará como resultado ~ 4% de tubulina etiquetada.
- Congele los 100 μL restantes de tubulina sin etiquetar en nitrógeno líquido (LN2) y guárdelo a -80 °C para usarlo en mezclas adicionales de tubulina.
- Tome la mezcla de tubulina etiquetada y alícuota en siete tubos con 15 μL cada uno. Cada alícuota se puede utilizar para una sola cámara experimental. Congela las alícuotas restantes y guárdalas a -80 °C para futuros experimentos.
3. Purificación MAP65
NOTA: MAP65 no está disponible comercialmente y, por lo tanto, necesita ser purificado para este trabajo. El protocolo ha sido elaborado previamente en varias publicaciones23,29.
- Transformar el plásmido MAP65 y el plásmido GFP-MAP65 en cepa de bacteria BL21 para la expresión de proteínas.
- Cultive las bacterias BL21 a una densidad óptica de 0.6-1 a 600 nm. Inducir la producción de proteínas utilizando el operador lac y cultivar las bacterias durante la noche.
- Pellet los cultivos y lisar las bacterias.
- Recoja el lisado después de la centrifugación e incube con perlas que tengan un ion de níquel disponible para unir la etiqueta 6x-histidina.
- Elute la proteína usando imidazol y desalarla.
- Congele la proteína con nitrógeno líquido y guárdela a -80 °C para su uso dentro de 1 año.
4. Montaje de cámaras de flujo
NOTA: Los experimentos se realizan en cámaras de flujo hechas de un portaobjetos de vidrio y vidrio de cubierta silanizada (Figura 3).
- Tome un portaobjetos de vidrio y límpielo con ddH2O, etanol y ddH2O en orden. Seque con una toallita de laboratorio sin pelusa entre cada enjuague.
- Utilice un trozo de cinta de doble cara para crear una ruta de flujo. Usando manos enguantadas, corte la cinta a ~ 25-30 mm de longitud. Divida la cinta longitudinalmente para crear dos tiras más delgadas. Coloque las dos tiras de cinta en la corredera con unos 5-8 mm entre ellas.
NOTA: Como el grosor de la cinta está estandarizado a aproximadamente 80-100 μm, el ancho del camino entre las piezas de cinta determinará el volumen en la cámara. - Coloque los cubrecubiertas silanizadas en la parte superior de la trayectoria del flujo. Selle la corredera y la cubierta a las tiras de cinta de doble cara presionando suavemente la región de la cinta con la parte posterior de un bolígrafo. Asegúrese de obtener un buen sello en toda el área; la cinta debe pasar de translúcida a transparente cuando el sello esté bien hecho.
- Retire la cinta adicional en los bordes, dejando solo 1 mm de la entrada de la cámara de flujo cortando la cinta con una cuchilla de afeitar.
- Etiquete la cámara con información sobre los parámetros experimentales, según sea necesario.
5. Experimentos tactoides
NOTA: Una vez que se generan todos los reactivos y suministros, se pueden utilizar para nuclear y polimerizar los tactoides de microtúbulos en la cámara de flujo.
- Recoger todos los reactivos a utilizar. Descongelarlos en hielo y almacenarlos en hielo mientras trabaja. Haga varias cámaras de flujo para los experimentos.
- Utilice una cámara de flujo para cada experimento. Cubra las superficies de la cámara de flujo con un cepillo de polímero fluyendo en 20 μL de surfactante de copolímero de bloque no iónico al 5% (Tabla de Materiales) disuelto en PEM-80, con pequeñas gotas en ambos extremos de la cámara para evitar la formación de burbujas de aire en el interior. Manténgalo en una cámara húmeda (es decir, placa de Petri con una toallita de laboratorio húmeda sin pelusa) hasta que esté listo para usar (al menos 5-7 min).
- En un tubo estéril, mezcle lo siguiente para crear la mezcla Tubulin-MAP: 9.5 μL de PEM-80; 4 μL de GMPCPP de 10 mM; 4 μL de Pluronic-F127 al 5%; 1 μL de TDT 1M; 1 μL de glucosa; 2 μL de polietilenglicol (PEG); 12 μL de mezcla de tubulina de 5 mg/ml (concentración final de 13,6 μM) de la etapa 2,5.; y 5,5 μL de stock de trabajo de MAP65 donde el 10% es GFP-MAP65 para visualización. Mantener en hielo mientras se mezcla.
NOTA: Se recomienda utilizar una pipeta de desplazamiento positivo para manipular la solución viscosa de PEG. Se pueden usar pipetas regulares después de cortar la punta para hacer que la abertura sea más grande; sin embargo, este método es menos preciso. - Mezclar 5x-6x mediante pipeteo.
- Justo antes de agregar a la cámara, agregue 1 μL de una solución premezclada de glucosa oxidasa (0.5 mg / ml) y catalasa (0.15 mg / ml) (Deoxy) en la mezcla Tubulin-MAP y mezcle 7x-8x. Divida el volumen total de la solución (40 μL) en dos porciones para utilizarlas en cámaras separadas.
- Fluya la mezcla Tubulin-MAP en cámaras. Como las cámaras ya tienen surfactante de copolímero de bloque no iónico en ellas, no se puede agregar más líquido sin eliminar el líquido viejo. Para hacer esto, use un trozo de papel de filtro o una toallita de laboratorio sin pelusa en el otro extremo de la cámara para eliminar el líquido a través de la acción capilar.
- Una vez que la muestra esté completamente dentro de la cámara, selle los dos extremos de la cámara con epoxi de 5 minutos y manténgala a 37 ° C durante ~ 30 minutos para nuclear y cultivar tactoides de microtúbulos.
6. Microscopía de fluorescencia
- Use un microscopio de fluorescencia para obtener imágenes de los tactoides.
NOTA: La microscopía de fluorescencia de reflexión interna total o la microscopía confocal de disco giratorio son buenas para eliminar la fluorescencia de fondo de la tubulina libre, pero los tactoides también son visibles por epifluorescencia regular e incluso microscopía de luz transmitida una vez que se forman, lo que hace que este procedimiento sea accesible sin equipo especializado. - Utilice un objetivo de apertura numérica (NA) de 1,2 NA o superior con un aumento de 60x o superior para recoger suficiente luz en fluorescencia. Estos objetivos a menudo requieren la inmersión en ddH2O o aceite.
- Grabe imágenes con cámara CMOS o CCD. Utilice un tamaño de píxel efectivo en la cámara de 108 nm.
NOTA: El tamaño de píxel depende de la cámara y del aumento utilizado, que en este caso fue de 60x o 100x con una apertura numérica alta (1.2 o 1.49 NA). Se pueden usar expansores de imagen adicionales antes de la cámara para lograr el tamaño de píxel necesario. - Mantenga la muestra a 37 °C utilizando una cámara ambiental ajustada a esta temperatura. Alternativamente, emplee otros calentadores de etapa, incluidos los calentadores de etapa de aire caliente y los collares objetivos de temperatura controlada con agua tibia circulante.
- Use fuentes de excitación que sean correctas para la fluorescencia necesaria. Para la rodamina tubulina, use un láser de 561 nm con al menos 1 mW de potencia en la muestra, y para GFP-MAP65, use un láser de 488 nm con al menos 1 mW de potencia en la muestra.
NOTA: Si usa microscopía de epifluorescencia de campo amplio, use un cubo de filtro de rodamina con excitación: 540 ± 12.5 nm, dicroico: 545 nm ± corte de 12.5 nm y emisión: paso largo de 575 nm, y un cubo de filtro GFP con excitación: 480 ± 15 nm, dicroico: 505 nm ± corte de 15 nm y emisión: paso largo de 515 nm. - Tome al menos 10 imágenes de diferentes áreas para obtener imágenes de más de 100 tactoides. Tome imágenes en los canales rojo y verde y guárdelas como imágenes tiff de 16 bits para su análisis. Asegúrese de que la potencia de iluminación y los tiempos de exposición sean tales que la escala de intensidad de la cámara no esté saturada.
7. Recuperación de fluorescencia después del fotoblanqueo (FRAP)
NOTA: Para estudiar la movilidad de los constituyentes internos de los tactoides, se utilizó FRAP. FRAP funciona fotoblanqueando una parte seleccionada de tubulina marcada con rodamina y tactoide MAP65 marcado con GFP y luego observando la recuperación de la fluorescencia con el tiempo en esa región. La tasa de recuperación depende de la rotación de la especie que se está fotoblanqueando. Esta tasa de rotación puede depender de la difusión y las reacciones de unión. Para map65 vinculante a los tactoides, se pueden estimar los tipos de cambio vinculantes. FRAP se realiza utilizando un sistema láser adicional de 405 nm que puede escanear el láser en cualquier forma. Hay muchas posibilidades para realizar FRAP, incluido el uso de la lámpara transmitida y la apertura para fotoblanquear un área local14.
- Seleccione un tactoide aislado en la cámara para crear una región de interés (ROI) que cubra partes de los tactoides y la solución circundante.
- Utilice un microscopio con un láser adicional de 405 nm para FRAP para fotobleach tanto la tubulina como el MAP65 simultáneamente. Alternativamente, se puede usar una lámpara brillante a través del tope de campo del iris14. Ajuste empíricamente la intensidad específica de los sistemas de fotoblanqueo para evitar dañar las proteínas durante el blanqueamiento.
- Grabe el tactoide como una película de serie temporal durante 30-60 s antes del fotoblanqueo para obtener información sobre la intensidad antes de la lejía. Graba los canales rojo y verde.
- Fotoblanquear el tactoide exponiendo el ROI con los láseres o la lámpara durante el tiempo que sea necesario para fotoblanquear sin dañar el tactoide. Determinar empíricamente la intensidad y el tiempo.
- Continúe grabando la película en ambos canales de color durante 5-10 minutos después del fotoblanqueo o hasta que la recuperación parezca haber alcanzado el equilibrio.
- Inspeccione visualmente el canal GFP-MAP65 para la recuperación.
8. Análisis de datos
NOTA: Se realizó un análisis cuantitativo de las imágenes de tactoides para conocer los efectos de los cambios ambientales impuestos a través de diferentes agentes de apiñamiento, las condiciones iónicas y la adición de otros factores.
- Caracterización de la forma tactoide
- Cuantificar la longitud y anchura de los tactoides a partir de las imágenes rojas y verdes tomadas con microscopía confocal.
- Abra las imágenes usando FIJI/ImageJ.
- Si los datos sin procesar se toman en 16 bits, ajuste el brillo y el contraste si es necesario. Seleccione Imagen > Ajustar > Brillo y Contraste para ajustar la imagen y poder ver el tactoide con claridad. Ajuste el brillo y el contraste sin aplicar la configuración para no alterar accidentalmente los datos de intensidad.
- Una vez que los tactoides sean claramente visibles, seleccione buenos tactoides para medir (Figura 4Bi). Asegúrese de que los tactoides sean claramente visibles sin superposición con ningún otro tactoide o agregado y que no estén curvados o doblados para poder usar herramientas de medición en línea recta.
- A continuación, compruebe que se ha establecido el tamaño de píxel correcto para las imágenes. Las imágenes del microscopio vienen con metadatos sobre el tamaño del píxel. Cuando utilice una cámara diferente que no tenga metadatos o sistemas de expansión de imágenes externas que puedan cambiar el tamaño de píxel efectivo esperado, ajuste el tamaño de píxel manualmente. En FIJI/ImageJ, vaya a Analizar > Establecer escala para establecer la conversión de píxeles correcta.
- Usando la herramienta Línea recta de la barra de herramientas en FIJI/ImageJ, haga clic en un extremo del tactoide y arrastre el cursor al otro extremo del tactoide (Figura 4Bii). Una vez seleccionado el ROI de la línea, seleccione Analizar > medir para medir la longitud. Si la longitud no se mide de forma predeterminada, asegúrese de establecer la medida para incluir la longitud en el cuadro de diálogo Analizar > Establecer medidas .
NOTA: Normalmente, cuando se mide con la herramienta Línea recta , se dará la longitud y el ángulo de la línea dibujada. Como ejemplo, la Figura 4Bii muestra una línea recta dibujada hacia el lado del tactoide para hacer visible este último, pero hacer la medición directamente sobre el tactoide. - Después de realizar la medición, utilice la herramienta Texto de la barra de herramientas para etiquetar el tactoide. Cree un cuadro de texto, agregue una etiqueta numérica y seleccione Editar > dibujar para fijar la etiqueta en la imagen. Guarde la imagen como un archivo ROI independiente.
NOTA: Etiquetar y guardar este archivo permite al investigador saber qué medida corresponde a qué tactoide a partir de los datos sin procesar. Asegúrese de medir cada tactoide una vez. - Después de medir los tactoides para toda la imagen, guarde los datos en la ventana Resultados en un archivo de texto delimitado por comas o tabulaciones (usando archivo > Guardar como) y abra los datos en un programa de hoja de cálculo para analizar los datos en números. Recopile todos los datos juntos (datos de imagen sin procesar, imagen ROI y archivo de texto de resultados) en una carpeta con una convención de nomenclatura adecuada para mantener todo organizado.
NOTA: Aunque las mediciones de la longitud del tacto se realizan a mano, dado que los anchos del tacto son estrechos, es mejor emplear un método diferente para medir el ancho del tactoide (ver más abajo) para reducir el error de medición. - Con ImageJ/FIJI, dibuje una región de línea con la herramienta Línea recta . Dibuje la línea como un bisector perpendicular al eje largo tactoide (Figura 4Bii).
- Seleccione Analizar > Perfil de trazado para crear el perfil de intensidad del bisector lineal (Figura 4Biii). Aparecerá una trama. Para recuperar y guardar los datos de la gráfica, seleccione el botón Lista en la parte inferior izquierda; esto genera la lista de archivos de texto de los datos de intensidad a lo largo de la longitud de la línea dibujada. Guarde el archivo de texto como un archivo .csv o .txt.
- Abra el archivo de texto en un programa de ajuste como MatLab, Python (sciPy) u otros programas. Ajuste los datos de intensidad con una función gaussiana de la forma:
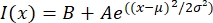 , donde I(x) es el valor de la escala de grises a lo largo de la longitud, x; B es el nivel de fondo; A es la amplitud del gaussiano; μ es la media o centro del gaussiano; y σ es la desviación estándar del gaussiano.
, donde I(x) es el valor de la escala de grises a lo largo de la longitud, x; B es el nivel de fondo; A es la amplitud del gaussiano; μ es la media o centro del gaussiano; y σ es la desviación estándar del gaussiano. - Informe 2σ como el ancho del tactoide. Estimar la intensidad de los microtúbulos en el tactoide calculando el área bajo el gaussiano (sin incluir el fondo).
NOTA: Si las imágenes están dentro del rango de intensidad lineal de la cámara y se toman con el mismo tiempo de exposición e intensidad de excitación, se pueden comparar intensidades integradas para estimar el número relativo de microtúbulos en el tactoide.
- Análisis FRAP
NOTA: Los experimentos para probar la movilidad de los microtúbulos y el MAP65 utilizaron FRAP para registrar el fotoblanqueo específico y la recuperación de la intensidad debido al movimiento molecular (Figura 5A). Los datos se cuantificaron a partir de los datos de la serie temporal de imágenes utilizando ImageJ/FIJI.- Utilice ImageJ/FIJI para abrir los datos de la película.
- Registre las pilas (datos de series temporales) a lo largo del tiempo para eliminar la deriva. Utilice el complemento StackReg junto con el complemento auxiliar TurboReg ; para obtener instrucciones sobre el uso de los complementos, consulte los enlaces web que se proporcionan en la Tabla de materiales. Seleccione la traducción para desplazar la posición de los fotogramas y así registrar las imágenes.
- Una vez que las imágenes están registradas para eliminar la deriva, gire la imagen para que el tactoide sea vertical u horizontal en el marco seleccionando Imagen > Transformar > Rotar. Seleccione el ángulo para girar y use Vista previa para determinar si el tactoide está lo suficientemente girado. Cuando la vista previa muestre que el tactoide es vertical u horizontal, seleccione Aceptar para girar todas las imágenes de la película.
- Utilice la herramienta de selección Rectángulo de la barra de herramientas para crear una sección rectangular sobre la región fotoblanqueada del tactoide. Registre la intensidad integrada del área de ROI para cada fotograma utilizando Image > Stacks > Measure Stack. Establezca el tipo de medición en Densidad integrada mediante Analizar > Establecer mediciones. Guarde los datos de intensidad analizados que se muestran en la ventana Resultados como un archivo de texto en formato .csv o .txt seleccionando Archivo > Guardar como.
NOTA: La Figura 5B muestra un ejemplo de los datos de intensidad de 16 bits en bruto medidos para los canales de microtúbulos y GFP-MAP65 en la región de lejía. - Como la intensidad general de las imágenes se desvanecerá con el tiempo a nivel mundial debido al fotoblanqueo causado por las imágenes, este fotoblanqueo global debe corregirse. Para ello, utilice el mismo tamaño de ROI (paso 8.2.4.) y muévalo a una región en el fondo de la imagen donde no se vean microtúbulos o MAP65. Mida la intensidad integrada de la pila como se describe en el paso 8.2.4. Guarde los resultados como un segundo archivo de texto.
NOTA: La Figura 5B muestra un ejemplo de los datos de intensidad sin procesar de 16 bits medidos para los canales de microtúbulos y GFP-MAP65 en la región de fondo. - Para corregir el desvanecimiento del fondo, divida la intensidad de la señal en el tactoide por la intensidad de fondo para el mismo punto de tiempo. Calcule Icorregido (t) como:
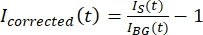 , donde IS (t) (la señal) es la medición realizada en la región blanqueada e IBG (t) (el ruido) es la medición realizada en la región de fondo (Figura 5C). Esto calcula la relación señal-ruido para cada fotograma y resta el ruido también.
, donde IS (t) (la señal) es la medición realizada en la región blanqueada e IBG (t) (el ruido) es la medición realizada en la región de fondo (Figura 5C). Esto calcula la relación señal-ruido para cada fotograma y resta el ruido también. - Luego, vuelva a escalar los datos para que oscilen entre cero y uno usando
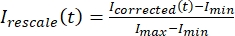 , donde Imin y Imax denotan, respectivamente, el mínimo global y el máximo de los datos corregidos por I durante todo el tiempo (Figura 5C).
, donde Imin y Imax denotan, respectivamente, el mínimo global y el máximo de los datos corregidos por I durante todo el tiempo (Figura 5C). - Ajuste estos datos a un exponencial decaimiento de la forma:
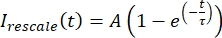 , donde A es la amplitud de la recuperación y τ es la escala de tiempo de la recuperación (Figura 5C).
, donde A es la amplitud de la recuperación y τ es la escala de tiempo de la recuperación (Figura 5C).
Con solo un pequeño número de componentes, dímeros de tubulina y reticulantes de microtúbulos, se pueden formar tactoides de microtúbulos (Figura 2A). Aunque este protocolo describe la incubación para nuclear y cultivar microtúbulos en una incubadora, la nucleación y el crecimiento se pueden observar directamente bajo el microscopio (que se completan en 30 min) (Figura 2B). La concentración de tubulina se mantiene constante a 13,6 μm y la unión MAP65-MT al 10%.
La figura 4 representa los datos correctos. Los tactoides deben ser visibles tanto con un láser de 561 nm en el canal de tubulina como con 488 nm en el canal MAP65, que se superponen perfectamente entre sí (Figura 4A). Un misterio del sistema ha sido que el ancho de los tactoides no parece variar bajo una variedad de cambios experimentales, incluido el cambio de las longitudes de los microtúbulos, la concentración de MAP65 y los agentes de apiñamiento (Figura 4B)22,28. La longitud es mucho más variable y depende tanto de las longitudes de los microtúbulos como de la concentración de MAP65 (Figura 4B)22,28.
Al realizar FRAP, se ha observado que la señal MAP65 se recupera, pero la señal de microtúbulos no se recupera (Figura 5). La recuperación en FRAP se debe a la movilidad y movimiento de los objetos etiquetados y fotoblanqueados. En el caso del MAP65, las moléculas oscurecidas se disocian y se alejan del microtúbulo y otras nuevas se mueven hacia la región (Figura 5). La unión MAP65 está en equilibrio, por lo que la tasa de unión y desunión es igual (medida en moléculas por segundo). Para los microtúbulos, no se observó recuperación, lo que implica que los microtúbulos no son capaces de abandonar el tactoide (Figura 5A, Bi, C). Además, no se observó la propagación de la región oscurecida, lo que sugiere que los microtúbulos están localmente inmóviles y no son un fluido dentro de la forma tactoide.
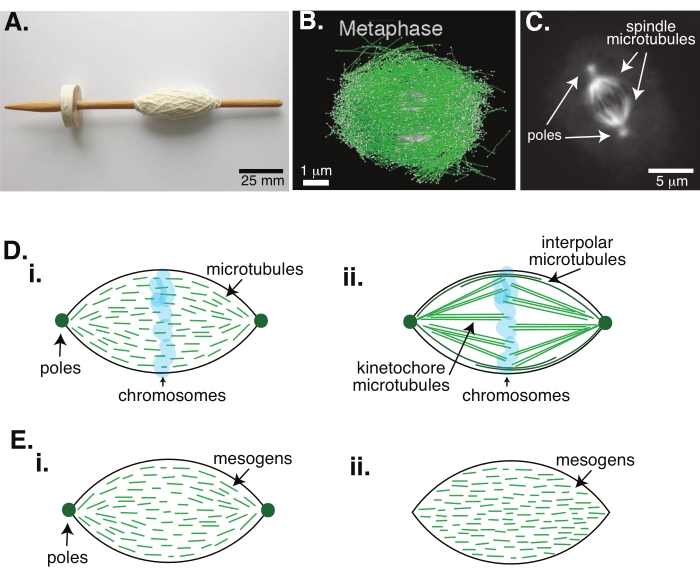
Figura 1: Diferentes modelos de formación de husos. Un huso mitótico es una máquina hecha de microtúbulos y sus proteínas y enzimas asociadas que alinea y separa los cromosomas en las dos nuevas células hijas durante la división celular. (A) Imagen de una réplica de husillo de caída de principios de la Edad Media con hilo fino de los Países Bajos. Esta figura ha sido modificada a partir de una imagen de Wikimedia por Peter van der Sluijs30. (B) Reconstrucción tridimensional de microtúbulos en diferentes etapas de la meiosis II de tipo silvestre. Los microtúbulos se muestran en verde y los cromosomas se muestran en gris. Barra de escala = 1 μm. Esta cifra ha sido modificada a partir de Lantzsch et al.31. (C) Imagen microscopía de los microtúbulos en un huso mitótico de una célula Sf9 en división. Los polos del huso y los microtúbulos del huso están marcados con una proteína verde fluorescente. Barra de escala = 5 μm. Esta cifra ha sido modificada a partir de Advani et al.32. (D) Diferentes modelos de cómo se organizan los microtúbulos del huso mitótico y meiótico. (i) Previamente observados para los husos meióticos creados a partir de extractos de huevos de Xenopus, se dedujo que los microtúbulos (verdes) eran cortos y dinámicos en todo el huso. Esto es similar a una organización tactoide bipolar dentro de un cristal líquido. (ii) El modelo canónico para la organización de microtúbulos dentro de un huso mitótico tiene dos tipos de microtúbulos: microtúbulos interpolares o polares (verde oscuro) que se entrecruzan en la zona media alrededor de los cromosomas y microtúbulos cinetocoros (verde claro) que se agrupan y estiran desde el polo hasta el cinetocoro para empujar y tirar de los cromosomas. En todas las imágenes, los cromosomas se muestran en azul transparente y los polos del huso se representan en verde oscuro. (E) Esquemas de mesógenos (líneas verdes) en un tactoide de cristal líquido para (i) tactoides bipolares y (ii) homogéneos. Los tactoides bipolares tienen dos polos al final del tactoide, y los mesógenos se reorientan para apuntar a esos polos. Los tactoides homogéneos tienen polos en el infinito, y los mesógenos no cambian de orientación a lo largo de la longitud del tactoide. Haga clic aquí para ver una versión más grande de esta figura.
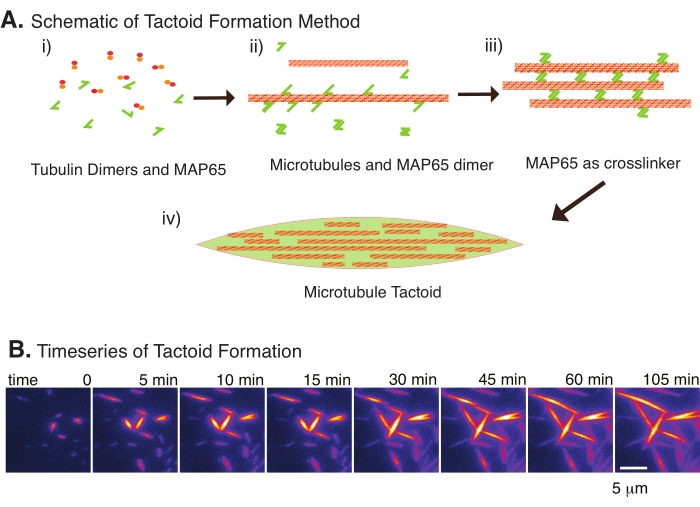
Figura 2: Condensación de microtúbulos. (A) Los microtúbulos pueden agruparse y reticularse mediante una variedad de métodos, incluidas especies iónicas, fuerzas de agotamiento causadas por agentes de apiñamiento y reticulantes de microtúbulos específicos, como MAP65. (i) Los dímeros de tubulina y las proteínas MAP65 se mezclan para nuclear y cultivar microtúbulos. (ii) Los microtúbulos se nuclean y crecen a partir de la tubulina, y MAP65 se une inmediatamente a los microtúbulos, otro monómero MAP65, o ambos y causa la agrupación. iii) Los microtúbulos de los haces reticulados nuclean y crecen. (iv) La configuración final es un tactoide de microtúbulos similar a un huso. (B) Series temporales de tactoides de microtúbulos que se nuclean y crecen durante 105 min. Barra de escala = 5 μm. Figura adaptada de Edozie et al.22. Haga clic aquí para ver una versión más grande de esta figura.
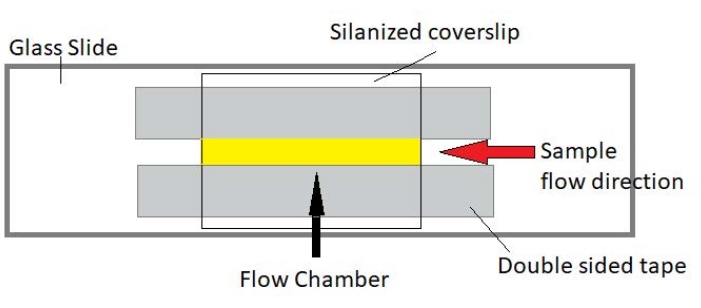
Figura 3: Montaje de la cámara de flujo. La cámara de flujo está hecha con un portaobjetos de vidrio, vidrio de cubierta silanizada y cinta permanente de doble cara. La región resaltada en amarillo es la trayectoria de flujo donde se fluye y se observa la muestra. El volumen de la cámara de flujo es de ~ 20 μL. El epoxi se utilizó para sellar los extremos de la cámara para evitar que la muestra se evapore durante las imágenes a largo plazo durante varias horas. Haga clic aquí para ver una versión más grande de esta figura.
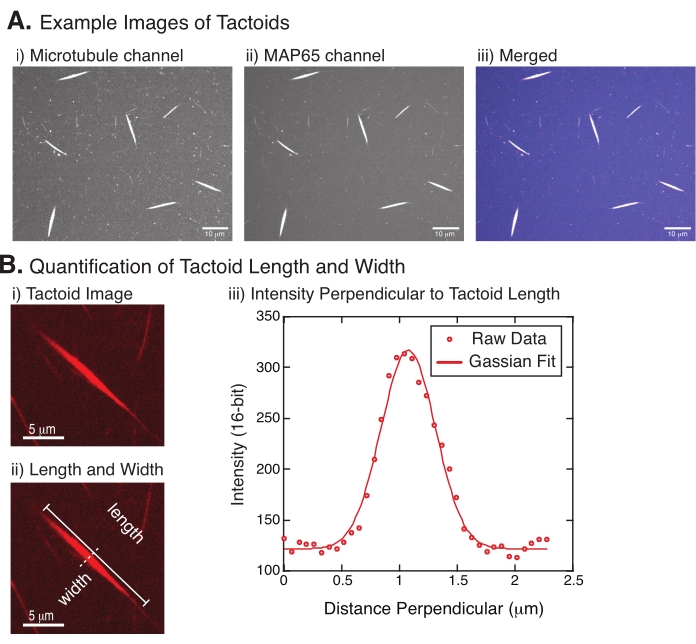
Figura 4: Imágenes tactoides y análisis de longitud y anchura. (A) Datos de ejemplo de tactoides formados como se describe y fotografiados utilizando confocales de disco giratorio que muestran (i) imágenes de canales de microtúbulos de tubulina marcadas con rodamina utilizando un láser de 561 nm, (ii) imágenes de canales GFP-MAP65 de GFP utilizando un láser de 488 nm, y (iii) imagen superpuesta combinada del canal de microtúbulos (magenta) y canal GFP-MAP65 (cian). Las regiones superpuestas se muestran como blancas y demuestran que los microtúbulos y MAP65 se colocalizan exactamente. Barra de escala = 10 μm para todas las imágenes en (A). (B) Cuantificación de la longitud y anchura del tacto. (i) Imagen de un tactoide para ser analizado sin etiquetas. Barra de escala = 5 μm. (ii) Misma imagen que en (i), donde se denotan las medidas de longitud (línea sólida con tapas de línea) y anchura (línea discontinua). Barra de escala = 5 μm. (iii) El ancho se midió tomando el perfil de intensidad a través del tactoide en el bisector perpendicular (línea discontinua) denotado en (ii). El perfil de intensidad se ajustó a una función gaussiana para revelar la amplitud y el ancho del tactoide. Haga clic aquí para ver una versión más grande de esta figura.
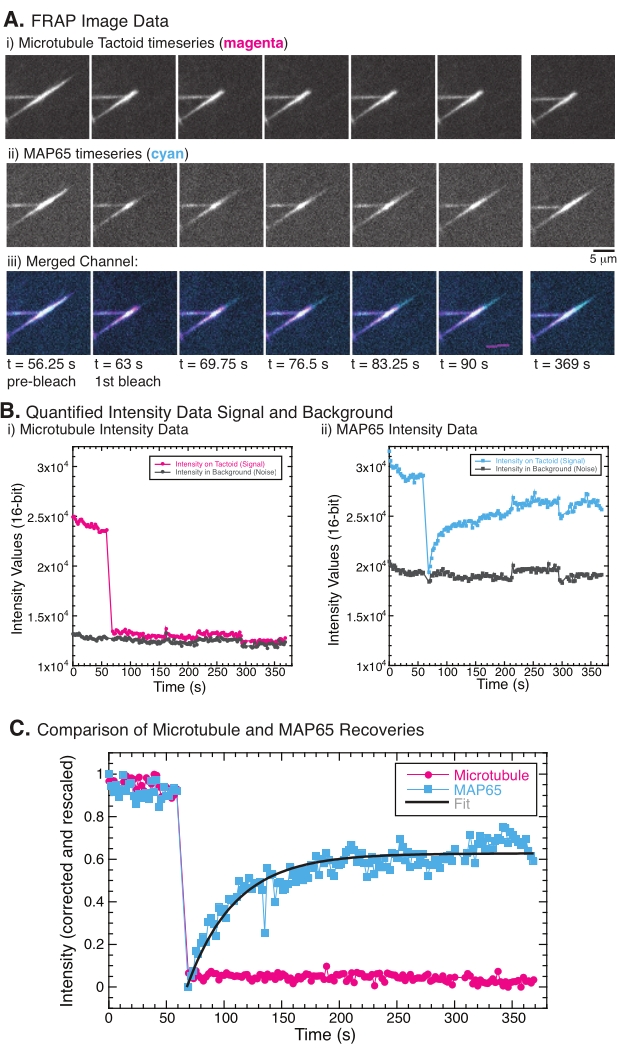
Figura 5: Datos y análisis representativos de FRAP. (A) Datos de series temporales de microscopía de (i) tactoide de microtúbulos y (ii) GFP-MAP65, y (iii) imagen superpuesta de ambos canales con microtúbulos en magenta y GFP-MAP65 en cian que fueron fotoblanqueados en el tiempo 63 s y observados durante 5 min. (B) Intensidad cuantificada de la (i) ) canal de microtúbulos en la región blanqueada (círculos magenta) y el fondo (círculos gris oscuros) y (ii) canal GFP-MAP65 en la región blanqueada (cuadrados cian) y el fondo (cuadrados gris oscuro). (C) Los datos se corrigieron para el ruido de fondo y se reescalaron para el canal de microtúbulos (círculos magenta) y el canal GFP-MAP65 (cuadrados cian). Los microtúbulos no se recuperan, pero el GFP-MAP65 sí lo hace y puede ajustarse (línea gris oscura) a una desintegración exponencial creciente para encontrar la amplitud y la escala de tiempo de recuperación. Haga clic aquí para ver una versión más grande de esta figura.
Los métodos aquí descritos se han utilizado en varios artículos para crear tactoides de microtúbulos (Figura 2)22,28. Estos experimentos son biológicamente relevantes para ayudar a descubrir los principios organizativos que controlan la forma y la estabilidad del huso mitótico o meiótico en la mayoría de los tipos de células. Además, los microtúbulos son mesógenos de cristal líquido modelo que pueden ayudar a aprender más sobre cómo los cristales líquidos nuclean y crecen las fases nemáticas a partir de las fases isotrópicas.
El procedimiento descrito aquí tiene varias ventajas para explorar la autoorganización de microtúbulos. En primer lugar, es altamente reproducible, ya que ha sido realizado en el laboratorio por muchos estudiantes, incluidos los estudiantes de secundaria, con poco conocimiento previo o capacitación antes de comenzar en el laboratorio. Los tactoides son birrefringentes22, lo que les permite ser vistos en luz transmitida además de la microscopía de fluorescencia, haciendo que este método sea accesible a muchos laboratorios y este procedimiento experimental adaptable a fines educativos, además de la investigación de alta gama. Finalmente, este proceso abre vías para continuar entendiendo y sondeando los sistemas biológicos en un enfoque reduccionista y despojado, lo que permite comprender cómo cada condición adicional, proteína o aditivo puede alterar la autoorganización de los tactoides y, tal vez, en última instancia, el huso. Los objetivos para una mejor biomímesis incluyen la actividad, la fluidez y la clasificación de la polaridad del filamento.
Puede haber varios factores que afectan el experimento dando resultados inesperados. Por ejemplo, si los tactoides no se forman (Figura 2) pero se observan patrones similares a los de un abanico, es probable que el MAP65 no esté presente o no se una a los microtúbulos22,28. Esto también debería ser obvio en el canal de fluorescencia MAP65 porque el GFP-MAP65 no estará unido a los microtúbulos.
Si los tactoides no se forman y el fondo aparece como motas en el vidrio, esto podría deberse al recubrimiento de la superficie. Una vez realizada, la silanización dura solo 1 mes en coverslips. Cuando desaparezca, la tubulina podrá unirse a la superficie expuesta de forma no específica. Este enlace se producirá en patrones impares.
Si los tactoides no se forman y la tubulina se observa en agregados de varias formas y tamaños, esto podría deberse a la tubulina de mala calidad. La tubulina se puede centrifugar para eliminar los agregados iniciales que pueden impulsar esta agregación fuera de la vía en lugar de la polimerización de microtúbulos. Si la superficie se une a la tubulina, también puede agotar la tubulina en la solución. Las bajas concentraciones de tubulina, por debajo de la concentración crítica para polimerizar microtúbulos, pueden dar lugar a agregados.
En los experimentos FRAP, si el canal MAP65 no muestra ninguna recuperación (Figura 5), es posible que el fotoblanqueo estuviera fotodestamizando los microtúbulos. El fotodaño provoca la destrucción localizada de los filamentos. Esto se puede verificar mediante un examen en el canal transmitido. Los tactoides de microtúbulos son visibles en el canal transmitido a través de un alto índice de desajuste de refracción con el agua circundante. El fotodaño inducido por la luz aparecerá como una marca de quemadura o pérdida de contraste en las imágenes de luz transmitidas en la ubicación del ROI sometido a fotoblanqueo. Si esto ocurre, la potencia del láser o de la luz debe reducirse para inhibir el fotodaño de las proteínas.
Hubo varios desafíos encontrados en este procedimiento y enfoque. Un problema es que las medidas de longitud se realizan actualmente a mano haciendo clic en la imagen. Este método, aunque sencillo, puede resultar en una alta incertidumbre. La medición de anchura que utiliza la sección transversal y el ajuste a un gaussiano es un mejor método para cuantificar el tamaño. Se podría emplear un método similar para la longitud. Un segundo problema es que, a veces, los tactoides, debido a que son tan largos y delgados, pueden doblarse. Esto hace que la cuantificación de la longitud sea más difícil. La longitud del contorno se puede cuantificar utilizando una línea segmentada, pero hay incertidumbre adicional cada vez que se agrega un segmento.
Desde una perspectiva científica, este enfoque tiene algunos otros desafíos para su uso como modelo para cristales líquidos o husos. El primer desafío ha sido la forma larga y delgada de los tactoides que crean los microtúbulos (Figura 3 y Figura 4). Como se señaló en publicaciones anteriores22, los tactoides de microtúbulos son tactoides homogéneos, no bipolares. Esto significa que los microtúbulos que componen la forma no se reorientan para apuntar hacia las puntas de la estructura. En cambio, todos los microtúbulos son paralelos al eje largo y los "polos" se encuentran en el infinito. Esto es muy diferente de los tactoides observados para cristales líquidos moleculares o incluso para actina o ADN que también pueden actuar como mesógenos de cristal líquido. En estos otros sistemas, los tactoides son bipolares y, cuando se ven en polarizadores cruzados, muestran los signos reveladores de reorientación de las varillas.
Un segundo gran desafío en este sistema es que los microtúbulos están inmóviles dentro del tactoide. Esto queda claro a partir de los experimentos y análisis de FRAP, ya que la recuperación de los microtúbulos es muy baja. Su naturaleza sólida hace que los tactoides de microtúbulos sean menos valiosos que los análogos de cristal líquido a gran escala. La fase nemática de un cristal líquido debe tener propiedades líquidas (fluidas) y cristalinas (organizadas). Aunque la forma parece adecuada para el huso, la inmovilidad hace que el sistema sea menos emocionante como un huso mitótico modelo. Por otro lado, este número brinda oportunidades para investigar cómo se pueden modificar los experimentos para crear más fluidez en el sistema.
Estos desafíos científicos ofrecen oportunidades emocionantes que permitirán nuevos conocimientos sobre el sistema. Para hacer que los tactoides de microtúbulos sean más bipolares, se podrían usar microtúbulos más cortos. Sin embargo, hay un desafío adicional, ya que los microtúbulos no tienen muchas proteínas de recubrimiento bien caracterizadas para controlar la longitud como lo hace la actina. El uso de nucleación y crecimiento requiere el uso de concentraciones muy altas de tubulina y GMPCPP para hacer microtúbulos cortos. La alta concentración de tubulina da como resultado un mayor número de filamentos en el sistema, lo que hace que sea más difícil separar los tactoides entre sí. La adición de nuevas tapadoras de microtúbulos, como DARPin33, puede ayudar con esta situación. El segundo problema de que los microtúbulos estén inmóviles podría mitigarse mediante la adición de proteínas motoras, como la quinesina-534, que son tetrámeros de motores utilizados en la mitosis. Alternativamente, se podrían usar dímeros artificiales de kinesina-1 dimérica15.
Otra forma de agregar más fluidez sería permitir que los microtúbulos realicen su inestabilidad dinámica, el crecimiento y la contracción de los microtúbulos. Actualmente, los microtúbulos que se siembran con filamentos GMPCPP estables y luego sufren inestabilidad dinámica son mucho más largos de lo deseado para formar un huso o tactoide, lo que daría como resultado organizaciones muy largas como ventiladores o paquetes. Por lo tanto, la adición de inestabilidad dinámica de microtúbulos tendría que hacerse con cuidado para preservar la forma tactoide. La adición de proteínas y enzimas asociadas que pueden controlar la longitud puede mitigar este problema. Por ejemplo, probablemente se necesitarían quinesinas despolimerizantes, como la kinesina-1335, o enzimas cortadoras, como la katanina36. Estos experimentos son complejos y difíciles, aunque serían muy perspicaces sin importar lo que revelen los resultados. Cualquiera que sea la dirección que tomen los experimentos futuros, la plataforma desarrollada aquí para crear tactoides de microtúbulos puede exponer nueva información sobre la base física de la organización de microtúbulos.
Los autores declaran que no tienen intereses financieros contrapuestos.
Los autores desean agradecer a todos los miembros del Ross Lab del verano de 2021, especialmente a K. Alice Lindsay, por su ayuda. Este trabajo fue apoyado por una subvención de NSF BIO-2134215 que apoyó a S. Sahu, N. Goodbee, H.B. Lee y J.L. Ross. Una subvención de la Fundación KECK (Rae Anderson, USD, PI principal) apoyó parcialmente a R. Branch y P. Chauhan
| Name | Company | Catalog Number | Comments |
| 2% Dichlorodimethylsilane | GE Healthcare | 118945 | A hydrophobic silane surface treatment. This can be resused upto three times and kept at room temperature for one year. |
| 5-minute epoxy | Bob Smith Industries | For sealing experimental chambers | |
| Acetone | Fisher | 32900HPLC | 100% |
| BL21 cells | Bio Labs | C2527I | Competent bacterial cells used to express MAP65 |
| Catalase | Sigma | C30-500MG | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C for upto one year. |
| Coverslips (22 mm x 22 mm or 22 mm x 30 mm) | Fisher | 12544-AP | For experimental chambers |
| Dithiothreitol | Sigma | 43815-5G | A small molecule that is used to break disulfide bonds and scavenge oxygen. A 1 M stock is made from powder (Sigma) in ddH2O. The solution is aliquoted and stored at -20C for upto one year. The aliquots are used 7-8 times then discarded. |
| EGTA | Sigma | E3889-10G | Tubulin buffer base ingredient |
| Eppendorf 0.5 ml tubes | Eppendorf | 05 402 18 | For holding experimental solutions |
| Ethanol | Fisher | 111000200 | 200 proof |
| Filter paper | Whatman | 1004-110 | For cleaning and for pulling solutions through experimental chambers |
| Fluorescence microscope with high NA objectives | Nikon | Ti-E, W2 Confocal | For imaging experiments |
| Freezer -20°C | Fisherbrand | 13986148 | For storing reagents |
| Freezer -80°C | Thermo scientific | 328223H01-C | For storing proteins. |
| Glass containers for silanization | Michaels | For preparing experimental chambers - treating cover glasses | |
| Glass slides | Fisher | 12544-4 | For experimental chambers |
| Glucose | Sigma | G7528-250G | Part of the oxygen scavenging system. A 300 mg/ml stock is made in ddH2O and stored in 10 μl aliquots in -20°C. |
| Glucose oxidase | Sigma | G2133-250KU | Part of the oxygen scavenging system. This is stored at 4°C for upto one year. |
| GMPCPP | Jenna Bioscience | NU-4055 | Slowly hydrolyzable analog of GTP used to polymerize and stabilize the microtubules by reducing the dynamic instability and spontaneous critical concentration for microtubule nucleation to get a consistent length. We purchase a 10 mM stock from Jena Biosciences and store it at -20°C for upto one year. |
| Heating element for microscope | Okolab stage top incubator | For imaging experiments | |
| Imidizole | Sigma | I2399 | Used to elute MAP65 protein from Nickel beads to purify MAP65 |
| Kimwipes | Fisher | 34155 | For cleaning and for pulling solutions through experimental chambers. |
| KOH | Sigma | P250-500 | 1 M in ddH2O, made fresh to prevent acidification over time. |
| Liquid Nitrogen | Airgas | NI 230LT22 | To drop freeze aliquots. This can also be used for storage (not recommended) |
| MAP65 protein | Ram Dixit | A microtubule-associated protein (MAP) with a molecular weight of 65 kD that is an antiparallel microtubule crosslinker. We have purified an unlabeled and GFP-labeled version of MAP65-1 from Arabidopsis thaliana. We mix the GFP-MAP65 with the unlabeled MAP65 such that 10% of the protein is labeled. The working stock is a 10.8 μM solution. This working solution is stored at 4°C and remade fresh every week. The protocol for purifying the MAP65 and GFP-MAP65 is given in our prior methods chapter (26). | |
| MgSO4 | Sigma | MKCJ940 | Tubulin buffer ingredient |
| NTA-Nickel beads | Qiagen | 30210 | Beads required to purify 6xHis tagged MAP65 proteins |
| Optomicroscan | Nikon | 405 nm laser system which can focus the laser in any desired shape of region of interest | |
| PEM80 | Neutral tubulin polymerizing base buffer for solution made from 80 mM K-PIPES, pH 6.8, 1 mM MgSO4, 1 mM EGTA, stored at 4°C for upto one year. | ||
| Permanent double-sided tape | 3M - Scotch | 34-8724-5691-7 | For experimental chambers |
| Petri dish | Fisher | FB0875713 | For humid chamber |
| PIPES | Sigma | P7643-100G | Tubulin buffer base ingredient |
| Pluronic-F127 | Sigma | P2443-250G | A block-copolymer with two hydrophilic polyethylene oxide (PEO) blocks on the ends and a hydrophobic center block of polyphenylene oxide (PPO) hydrophobic surface coating we use to prevent protein binding to the surface. Pluronic-F127 is purchased as a powder (Sigma) and dissolved to a 5% (w/v) solution in ddH2O overnight. Once dissolved, the solution can be stored at room temperature for upto one year. |
| Polyethylene Glycol | Affymetrix Inc | 19966 500 GM | A crowding agent used to create depletion forces to bring tactoids to the surface and help to organize microtubules. We create a 5% (w/v) solution of 100 kDa PEG in PEM80. The solution is stored in 4°C for upto one year and placed on the rocker before use. It is viscous, so we recommend using a positive displacement pipette to work with it. |
| Positive displacement pipette | Eppendorf | For pipetting viscous liquids | |
| Racks for coverslips | Electron Microscopy Sciences | 72240 | For preparing experimental chambers - treating cover glasses |
| Refrigerator 4°C | Fisherbrand | For storing reagents | |
| Tubulin Labeled | Cytoskeleton | TL590M | lyophilized rhodamine-labeled tubulin from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| Tubulin protein | Cytoskeleton | T240 | lyophilized tubulin 99% pure unlabeled from pig brain is purchased (Cytoskeleton) and stored in -80°C until hydrated in PEM80 and used for upto one year. |
| UV-Ozone | Jelight | Model 342 | For preparing experimental chambers - treating cover glasses |
| Stackreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/stackreg/ | plugin for registering time series data to remove drift |
| Turboreg | Biomedical Imaging Group | http://bigwww.epfl.ch/thevenaz/turboreg/ | plugin for registering time series data to remove drift |
- Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C., Janke, C. Microtubule-associated proteins: structuring the cytoskeleton. Trends in Cell Biology. 29 (10), 804-819 (2019).
- Severson, A. F., von Dassow, G., Bowerman, B. Oocyte meiotic spindle assembly and function. Current Topics in Developmental Biology. 116, 65-98 (2016).
- Brugués, J., Needleman, D. Physical basis of spindle self-organization. Proceedings of the National Academy of Sciences of the United States of America. 111 (52), 18496-18500 (2014).
- Brugués, J., Nuzzo, V., Mazur, E., Needleman, D. J. Nucleation and transport organize microtubules in metaphase spindles. Cell. 149 (3), 554-564 (2012).
- Gatlin, J. C., et al. Spindle fusion requires dynein-mediated sliding of oppositely oriented microtubules. Current Biology. 19 (4), 287-296 (2009).
- Goshima, G., Mayer, M., Zhang, N., Stuurman, N., Vale, R. D. Augmin: A protein complex required for centrosome-independent microtubule generation within the spindle. Journal of Cell Biology. 181 (3), 421-429 (2008).
- Inoue, S., Sato, H. Cell motility by labile association of the nature of mitotic spindle fibers and their role in chromosome movement molecules. Journal of General Physiology. 50 (6), 259-292 (1967).
- Allenspach, A. L., Roth, L. E. Structural variations during mitosis in the chick embryo. Journal of Cell Biology. 33 (1), 179-196 (1967).
- Theg, D. E. Cytoplasmic microtubules in different animal cells. Journal of Cell Biology. 23 (2), 265-275 (1964).
- Mcdonald, K., Pickett-Heaps, J. D., Mcintosh, J. R., Tippit, D. H. On the mechanism of anaphase spindle elongation in Diatoma vulgare. Journal of Cell Biology. 9, 377-388 (1977).
- Chaikin, P. M., Lubensky, T. C. . Principles of Condensed Matter Physics. , (1995).
- Hamon, L., Savarin, P., Curmi, P. A., Pastré, D. Rapid assembly and collective behavior of microtubule bundles in the presence of polyamines. Biophysical Journal. 101 (1), 205 (2011).
- Needleman, D. J., et al. Higher-order assembly of microtubules by counterions: From hexagonal bundles to living necklaces. Proceedings of the National Academy of Sciences. 101 (46), 16099-16103 (2004).
- Ross, J. L., Fygenson, D. K. Mobility of taxol in microtubule bundles. Biophysical Journal. 84, 3959-3967 (2003).
- Sanchez, T., Welch, D., Nicastro, D., Dogic, Z. Cilia-like beating of active microtubule bundles. Science. 333 (6041), 456-459 (2011).
- Brandt, R., Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. Journal of Biological Chemistry. 268 (5), 3414-3419 (1993).
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Current Opinion in Cell Biology. 6 (1), 74-81 (1994).
- Kanai, Y., et al. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. The Journal of Cell Biology. 109 (3), 1173 (1989).
- MacRae, T. H. Microtubule organization by cross-linking and bundling proteins. Biochimica et Biophysica Acta (BBA). Protein Structure and Molecular Enzymology. 1160 (2), 145-155 (1992).
- She, Z. Y., Wei, Y. L., Lin, Y., Li, Y. L., Lu, M. H. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biological Reviews. 94 (6), 2033-2048 (2019).
- Walczak, C. E., Shaw, S. L. A MAP for Bundling Microtubules. Cell. 142 (3), 364-367 (2010).
- Edozie, B., et al. Self-organization of spindle-like microtubule structures. Soft Matter. 15 (24), 4797-4807 (2019).
- Tulin, A., McClerklin, S., Huang, Y., Dixit, R. Single-molecule analysis of the microtubule cross-linking protein MAP65-1 reveals a molecular mechanism for contact-angle-dependent microtubule bundling. Biophysical Journal. 102 (4), 802-809 (2012).
- Chan, J., Jensen, C. G., Jensen, L. C. W., Bush, M., Lloyd, C. W. The 65-kDa carrot microtubule-associated protein forms regularly arranged filamentous cross-bridges between microtubules. Proceedings of the National Academy of Sciences of the United States of America. 96 (26), 14931-14936 (1999).
- Scheff, D. R., et al. Tuning shape and internal structure of protein droplets via biopolymer filaments. Soft Matter. 16 (24), 5659-5668 (2020).
- Weirich, K. L., et al. Liquid behavior of cross-linked actin bundles. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2131-2136 (2017).
- Weirich, K. L., Dasbiswas, K., Witten, T. A., Vaikuntanathan, S., Gardel, M. L. Self-organizing motors divide active liquid droplets. Proceedings of the National Academy of Sciences of the United States of America. 166 (23), 11125-11130 (2019).
- Sahu, S., Herbst, L., Quinn, R., Ross, J. L. Crowder and surface effects on self-organization of microtubules. Physical Review E. 103 (6), 062408 (2021).
- Stanhope, K. T., Ross, J. L. Microtubules, MAPs, and motor patterns. Methods in Cell Biology. 128, 23-38 (2015).
- Early Middle Ages drop single.jpg. Wikimedia Commons Available from: https://commons.wikimedia.org/wiki/File:Early_middle_ages_drop_spingle.jpg (2022)
- Lantzsch, I., et al. Microtubule reorganization during female meiosis in c. Elegans. eLife. 10, 58903 (2021).
- Advani, S., Maresca, T. J., Ross, J. L. Creation and testing of a new, local microtubule-disruption tool based on the microtubule-severing enzyme, katanin p60. Cytoskeleton. 75 (12), 531-544 (2018).
- Pecqueur, L., et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proceedings of the National Academy of Sciences of the United States of America. 109 (30), 12011-12016 (2012).
- Valentine, M. T., Fordyce, P. M., Krzysiak, T. C., Gilbert, S. P., Block, S. M. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nature Cell Biology. 8 (5), 470-476 (2006).
- Wagenbach, M., Domnitz, S., Wordeman, L., Cooper, J. A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. Journal of Cell Biology. 183 (4), 617-623 (2008).
- McNally, F. J., Vale, R. D. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 75 (3), 419-429 (1993).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved